

Abstract
Infections by Candida albicans and related fungal pathogens pose a serious health problem for immunocompromised patients. Azole drugs, the most common agents used to combat infections, target the sterol biosynthetic pathway. Adaptation to azole therapy develops as drug-stressed cells compensate by upregulating several genes in the pathway, a process mediated in part by the Upc2 transcription factor. We have implemented a cell-based high-throughput screen to identify small-molecule inhibitors of Upc2-dependent induction of sterol gene expression in response to azole drug treatment. The assay is designed to identify not only Upc2 DNA binding inhibitors but also compounds impeding the activation of gene expression by Upc2. An AlphaScreen assay was developed to determine whether the compounds identified interact directly with Upc2 and inhibit DNA binding. Three compounds identified by the cell-based assay inhibited Upc2 protein level and UPC2-LacZ gene expression in response to a block in sterol biosynthesis. The compounds were growth inhibitory and attenuated antifungal-induced sterol gene expression in vivo. They did so by reducing the level of Upc2 protein and Upc2 DNA binding in the presence of drug. The mechanism by which the compounds restrict Upc2 DNA binding is not through a direct interaction, as demonstrated by a lack of DNA binding inhibitory activity using the AlphaScreen assay. Rather, they likely inhibit a novel pathway activating Upc2 in response to a block in sterol biosynthesis. We suggest that the compounds identified represent potential precursors for the synthesis of novel antifungal drugs.
INTRODUCTION
Candida species cause systemic infections in the immunocompromised, such as HIV patients, those receiving cancer chemotherapy, and organ transplant patients (1–5). Systemic fungal infections are a mortality factor for individuals with HIV (4), and affect the quality of life of elderly diabetics (6, 7). The fungal sterol biosynthetic pathway is the target for many of the commonly administered antifungal drugs (see Fig. 1A) (8–11). There are several classes of sterol pathway targeting drugs, the most common being the azoles, which includes the N-substituted imidazoles fluconazole and miconazole and the new triazole derivatives itraconazole and posaconazole (12). In addition, there are allylamines (terbinafine) (13) and morpholine classes of drugs (fenpropimorph) (14), as well as sterol binders (amphotericin B and nystatin) (15). All are intended to reduce sterol levels or sequester sterol. Finally, there are the echinocandins that inhibit β-glucan synthase, which blocks cell wall biosynthesis (16, 17).
FIG 1.

(A) Schematic depicting the yeast ergosterol pathway. The arrows represent individual enzymatic steps in the biosynthetic pathway. Steps inhibited by an antifungal compound are indicated. (B) Schematic of the critical path used for identifying compound inhibitors of Upc2. All compounds isolated that were positive in the UPC-LacZ assay for inhibition were tested in the PGK1-LacZ assay. The compounds that had positive results in the whole-cell assay that did not inhibit PGK1-LacZ activity were screened in the AlphaScreen assay. Compounds carried forward were assayed for inhibiting Upc2-dependent transcription factor activity. Positive compounds remaining were tested for inhibitory synergy with fluconazole. (C) Schematic of the UPC2 promoter indicating the location of each SRE. WT, wild type; MUT, mutant.
Saccharomyces cerevisiae, Candida albicans, and Candida glabrata produce ergosterol as the end product of sterol biosynthesis rather than cholesterol (18). The ergosterol biosynthetic pathway is regulated under various growth conditions by multiple transcription factors (TFs) (19–24). Upc2 is a member of the fungus-conserved Zn2-Cys6 binuclear cluster TFs (25). There is a homolog of S. cerevisiae Upc2 (ScUpc2), ScEcm22 (26, 27), a single Candida albicans ortholog, C. albicans Upc2 (CaUpc2) (28), and two C. glabrata Upc2 (CgUpc2) orthologous isoforms in C. glabrata (29). Upc2 induces ergosterol biosynthetic gene expression in response to azole antifungal drug treatment (30). It does so by binding to yeast sterol response elements (SREs) in the promoters of sterol genes (26, 30–35). Strains lacking Upc2 and Ecm22 are sensitive to antifungal treatment and are themselves growth compromised (28). Clinical isolates with gain-of-function mutations in CaUPC2 are resistant to azole therapies (35–38).
There are multiple reviews discussing the feasibility of HTS (high-throughput screen) identification of compounds directly interacting with TFs and their therapeutic use (39–42). Although this area has met with limited success, many of the compounds isolated show surprising selectivity, and in some cases in vivo efficacy (43–46). The Upc2 transcription factor is a critical regulator of the antifungal drug response, and as such represents an excellent target for antifungal drug discovery. Antifungal resistance requires induced sterol gene expression. Upc2 inhibition would eliminate the most downstream event required for resistance, thus circumventing a number of present resistant mechanisms.
We have developed two independent high-throughput screens aimed at identifying small-molecule Upc2 inhibitors. One is a homogenous whole-cell assay that screens for compounds reducing fluconazole-induced UPC2-LacZ gene expression. The second is an in vitro AlphaScreen assay (PerkinElmer) designed to determine whether compounds directly or indirectly inhibit Upc2 DNA binding. Our efforts have resulted in the identification of three small-molecule compounds inhibiting Upc2-dependent transcriptional signaling in vivo, while also reducing the level of Upc2 that is observed in response to a block in sterol biosynthesis (47).
MATERIALS AND METHODS
Yeast strains and miscellaneous methods.
Saccharomyces cerevisiae strains were constructed in the W303 strain background (MATa ura3-52 leu2 his3 lys2 ade2). The Candida albicans strain used was BWP17 (ura3/ura3 arg4/arg4 his1/his1). The wild-type Candida glabrata strain is 66032 (American Tissue Culture Collection). Strains were grown in YEPD (1% yeast extract, 2% Bacto peptone, 2% glucose) or in synthetic minimal medium (0.67% yeast nitrogen base [Difco]) supplemented with the appropriate amino acids and adenine. For the screen, fluconazole was added directly to liquid media. Yeast transformation was performed using the procedure described by Ito et al. (48). For routine propagation of plasmids, Escherichia coli XL1-Blue cells were used and grown in LB medium supplemented with ampicillin (150 μg/ml). Fluconazole and dimethyl sulfoxide were obtained from Sigma-Aldrich (St. Louis, MO, USA). Microtiter plates were purchased from Greiner Bio-One (Monroe, NC, USA), and cell strainers (70 μm) were purchased from Becton, Dickinson (Franklin Lakes, NJ, USA). VB00075177 (compound 1), VB00075853 (compound 2), and VB00075845 (compound 3) were purchased from TimTec for rescreening.
Plasmid constructions for HTS.
The yeast integrating plasmid, YIp353, was used to construct all plasmids. YIp353-UPC2-LacZ contains a wild-type ScUPC2-LacZ promoter fusion containing two SREs that ScUpc2 binds to in order to regulate fluconazole-induced transcription (67). Seven hundred base pairs of the ScUPC2 promoter drive the expression of β-galactosidase (49). YIp353-sre-UPC2-LacZ contains a ScUPC2 promoter deleted for both SREs, which were deleted by site-directed mutagenesis. The plasmid YIp353-PGK1-LacZ contains the wild-type ScPGK1 (phosphoglycerate kinase 1) promoter. Site-directed mutagenesis was performed using the QuikChange Multi site-directed mutagenesis kit (Stratagene, La Jolla, CA), and mutations were verified by DNA sequencing. YIp353 constructs were integrated at the endogenous URA3 locus by digestion with StuI and transformation into a wild-type S. cerevisiae strain (YJN16).
Compound libraries.
Ten small-molecule compound libraries were purchased from commercial sources. FDA-approved drug libraries included the Prestwick Collection (Prestwick Chemical, Illkirch, France; 1,120 compounds) and the Enzo FDA-approved compound library (Enzo Life Sciences, Plymouth Meeting, PA, USA; 640 compounds). The Natural Products Library was purchased from TimTec (Newark, DE, USA; 640 compounds). The kinase-targeted library Acti-Targ-K (960 compounds) was obtained from TimTec. Diversity compound sets were as follows: Maybridge Micro HitFinder (Thermo Fisher Scientific, Waltham, MA, USA; 14,400 compounds), Chembridge DIVERSet (ChemBridge, San Diego, CA, USA; 10,000 compounds), TimTec Diversity Set and ActiProbe 25 (TimTec; 10,000 and 25,000 compounds, respectively), and 15,280 compounds from ChemDiv (San Diego, CA, USA). Compounds were screened at a concentration of 5 to 10 μM.
Ultrahigh-throughput screen (uHTS).
S. cerevisiae strain YJN3672 (W303 ura3::URA3::UPC2-LacZ) was grown to log phase (optical density at 600 nm [OD600] of 0.8 to 1.2) in minimal yeast medium lacking uracil. Cells were diluted 1:20 into the final assay volume of 4 μl. Test compounds, dried in wells of solid white 1,536-well microtiter plates, were redissolved for 30 min at 30°C in 2 μl/well of a 1:10 dilution of log-phase cells. Fluconazole, diluted from a 60-mg/ml dimethyl sulfoxide stock into culture medium, was then added to a concentration of 35 μg/ml, in a final assay volume of 4 μl, and incubation was continued for 4 h at 30°C. A fluconazole concentration of 35 μg/ml was used to stimulate the activity of cells at a concentration where sterol-responsive activity was at 80% of maximum. A cutoff level of 50% of control was used for choosing compounds inhibiting Upc2-dependent β-galactosidase activity, at a screening concentration of 8 μM. β-Galactosidase detection was performed using BetaGlo (Promega, Madison, WI, USA). After 30 min at room temperature, luminescence was detected in the EnVision multimode plate reader (PerkinElmer, Waltham, MA, USA). Curve fitting was performed using GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA) or ActivityBase software (IDBS, Guildford, United Kingdom).
Mammalian cell toxicity assay.
HEK293 cells were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, sodium bicarbonate, penicillin, and streptomycin and were grown in 5% CO2 at 37°C. The cells were seeded at 2 × 105 cells/ml in 4 μl of medium and test compound in solid white 1,536-well microtiter plates (Greiner Bio One, Monroe, NC). The plates were incubated for 48 h. The plates were equilibrated to room temperature before using Cell Titer-Glo luminescent cell viability assay according to the manufacturer's directions (Promega, USA). Luminescence was detected using the EnVision 2103 multilabel reader (PerkinElmer, USA).
Expression and purification of GST-Upc2 DNA binding domain fusion proteins.
The UPC2 gene fragments encoding the DNA binding domains from Candida albicans, S. cerevisiae, and C. glabrata were amplified by PCR using chromosomal DNA as a template. The PCR products were ligated into the E. coli expression vector pGEX-4T3, using BamHI and EcoRI, to create pGEX-CaUPC2, pGEX-ScUPC2, and pGEX-CgUPC2. All sequences were fused in frame to vector-derived sequences encoding glutathione S-transferase (GST), followed by a stop codon. DNA sequencing was performed to confirm that no mutations were introduced into the sequences, and the vectors were transformed into E. coli strain BL21 for expression of the fusion proteins.
To induce expression of the fusion proteins, transformed E. coli BL21 cells were grown to mid-log phase (A600 of ∼0.5) at 37°C in Luria broth containing 100 μg/ml ampicillin and 150 μM ZnSO4. Isopropyl-β-d-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM, and cultures were incubated with shaking at 37°C for an additional 4 h. Soluble cytoplasmic protein was extracted from cells using BugBuster master mix (EMD Chemicals, Gibbstown, NJ) according to the manufacturer's instructions, except that the BugBuster was supplemented with 10 μM ZnSO4. GST fusion proteins were purified using a GSTrap column (GE Healthcare, Parsippany, NJ), following the manufacturer's instructions, except that all buffers contained 10 μM ZnSO4.
AlphaScreen DNA binding assay.
AlphaScreen is a bead-based binding assay technology. Donor beads contain a photosensitizer that converts ambient oxygen to a short-lived form of singlet oxygen upon illumination at 680 nm. Singlet oxygen diffuses up to 200 nm in solution before it decays. Interaction between GST-CaUpc2 and SRE brings the donor beads into close proximity with the acceptor beads, allowing singlet oxygen to activate a thioxene derivative in the acceptor beads, leading to the emission of light between 520 and 620 nm. In the absence of acceptor beads or in the presence of acceptor beads and a small molecule inhibiting DNA binding, the singlet oxygen decays to the ground state with no light emission. Donor beads can release up to 60,000 singlet oxygen molecules per second, resulting in signal amplification. Because signal detection is performed in a time-resolved manner and at a lower wavelength than excitation, interference from fluorescent test compounds is low.
Binding of the CaUpc2 DNA binding domain to DNA was detected using streptavidin-coated AlphaScreen donor beads (PerkinElmer, Waltham, MA), which capture biotinylated DNA, and anti-GST-coated AlphaScreen acceptor beads, which bind to the GST-CaUpc2 fusion protein. The DNA probe sequence used for the screening assay was: SRE, 5′-biotin-TEG-CTGTATTGTCGTATAAAAGTGG-3′ and 5′-CCACTTTTATACGACAATACAG-3′, where TEG is a triethylene glycol spacer (C. albicans ERG2 consensus). The probe was hybridized to form a biotinylated, double-stranded oligonucleotide by combining them at a concentration of 50 μM each in a buffer consisting of 100 mM potassium acetate (KOAc) and 30 mM HEPES (pH 7.4). The mixture was heated to 91 to 95°C for 2 min and allowed to equilibrate to 25°C.
For this assay, fusion protein was diluted to 8 nM (0.34 μg/ml) in an assay buffer consisting of 25 mM HEPES (pH 7.0), 200 nM NaCl, 0.1% Tween 20, and 3 μM ZnSO4. Protein was then combined with an equal volume of acceptor beads, which had been diluted to 80 μg/ml in assay buffer, and 4 μl/well of the mixture was transferred to a white, 1,536-well assay plate. The plate was incubated for 30 min at room temperature. Biotinylated, double-stranded DNA was diluted to 40 nM in assay buffer and combined with an equal volume of 80 μg/ml donor beads in assay buffer. Four microliters of this mixture was added to each well of the assay plate, and the plate was incubated for an additional hour at room temperature in the dark. The signal was detected using a PerkinElmer EnVision microplate reader.
IC50 determination.
Yeast cultures were diluted to 5 × 103 cells per ml in YEPD medium. Cells (100 μl) were distributed to wells of a 96-well flat-bottom plate, except for row A, which received 200 μl. Drug was added to row A at the desired concentration and then serially 2-fold diluted to rows B through F; row G served as a drug-free control, and row H served as a cell free control. The plates were incubated at 35°C for 24 h. Absorbance at 620 nm was read with a microplate reader (FluorStar Galaxy, BMG); background due to medium was subtracted from all readings. The IC50 was defined as the lowest concentration inhibiting growth at least 50% relative to the drug-free control.
Chromatin immunoprecipitation (ChIP) assays.
Yeast cells from a single colony were grown in YEPD liquid medium at 30°C overnight. Exponentially growing mother cultures were back-diluted to 5 × 105 cells/ml for S. cerevisiae cells and 1 × 105 cells/ml for C. albicans or C. glabrata cells. Each compound was individually added to diluted cultures at IC50 levels, and cells were incubated for 30 min at 30°C. Fluconazole at 50 μg/ml was added directly to cultures and incubated for additional 6 h at 30°C. Compounds and fluconazole were dissolved in dimethyl sulfoxide (DMSO) (Sigma Chemicals).
After incubation with fluconazole, cells were treated with 1% formaldehyde for 2 h at room temperature. Cross-linking was stopped by the addition of 1× TBS (Tris-buffered saline) (1× TBS is 20 mM Tris-HCl [pH 7.6] and 150 mM NaCl). The cells were pelleted, washed with 1× TBS, and again pelleted. The cells were then lysed using lysis buffer (50 mM HEPES [pH 7.5], 140 mM NaCl, 1 mM EDTA, 0.1% Triton X-100, 0.1% sodium deoxycholate, and a protease cocktail) and cold glass beads and spun down to remove cellular debris. Two hundred fifty milligrams of the resulting supernatant was used for immunoprecipitating ScUpc2-MYC-bound to each SRE for 1 day at 4°C, using 20 μl of anti-MYC monoclonal antibody. Immunoprecipitated ScUpc2-SRE complexes were isolated after 1 h at 4°C using protein G Sepharose/agarose beads. The beads were washed with lysis buffer containing 500 mM NaCl, and finally with a solution of 10 mM Tris-HCl (pH 8.0), 0.25 M LiCl, 0.5% NP-40, and 0.5% sodium deoxycholate. ScUpc2-DNA complexes were eluted from beads using a solution of 50 mM Tris-HCl (pH 8.0), 10 mM EDTA, and 1% SDS. Cross-linking was reversed using 5 M NaCl at 65°C for 6 h. Samples were then diluted with a solution of 500 mM Tris-HCl (pH 8.0), 10 mM EDTA, and 0.67% SDS. Samples were treated with 250 μg proteinase K for 1 h at 37°C. DNA was eluted from samples using the DNeasy kit (Qiagen). Purified DNA and PCR amplification were used to quantitate the concentration of ScUpc2-MYC to each SRE.
Quantitative reverse transcription-PCR (qRT-PCR) analyses.
Cells were grown to exponential log phase at 30°C. Total RNA was isolated as described previously (50). Total RNA was resuspended in diethyl pyrocarbonate (DEPC)-treated water. Fifty nanograms of RNA, 11.5 μl of the SYBR green master mix (Quanta), and 5 μM primer sets were loaded in triplicate onto a 96-well plate. RT-PCR amplification and analysis were completed using the Stratagene Max-Pro (Mx3000P) software version 4.0.
Western blot analysis.
Yeast strains were grown at 30°C in Ura− medium (medium lacking uracil) to exponential log phase. Cells were treated with compound alone or treated with compound in the presence of fluconazole for 16 h. Cell extracts were then isolated, and the ScUpc2-MYC level was detected using Western blot analysis. Briefly, cells were disrupted with cold glass beads and lysis buffer [Tris base (pH 7.9) containing (NH4)2SO4, MgSO4, glycerol, and EDTA]. Lysed cells were centrifuged at 3,300 rpm for 5 min at 4°C to obtain a total cell extract. Thirty micrograms of total cell extract was resolved by SDS-PAGE and transferred onto a nitrocellulose membrane. Western blot analysis was performed using anti-MYC and anti-GAPDH (antibody against glyceraldehyde-3-phosphate dehydrogenase) antibodies at a 1:1,000 dilution in TBS plus 1% nonfat milk. Secondary antibodies were horseradish peroxidase-conjugated IgG secondary antibodies diluted to 1:2,000 in TBS plus 1% nonfat milk.
RESULTS
A whole-cell HTS designed to identify compounds inhibiting ScUpc2-dependent transcription.
A high-throughput screening campaign was undertaken, which was designed to identify small-molecule compounds targeting the Upc2 transcription factor. The budding yeast Saccharomyces cerevisiae was used as a model. S. cerevisiae is more amenable to genetic manipulation than C. albicans or C. glabrata is, which helped in engineering a strain suitable for a high-throughput cell-based assay (51). We are aware that a potential limitation in using the S. cerevisiae LacZ strain for screening, rather than a C. albicans LacZ strain, is that compounds directly inhibiting only CaUpc2-SRE binding may not be identified. However, based on the km values determined using in vitro AlphaScreen assays (see below), it was believed the compounds identified would inhibit ScUpc2 and CaUpc2 DNA binding (Fig. 1B).
An S. cerevisiae strain was constructed containing an integrated copy of a ScUPC2-LacZ promoter fusion (Fig. 1C). The goal of the whole-cell screen was to identify compounds reducing fluconazole-induced ScUPC2-LacZ promoter activity. A second strain was constructed; this strain carried an integrated copy of an ScPGK1-LacZ promoter fusion lacking SREs. This strain was used in a counterscreen designed to eliminate compounds inhibiting general RNA polymerase II transcription (Fig. 1B).
The fluconazole concentration to be used in the whole-cell assay was first determined. Cells were assayed for ScUPC2-LacZ-dependent β-galactosidase activity in the presence of various concentrations of fluconazole. β-Galactosidase activity showed a dose dependence on the concentration of fluconazole present (Fig. 2, black circles). An Scupc2 Scecm22 ScUPC2-LacZ null strain had no observable fluconazole-induced SRE activity, as both the ScUpc2 and ScEcm22 transcription factors that bind to SREs were absent (not shown). A wild-type strain harboring a ScUPC2-LacZ promoter where the SREs were mutated (YIp353-sre-ScUPC2-LacZ) lacked β-galactosidase activity (Fig. 2, white circles). Thus, any fluconazole-dependent activity observed was SRE dependent and required ScUpc2 for activity.
FIG 2.
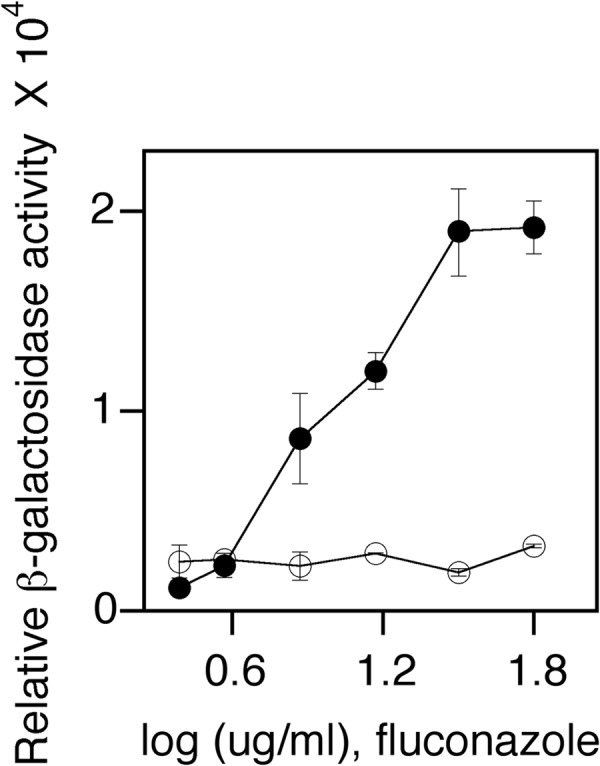
Cells expressing an UPC2-LacZ promoter fusion show a dose-dependent increase in β-galactosidase activity in response to increasing concentrations of fluconazole (in micrograms per milliliter). Cells were diluted 1:20 into the final assay volume of 4 μl. Test compounds in wells of solid white 1,536-well microtiter plates were redissolved in medium containing cells. Increasing concentrations of fluconazole were then added in a final assay volume of 4 μl, and cells were incubated for 4 h at 30°C. β-Galactosidase activity was performed using BetaGlo (Promega, Madison, WI, USA). β-Galactosidase activity of the UPC2-LacZ wild-type strain (solid black circles) and ecm22 upc2 UPC2-LacZ strain (white circles) are shown.
The commercially available Prestwick small-compound collection was initially screened (52). The Prestwick collection contains 1,200 FDA-approved small-molecule compounds. Z' values for the whole-cell assay were found to be between 0.6 and 0.8. A Z' calculation quantifies the suitability of an assay for use in a high-throughput screen. A Z' value above 0.5 indicates an accuracy sufficient for screening.
Not unexpectedly, a number of compounds isolated enhanced fluconazole-dependent UPC2-LacZ promoter activity. Many were representatives of the azole, statin, and allylamine families. Identifying synergistic activators indicates that the whole-cell assay is a valid method to identify Upc2 transcription factor inhibitors. Eleven Prestwick compounds that inhibited fluconazole-induced ScUPC2-LacZ activity were isolated; IC50s were between ∼3.0 and 20 μM, and in all but one case were >50 μM for ScPGK1-LacZ promoter activity. The 11 compounds included those with antifungal activities, and several nonsteroidal anti-inflammatory drugs (NSAIDs) and steroid molecules.
A 76,000-compound diversity set collection was next screened (see Materials and Methods). The eight compounds identified inhibited fluconazole-induced ScUPC2-LacZ promoter activity; IC50s were between 0.3 and 4 μM (Table 1), while IC50s for the PGK1-LacZ promoter were between 20 and 60 μM. Molecular structures are shown in Fig. 3. Among the eight compounds, compounds 1, 2, and 3 were closely related. Several other analogs containing the same 2-phenyl-3-nitro-2H-chromene core showed similar activity. However, not all 3-nitro-2H-chromene compounds were active, indicating that the core was not solely responsible for activity.
TABLE 1.
Hits from whole-cell uHTS screen using the Venenum 76,000 diversity small compound collection
| Compound | IC50 (μM) on the following promoter activity: |
AlphaScreen assay % control at 30 μM | |
|---|---|---|---|
| ScUPC2-LacZ (n = 4) | ScPGK1-LacZ (n = 2) | ||
| VB00075177 | 0.29 ± 0.07 | No inhibition at 30 μM | 99 ± 10 |
| VB00075853 | 0.63 ± 0.06 | >20 μM | 51 ± 5 |
| VB00075845 | 0.92 ± 0.10 | >20 μM | 51 ± 3 |
| VB00049027 | 1.5 ± 1.0 | No inhibition at 60 μM | 83 ± 7 |
| VB00008679 | 2.5 ± 0.5 | No inhibition at 60 μM | 90 ± 10 |
| VB00073293 | 2.6 ± 0.7 | No inhibition at 60 μM | 94 ± 12 |
| VB00059166 | 3.3 ± 0.5 | No inhibition at 60 μM | 84 ± 6 |
| VB00068594 | 4.2 ± 0.7 | No inhibition at 60 μM | 69 ± 3 |
FIG 3.
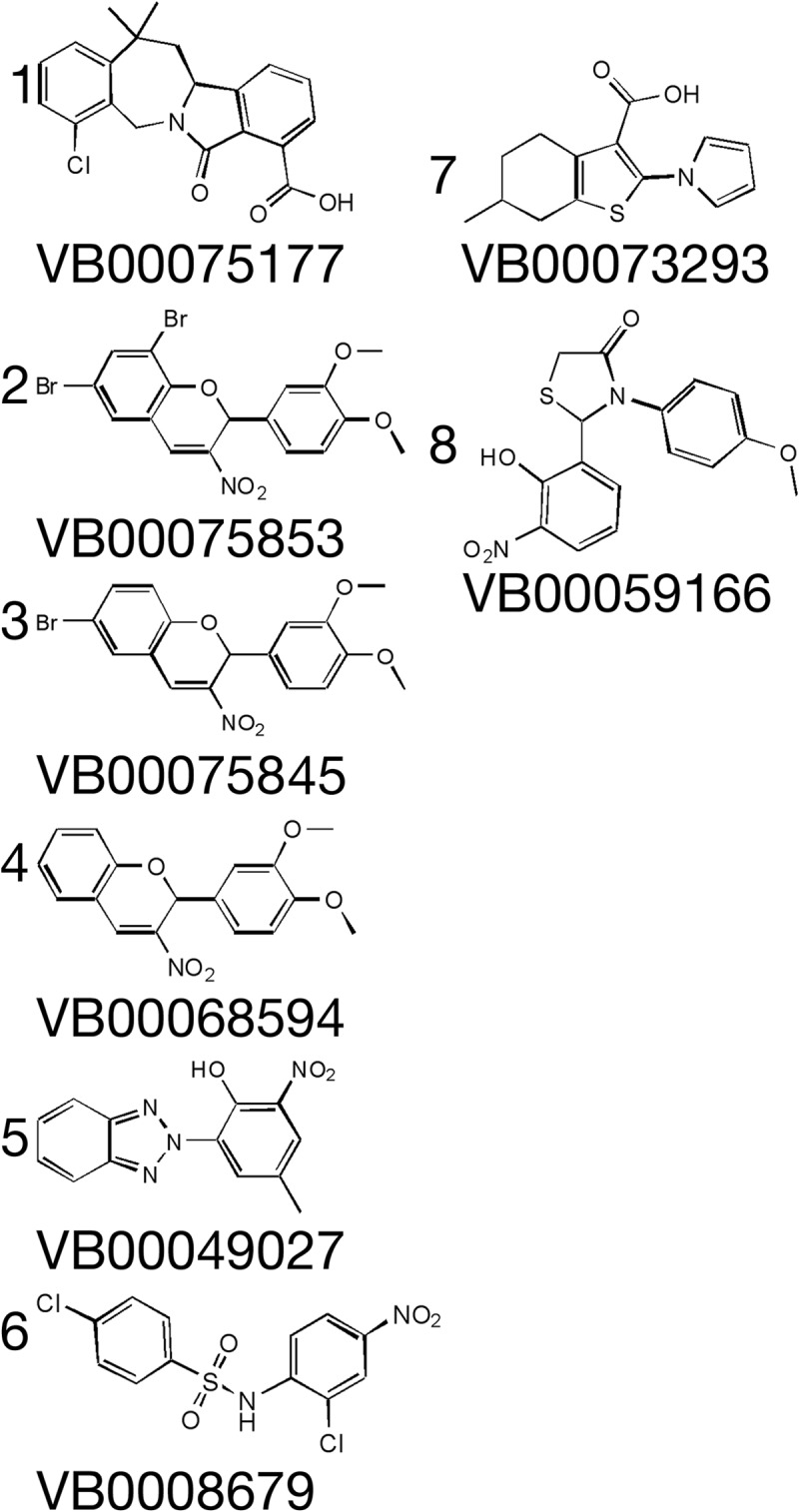
Molecular structures of the identified compounds The molecular structures and VB numbers of compounds 1 to 8 are shown. The compounds were isolated using the commercial small-molecule collection.
AlphaScreen assay designed to validate compounds as direct Upc2 DNA binding inhibitors.
A whole-cell reporter assay cannot distinguish between direct and indirect inhibition of DNA binding. Thus, an in vitro AlphaScreen assay that detects the direct interaction of C. albicans CaUpc2 with its SRE was developed. The assay was set up as described in Materials and Methods.
The CaUpc2 concentration that would be used in the assay was determined. The binding domain of CaUpc2 (amino acids [aa] 1 to 170) was used and purified as a GST fusion protein. Binding to the consensus SRE from CaERG2 was determined (31). An apparent Km of 50 nM was calculated for CaUpc2 (Fig. 4A). Similar Km values were obtained for the DNA binding domains of ScUpc2 and CgUpc2 (Fig. 4B and C).
FIG 4.

Upc2 DNA binding in the AlphaScreen assay shows a dose-dependent increase in activity in response to an increasing concentration of protein. Increasing concentrations of fusion proteins were diluted in assay buffer. Protein was combined with an equal volume of acceptor beads, which had been diluted in assay buffer. Part of the mixture (4 μl/well) was transferred to a white, 1,536-well assay plate. The plate was incubated for 30 min at room temperature. Biotinylated, double-stranded DNA was diluted in assay buffer and combined with an equal volume of donor beads in assay buffer. Four microliters of this mixture was added to each well, and the plate was incubated for an additional hour at room temperature in the dark. The signal was detected using a PerkinElmer EnVision microplate reader. GST-CaUpc2, glutathione S-transferase (GST) fused to the C. albicans Upc2 DNA binding domain; GST-ScUpc2, S. cerevisiae Upc2 DNA binding domain; GST-CgUpc2, C. glabrata Upc2 DNA binding domain.
Determining Upc2 DNA binding inhibitory activity.
The 11 Prestwick collection hits from the ScUPC2-LacZ assay were tested. None inhibited DNA binding, indicating their direct target(s) is not CaUpc2. The entire Prestwick collection was screened and resulted in the identification of 11 compounds. These compounds were given low priority as precursors for a hit-to-lead program based on their molecular structures and mode of action.
The eight hits identified from the ScUPC2-LacZ commercial library screen were tested. Each compound did not inhibit CaUpc2 DNA binding. None harbored activity against the ScPGK-LacZ promoter, and they were not cytotoxic using a HEK293 cell proliferation assay. Based on this data, the compounds identified are most likely regulating a signaling pathway required for Upc2 activation, rather than directly inhibiting Upc2 DNA binding.
On the basis of the fact that all compounds tested did not inhibit DNA binding, we chose to carry forward three compounds showing the lowest IC50s in the UPC2-LacZ cell assay (VB00075177, VB00075853, and VB00075845, referred to as compounds 1, 2, and 3, respectively). The compounds were tested for the ability to inhibit Scecm22 cell growth; these cells contain only ScUpc2. The three compounds inhibited cell growth at low micromolar concentrations. The IC50s were determined for the three compounds and were 1.2 μM ± 0.3 μM for compound 1, 2.5 μM ± 0.2 μM for compound 2, and 0.3 μM ± 0.1 μM for compound 3. These compounds inhibited wild-type S. cerevisiae and C. glabrata strains with similar IC50s but had no effect on wild-type C. albicans cell growth.
Each compound blocks fluconazole-induced UPC2 promoter activity.
Compounds were tested for the ability to inhibit ScUpc2-dependent transcription in the presence of fluconazole. The expression of ScERG11 and ScERG25 is induced in the presence of fluconazole through Upc2 activation (53–55). ScUPC2 expression is also induced (47), while ScPGK1 expression remains at basal level. As expected, Scecm22 null cells grown in the presence of fluconazole alone induced the expression of ScUPC2/ERG11/ERG25 (∼2- to 4-fold) (Fig. 5A to C, without FLC versus with FLC). The expression level of ScPGK1-LacZ did not change (Fig. 5D). The addition of each compound abolished fluconazole-induced ScERG25/ScERG11/ScUPC2 expression, while having no effect on ScPKG1-LacZ basal expression.
FIG 5.

Compounds 1, 2, and 3 inhibit fluconazole-induced Upc2-dependent activity. Cells were grown to exponential phase in the absence (−) and presence (+) of fluconazole (FLC) and each compound (compound 1, 2, or 3) for 6 h at 30°C. Total RNA was extracted, and the mRNA expression levels of ERG11, ERG25, UPC2, and PGK1 from S. cerevisiae (Sc) were determined by qRT-PCR.
Compounds were tested for inhibiting fluconazole-induced gene expression in the pathogenic fungi, C. albicans and C. glabrata. No effects on C. albicans expression were seen in the presence of each compound and fluconazole. The addition of each compound to C. glabrata cells attenuated fluconazole-induced expression (Fig. 6A to D). Thus, the three compounds inhibit sterol gene expression in S. cerevisiae and C. glabrata.
FIG 6.

Compounds 1, 2, and 3 inhibit fluconazole-induced CgUpc2-dependent activity. Cells were grown to exponential phase in the absence (−) and presence (+) of fluconazole and each compound (compound 1, 2, or 3) for 6 h at 30°C. Total RNA was extracted, and the mRNA expression levels of ERG11, ERG25, UPC2, and PGK1 from C. glabrata (Cg) were determined by qRT-PCR.
Fluconazole-dependent ScUpc2 protein expression is decreased in the presence of compound.
ScUpc2 protein level was determined to see whether loss of ScUPC2 transcription resulted in loss of protein. An increase in ScUpc2-MYC protein expression was seen in the presence of fluconazole (Fig. 7, con versus FLC) that was eliminated by addition of each compound (Fig. 7, con versus FLC + 1, FLC + 2, or FLC + 3).
FIG 7.
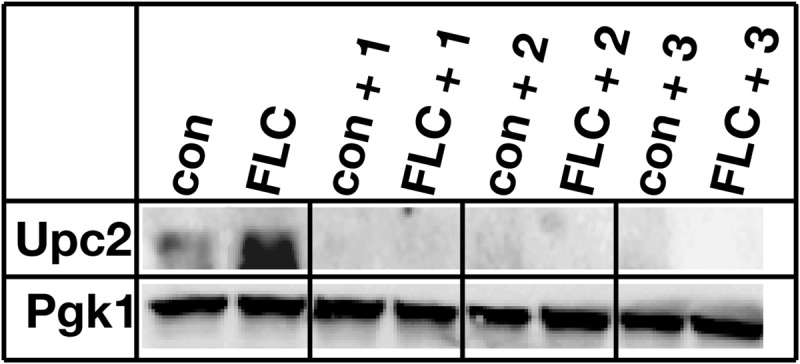
Compounds 1, 2, and 3 decrease Upc2 protein expression in the presence of fluconazole. Wild-type S. cerevisiae cells were grown to exponential phase in the absence and presence of fluconazole (FLC) and each compound (compound 1, 2, or 3) for 6 h at 30°C (con, control). Cell lysates were then prepared, and proteins were resolved by SDS-PAGE. The protein level of ScUpc2 was visualized by Western blot analysis using rabbit anti-ScUpc2 polyclonal antibodies and horseradish peroxidase (HRP)-conjugated anti-rabbit IgG polyclonal antibodies.
Each compound reduces ScUpc2 binding to endogenous SRE sites.
The level of in vivo ScUpc2 SRE binding was determined in the presence of each compound. The ScUPC2 promoter contains two SREs (SRE1, positions −585 to −379; SRE2, positions 359 to 353). ChIP was used to determine the level of ScUpc2 binding to each SRE. A low level of bound ScUpc2 was seen at both SREs in the absence of fluconazole (Fig. 8). Upon the addition of fluconazole, a 3.5-fold increase in binding was seen to SRE2 (Fig. 8, bottom panel). No fold increase over basal binding was seen for SRE1 (Fig. 8, top panel). Each compound abolished the increase seen for SRE2.
FIG 8.
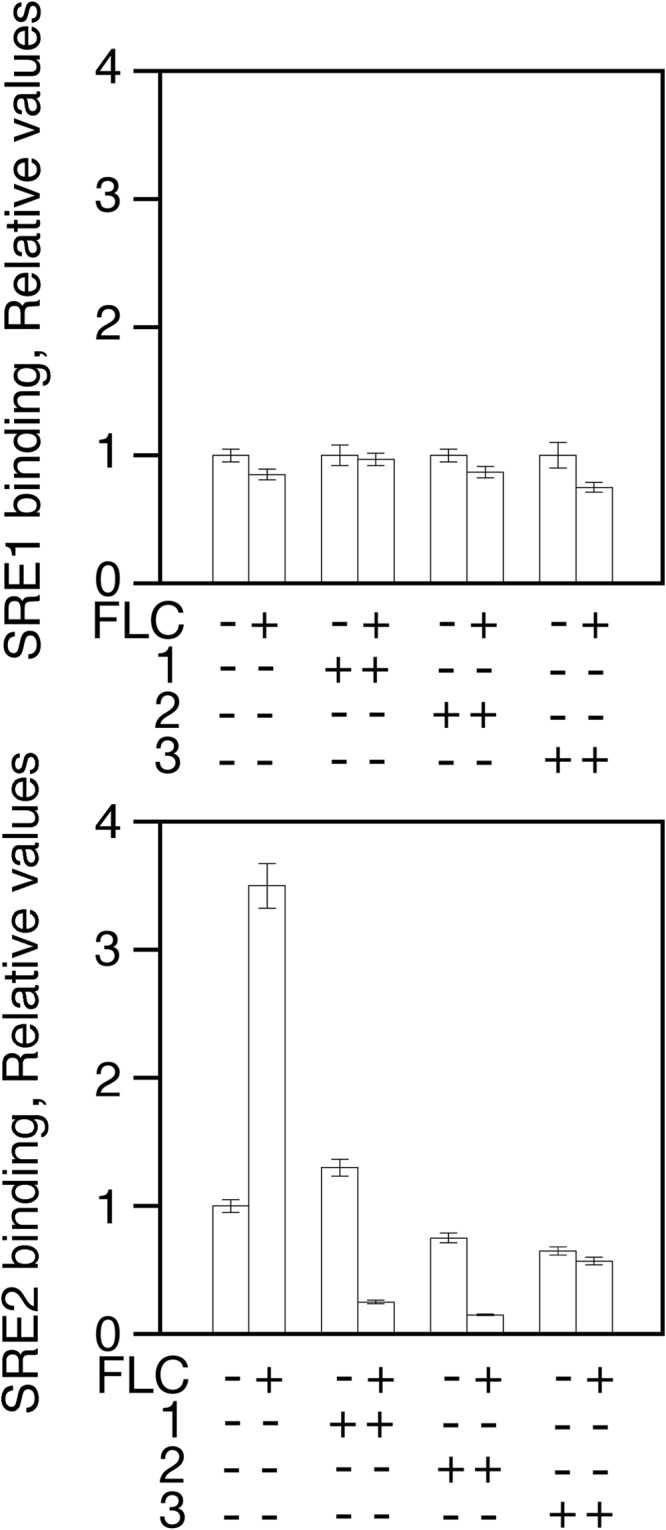
Compounds 1, 2, and 3 decrease Upc2 SRE binding in the presence of fluconazole. Wild-type S. cerevisiae cells were grown to exponential phase in the absence and presence of fluconazole and each compound for 6 h at 30°C. ScUpc2 was immunoprecipitated from cross-linked cells using rabbit anti-ScUpc2 polyclonal antibodies. Cross-linked ScUpc2-bound DNA was isolated and quantitated by qRT-PCR using primers specific for SRE1 or SRE2.
DISCUSSION
Many resistance mechanisms stimulate the overexpression of ERG genes, including gain-of-function mutations in UPC2 (35, 37, 38, 56). The UPC2 mutations responsible have been found in resistant clinical isolates, and resistance has been recapitulated in laboratory strains (37, 38). Upc2 is the most downstream factor regulating a pathway responsible for induction of ERG gene expression in the presence of azole antifungals. It represents an excellent target for compound discovery, as inhibition would circumvent multiple resistance mechanisms upregulating ERG genes (54).
A whole-cell HTS assay that was designed to identify compounds attenuating ScUpc2-dependent transcription was developed. An in vitro AlphaScreen assay was implemented to determine whether the compounds were true Upc2 DNA binding inhibitors. All compounds identified reduced Upc2 activity but did not inhibit DNA binding. Thus, the three compounds most likely attenuate a signaling pathway(s) regulating Upc2 function in response to a block in sterol biosynthesis. To our knowledge, these are the first small molecule Upc2 pathway inhibitors identified.
The compounds identified inhibited cell growth but did not kill cells. This seems reasonable since Scecm22 Scupc2 double null cells are viable, although their growth is compromised (28, 29). Vegetative cell growth was inhibited in the absence of fluconazole, suggesting that Upc2 may regulate basal gene expression. A genome-wide ChIP study showed that CaUpc2 bound to promoters involved in ribosomal subunit biosynthesis, general transcription factor activities, and sulfur amino acid metabolism (31). The fact that wild-type S. cerevisiae cells were growth inhibited suggests the compounds regulate both ScUpc2 and ScEcm22. This is interesting, in light of the fact that ScUpc2 and ScEcm22 activities may not be completely redundant, as Ecm22 binding to SREs is reduced in the presence of fluconazole, while ScUpc2 binding is increased (47).
C. glabrata is more evolutionarily similar to S. cerevisiae than is C. albicans (57). C. glabrata contributes significantly to systemic infections in immunocompromised patients (58–60). Infections caused by C. glabrata are on the rise, due to acquisition of azole drug resistance (61, 62). C. glabrata cells did respond to drug treatment. We did not observe the same for C. albicans. CaUpc2 is highly orthologous to ScUpc2 and ScEcm22, with BLAST search values of e−112 and 2e−87, respectively (28). In addition, there is 66% and 64% homology within the N-terminal conserved Zn2-Cys6 binuclear cluster and C-terminal transmembrane domain of each protein, respectively (28). Resistant C. albicans strains have conserved CaUPC2 gain-of-function mutations that are seen in ScUPC2 alleles conferring resistance (35, 36, 38).
Based on this degree of homology and conservation of function, it is somewhat surprising that growth inhibition was not observed. Again, one possibility is that an S. cerevisiae strain carrying an ScUPC2-LacZ gene was used, rather than a C. albicans strain harboring a CaUPC2-LacZ gene. Compounds inhibiting CaUpc2 alone could have possibly been missed. Another possibility is that C. albicans uses other transcription factors to regulate ERG gene expression in addition to CaUpc2 (32, 63). The CaTac1 transcription factor induces the expression of efflux pump genes needed for antifungal drug excretion (64–66). CaUpc2-dependent and -independent factors regulating CaUPC2 expression also exist (32, 63). It is possible that targeting several pathways may be necessary to cause C. albicans growth inhibition. In any event, the compounds identified may serve as initial precursors for the chemical optimization of novel antifungal agents with widespread use in treating a number of serious systemic fungal infections.
ACKNOWLEDGMENTS
We appreciate the many discussions with James Merritt and colleagues (Kean University), Eli Mordechai and Martin Adelson, and the Target Biology, HTS, and Medicinal Chemistry groups of Venenum.
We acknowledge and are grateful for the financial support of the Genesis Biotechnology Group.
Footnotes
Published ahead of print 21 October 2013
REFERENCES
- 1.Odds FC, Webster CE, Mayuranathan P, Simmons PD. 1988. Candida concentrations in the vagina and their association with signs and symptoms of vaginal candidosis. J. Med. Vet. Mycol. 26:277–283. 10.1080/02681218880000391 [DOI] [PubMed] [Google Scholar]
- 2.Krcmery V, Barnes AJ. 2002. Non-albicans Candida spp. causing fungaemia: pathogenicity and antifungal resistance. J. Hosp. Infect. 50:243–260. 10.1053/jhin.2001.1151 [DOI] [PubMed] [Google Scholar]
- 3.Pfaller MA, Jones RN, Doern GV, Sader HS, Messer SA, Houston A, Coffman S, Hollis RJ. 2000. Bloodstream infections due to Candida species: SENTRY antimicrobial surveillance program in North America and Latin America, 1997–1998. Antimicrob. Agents Chemother. 44:747–751. 10.1128/AAC.44.3.747-751.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vazquez JA, Sobel JD, Peng G, Steele-Moore L, Schuman P, Holloway W, Neaton JD. 1999. Evolution of vaginal Candida species recovered from human immunodeficiency virus-infected women receiving fluconazole prophylaxis: the emergence of Candida glabrata? Terry Beirn Community Programs for Clinical Research in AIDS (CPCRA). Clin. Infect. Dis. 28:1025–1031 [DOI] [PubMed] [Google Scholar]
- 5.Wai PH, Ewing CA, Johnson LB, Lu AD, Attinger C, Kuo PC. 2001. Candida fasciitis following renal transplantation. Transplantation 72:477–479. 10.1097/00007890-200108150-00019 [DOI] [PubMed] [Google Scholar]
- 6.Wheat LJ. 1980. Infection and diabetes mellitus. Diabetes Care 3:187–197. 10.2337/diacare.3.1.187 [DOI] [PubMed] [Google Scholar]
- 7.de Leon EM, Jacober SJ, Sobel JD, Foxman B. 2002. Prevalence and risk factors for vaginal Candida colonization in women with type 1 and type 2 diabetes. BMC Infect. Dis. 2:1. 10.1186/1471-2334-2-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mukherjee PK, Sheehan D, Puzniak L, Schlamm H, Ghannoum MA. 2011. Echinocandins: are they all the same? J. Chemother. 23:319–325 [DOI] [PubMed] [Google Scholar]
- 9.Lass-Florl C. 2011. Triazole antifungal agents in invasive fungal infections: a comparative review. Drugs 71:2405–2419. 10.2165/11596540-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 10.Traunmuller F, Popovic M, Konz KH, Smolle-Juttner FM, Joukhadar C. 2011. Efficacy and safety of current drug therapies for invasive aspergillosis. Pharmacology 88:213–224. 10.1159/000331860 [DOI] [PubMed] [Google Scholar]
- 11.Cartwright RY. 1975. Antifungal drugs. J. Antimicrob. Chemother. 1:141–162. 10.1093/jac/1.2.141 [DOI] [PubMed] [Google Scholar]
- 12.Ostrosky-Zeichner L, Casadevall A, Galgiani JN, Odds FC, Rex JH. 2010. An insight into the antifungal pipeline: selected new molecules and beyond. Nat. Rev. Drug Discov. 9:719–727. 10.1038/nrd3074 [DOI] [PubMed] [Google Scholar]
- 13.Krishnan-Natesan S. 2009. Terbinafine: a pharmacological and clinical review. Expert Opin. Pharmacother 10:2723–2733. 10.1517/14656560903307462 [DOI] [PubMed] [Google Scholar]
- 14.Marcireau C, Guilloton M, Karst F. 1990. In vivo effects of fenpropimorph on the yeast Saccharomyces cerevisiae and determination of the molecular basis of the antifungal property. Antimicrob. Agents Chemother. 34:989–993. 10.1128/AAC.34.6.989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vanden Bossche H, Willemsens G, Marichal P. 1987. Anti-Candida drugs–the biochemical basis for their activity. Crit. Rev. Microbiol. 15:57–72. 10.3109/10408418709104448 [DOI] [PubMed] [Google Scholar]
- 16.Debono M, Gordee RS. 1994. Antibiotics that inhibit fungal cell wall development. Annu. Rev. Microbiol. 48:471–497. 10.1146/annurev.mi.48.100194.002351 [DOI] [PubMed] [Google Scholar]
- 17.Kurtz MB, Douglas CM. 1997. Lipopeptide inhibitors of fungal glucan synthase. J. Med. Vet. Mycol. 35:79–86. 10.1080/02681219780000961 [DOI] [PubMed] [Google Scholar]
- 18.Lees ND, Bard M, Kirsch DR. 1997. Biochemistry and molecular biology of sterol synthesis in Saccharomyces cerevisiae, p 85–99 In Parish EJ, Nes WD. (ed), Biochemistry and function of sterols. CRC Press, Boca Raton, FL: [PubMed] [Google Scholar]
- 19.Pfeifer K, Arcangioli B, Guarente L. 1987. Yeast HAP1 activator competes with the factor RC2 for binding to the upstream activation site UAS1 of the CYC1 gene. Cell 49:9–18. 10.1016/0092-8674(87)90750-1 [DOI] [PubMed] [Google Scholar]
- 20.Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. 2000. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 11:4241–4257. 10.1091/mbc.11.12.4241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alarco AM, Balan I, Talibi D, Mainville N, Raymond M. 1997. AP1-mediated multidrug resistance in Saccharomyces cerevisiae requires FLR1 encoding a transporter of the major facilitator superfamily. J. Biol. Chem. 272:19304–19313. 10.1074/jbc.272.31.19304 [DOI] [PubMed] [Google Scholar]
- 22.Grishin AV, Rothenberg M, Downs MA, Blumer KJ. 1998. Mot3, a Zn finger transcription factor that modulates gene expression and attenuates mating pheromone signaling in Saccharomyces cerevisiae. Genetics 149:879–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hongay C, Jia N, Bard M, Winston F. 2002. Mot3 is a transcriptional repressor of ergosterol biosynthetic genes and is required for normal vacuolar function in Saccharomyces cerevisiae. EMBO J. 21:4114–4124. 10.1093/emboj/cdf415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evangelista CC, Jr, Rodriguez Torres AM, Limbach MP, Zitomer RS. 1996. Rox3 and Rts1 function in the global stress response pathway in baker's yeast. Genetics 142:1083–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacPherson S, Larochelle M, Turcotte B. 2006. A fungal family of transcriptional regulators: the zinc cluster proteins. Microbiol. Mol. Biol. Rev. 70:583–604. 10.1128/MMBR.00015-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vik A, Rine J. 2001. Upc2p and Ecm22p, dual regulators of sterol biosynthesis in Saccharomyces cerevisiae. Mol. Cell. Biol. 21:6395–6405. 10.1128/MCB.21.19.6395-6405.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shianna KV, Dotson WD, Tove S, Parks LW. 2001. Identification of a UPC2 homolog in Saccharomyces cerevisiae and its involvement in aerobic sterol uptake. J. Bacteriol. 183:830–834. 10.1128/JB.183.3.830-834.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Silver PM, Oliver BG, White TC. 2004. Role of Candida albicans transcription factor Upc2p in drug resistance and sterol metabolism. Eukaryot. Cell 3:1391–1397. 10.1128/EC.3.6.1391-1397.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagi M, Nakayama H, Tanabe K, Bard M, Aoyama T, Okano M, Higashi S, Ueno K, Chibana H, Niimi M, Yamagoe S, Umeyama T, Kajiwara S, Ohno H, Miyazaki Y. 2011. Transcription factors CgUPC2A and CgUPC2B regulate ergosterol biosynthetic genes in Candida glabrata. Genes Cells 16:80–89. 10.1111/j.1365-2443.2010.01470.x [DOI] [PubMed] [Google Scholar]
- 30.MacPherson S, Akache B, Weber S, De Deken X, Raymond M, Turcotte B. 2005. Candida albicans zinc cluster protein Upc2p confers resistance to antifungal drugs and is an activator of ergosterol biosynthetic genes. Antimicrob. Agents Chemother. 49:1745–1752. 10.1128/AAC.49.5.1745-1752.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Znaidi S, Weber S, Al-Abdin OZ, Bomme P, Saidane S, Drouin S, Lemieux S, De Deken X, Robert F, Raymond M. 2008. Genomewide location analysis of Candida albicans Upc2p, a regulator of sterol metabolism and azole drug resistance. Eukaryot. Cell 7:836–847. 10.1128/EC.00070-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoot SJ, Brown RP, Oliver BG, White TC. 2010. The UPC2 promoter in Candida albicans contains two cis-acting elements that bind directly to Upc2p, resulting in transcriptional autoregulation. Eukaryot. Cell 9:1354–1362. 10.1128/EC.00130-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cohen BD, Sertil O, Abramova NE, Davies KJ, Lowry CV. 2001. Induction and repression of DAN1 and the family of anaerobic mannoprotein genes in Saccharomyces cerevisiae occurs through a complex array of regulatory sites. Nucleic Acids Res. 29:799–808. 10.1093/nar/29.3.799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abramova NE, Cohen BD, Sertil O, Kapoor R, Davies KJ, Lowry CV. 2001. Regulatory mechanisms controlling expression of the DAN/TIR mannoprotein genes during anaerobic remodeling of the cell wall in Saccharomyces cerevisiae. Genetics 157:1169–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dunkel N, Liu TT, Barker KS, Homayouni R, Morschhauser J, Rogers PD. 2008. A gain-of-function mutation in the transcription factor Upc2p causes upregulation of ergosterol biosynthesis genes and increased fluconazole resistance in a clinical Candida albicans isolate. Eukaryot. Cell 7:1180–1190. 10.1128/EC.00103-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flowers SA, Barker KS, Berkow EL, Toner G, Chadwick SG, Gygax SE, Morschhauser J, Rogers PD. 2012. Gain-of-function mutations in UPC2 are a frequent cause of ERG11 upregulation in azole-resistant clinical isolates of Candida albicans. Eukaryot. Cell 11:1289–1299. 10.1128/EC.00215-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoot SJ, Smith AR, Brown RP, White TC. 2011. An A643V amino acid substitution in Upc2p contributes to azole resistance in well-characterized clinical isolates of Candida albicans. Antimicrob. Agents Chemother. 55:940–942. 10.1128/AAC.00995-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heilmann CJ, Schneider S, Barker KS, Rogers PD, Morschhauser J. 2010. An A643T mutation in the transcription factor Upc2p causes constitutive ERG11 upregulation and increased fluconazole resistance in Candida albicans. Antimicrob. Agents Chemother. 54:353–359. 10.1128/AAC.01102-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arndt HD. 2006. Small molecule modulators of transcription. Angew. Chem. Int. Ed. Engl. 45:4552–4560. 10.1002/anie.200600285 [DOI] [PubMed] [Google Scholar]
- 40.Berg T. 2008. Signal transducers and activators of transcription as targets for small organic molecules. Chembiochem 9:2039–2044. 10.1002/cbic.200800274 [DOI] [PubMed] [Google Scholar]
- 41.Bernhardt WM, Warnecke C, Willam C, Tanaka T, Wiesener MS, Eckardt KU. 2007. Organ protection by hypoxia and hypoxia-inducible factors. Methods Enzymol. 435:221–245 [DOI] [PubMed] [Google Scholar]
- 42.Roman-Blas JA, Jimenez SA. 2008. Targeting NF-kappaB: a promising molecular therapy in inflammatory arthritis. Int. Rev. Immunol. 27:351–374. 10.1080/08830180802295740 [DOI] [PubMed] [Google Scholar]
- 43.Ratan RR, Siddiq A, Aminova L, Langley B, McConoughey S, Karpisheva K, Lee HH, Carmichael T, Kornblum H, Coppola G, Geschwind DH, Hoke A, Smirnova N, Rink C, Roy S, Sen C, Beattie MS, Hart RP, Grumet M, Sun D, Freeman RS, Semenza GL, Gazaryan I. 2008. Small molecule activation of adaptive gene expression: tilorone or its analogs are novel potent activators of hypoxia inducible factor-1 that provide prophylaxis against stroke and spinal cord injury. Ann. N. Y. Acad. Sci. 1147:383–394. 10.1196/annals.1427.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jones DT, Harris AL. 2006. Identification of novel small-molecule inhibitors of hypoxia-inducible factor-1 transactivation and DNA binding. Mol. Cancer Ther. 5:2193–2202. 10.1158/1535-7163.MCT-05-0443 [DOI] [PubMed] [Google Scholar]
- 45.Bialkowska AB, Du Y, Fu H, Yang VW. 2009. Identification of novel small-molecule compounds that inhibit the proproliferative Kruppel-like factor 5 in colorectal cancer cells by high-throughput screening. Mol. Cancer Ther. 8:563–570. 10.1158/1535-7163.MCT-08-0767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wei W, Chua MS, Grepper S, So S. 2010. Small molecule antagonists of Tcf4/beta-catenin complex inhibit the growth of HCC cells in vitro and in vivo. Int. J. Cancer 126:2426–2436. 10.1002/ijc.24810 [DOI] [PubMed] [Google Scholar]
- 47.Davies BSJ, Wang HS, Rine J. 2005. Dual activators of the sterol biosynthetic pathway of Saccharomyces cerevisiae: similar activation/regulatory domains but different response mechanisms. Mol. Cell. Biol. 25:7375–7385. 10.1128/MCB.25.16.7375-7385.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ito H, Fukuda Y, Murata K, Kimura A. 1983. Transformation of intact yeast cells treated with alkali cations. J. Bacteriol. 153:163–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guarente L. 1983. Yeast promoters and lacZ fusions designed to study expression of cloned genes in yeast. Methods Enzymol. 101:181–191. 10.1016/0076-6879(83)01013-7 [DOI] [PubMed] [Google Scholar]
- 50.Nolt JK, Rice LM, Gallo-Ebert C, Bisher ME, Nickels JT. 2011. PP2A (Cdc) is required for multiple events during meiosis I. Cell Cycle 10:1420–1434. 10.4161/cc.10.9.15485 [DOI] [PubMed] [Google Scholar]
- 51.Botstein D, Fink GR. 2011. Yeast: an experimental organism for 21st century biology. Genetics 189:695–704. 10.1534/genetics.111.130765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Langer T, Hoffmann R, Bryant S, Lesur B. 2009. Hit finding: towards ‘smarter' approaches. Curr. Opin. Pharmacol. 9:589–593. 10.1016/j.coph.2009.06.001 [DOI] [PubMed] [Google Scholar]
- 53.Germann M, Gallo C, Donahue T, Shirzadi R, Stukey J, Lang S, Ruckenstuhl C, Oliaro-Bosso S, McDonough V, Turnowsky F, Baliiano G, Nickels JT., Jr 2005. Characterizing sterol defect suppressors uncovers a novel transcriptional signaling pathway regulating zymosterol biosynthesis. J. Biol. Chem. 280:35904–35913. 10.1074/jbc.M504978200 [DOI] [PubMed] [Google Scholar]
- 54.Oliver BG, Song JL, Choiniere JH, White TC. 2007. cis-Acting elements within the Candida albicans ERG11 promoter mediate the azole response through transcription factor Upc2p. Eukaryot. Cell 6:2231–2239. 10.1128/EC.00331-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Turi TG, Loper JC. 1992. Multiple regulatory elements control expression of the gene encoding the Saccharomyces cerevisiae cytochrome P450 lanosterol 14 alpha-demethylase (ERG11). J. Biol. Chem. 267:2046–2056 [PubMed] [Google Scholar]
- 56.Crowley JH, Leak FW, Jr, Shianna KV, Tove S, Parks LW. 1998. A mutation in a purported regulatory gene affects control of sterol uptake in Saccharomyces cerevisiae. J. Bacteriol. 180:4177–4183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Herrero E. 2005. Evolutionary relationships between Saccharomyces cerevisiae and other fungal species as determined from genome comparisons. Rev. Iberoam. Micol. 22:217–222. 10.1016/S1130-1406(05)70046-2 [DOI] [PubMed] [Google Scholar]
- 58.Katiyar S, Pfaller M, Edlind T. 2006. Candida albicans and Candida glabrata clinical isolates exhibiting reduced echinocandin susceptibility. Antimicrob. Agents Chemother. 50:2892–2894. 10.1128/AAC.00349-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wingard JR. 1995. Importance of Candida species other than C. albicans as pathogens in oncology patients. Clin. Infect. Dis. 20:115–125. 10.1093/clinids/20.1.115 [DOI] [PubMed] [Google Scholar]
- 60.Li L, Redding S, Dongari-Bagtzoglou A. 2007. Candida glabrata: an emerging oral opportunistic pathogen. J. Dent. Res. 86:204–215. 10.1177/154405910708600304 [DOI] [PubMed] [Google Scholar]
- 61.Pfaller MA, Castanheira M, Lockhart SR, Ahlquist AM, Messer SA, Jones RN. 2012. Frequency of decreased susceptibility and resistance to echinocandins among fluconazole-resistant bloodstream isolates of Candida glabrata. J. Clin. Microbiol. 50:1199–1203. 10.1128/JCM.06112-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pfaller MA. 2012. Antifungal drug resistance: mechanisms, epidemiology, and consequences for treatment. Am. J. Med. 125:S3–S13. 10.1016/j.amjmed.2011.11.001 [DOI] [PubMed] [Google Scholar]
- 63.Hoot SJ, Oliver BG, White TC. 2008. Candida albicans UPC2 is transcriptionally induced in response to antifungal drugs and anaerobicity through Upc2p-dependent and -independent mechanisms. Microbiology 154:2748–2756. 10.1099/mic.0.2008/017475-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sasse C, Dunkel N, Schafer T, Schneider S, Dierolf F, Ohlsen K, Morschhauser J. 2012. The stepwise acquisition of fluconazole resistance mutations causes a gradual loss of fitness in Candida albicans. Mol. Microbiol. 86:539–556. 10.1111/j.1365-2958.2012.08210.x [DOI] [PubMed] [Google Scholar]
- 65.Coste AT, Karababa M, Ischer F, Bille J, Sanglard D. 2004. TAC1, transcriptional activator of CDR genes, is a new transcription factor involved in the regulation of Candida albicans ABC transporters CDR1 and CDR2. Eukaryot. Cell 3:1639–1652. 10.1128/EC.3.6.1639-1652.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.MacCallum DM, Coste A, Ischer F, Jacobsen MD, Odds FC, Sanglard D. 2010. Genetic dissection of azole resistance mechanisms in Candida albicans and their validation in a mouse model of disseminated infection. Antimicrob. Agents Chemother. 54:1476–1483. 10.1128/AAC.01645-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gallo-Ebert C, Donigan M, Liu HY, Pascual F, Manners M, Pandya D, Swanson R, Gallagher D, Chen W, Carman GM, Nickels JT., Jr The yeast anaerobic response element AR1b regulates antifungal drug-dependent sterol gene expression. J. Biol. Chem., in press [DOI] [PMC free article] [PubMed] [Google Scholar]