

Abstract
The implementation of new antiretroviral therapies targeting transcription of early viral proteins in postintegrated HIV-1 can aid in overcoming current therapy limitations. Using high-throughput screening assays, we have previously described a novel Tat-dependent HIV-1 transcriptional inhibitor named 6-bromoindirubin-3′-oxime (6BIO). The screening of 6BIO derivatives yielded unique compounds that show potent inhibition of HIV-1 transcription. We have identified a second-generation derivative called 18BIOder as an inhibitor of HIV-1 Tat-dependent transcription in TZM-bl cells and a potent inhibitor of GSK-3β kinase in vitro. Structurally, 18BIOder is half the molecular weight and structure of its parental compound, 6BIO. More importantly, we also have found a different GSK-3β complex present only in HIV-1-infected cells. 18BIOder preferentially inhibits this novel kinase complex from infected cells at nanomolar concentrations. Finally, we observed that neuronal cultures treated with Tat protein are protected from Tat-mediated cytotoxicity when treated with 18BIOder. Overall, our data suggest that HIV-1 Tat-dependent transcription is sensitive to small-molecule inhibition of GSK-3β.
INTRODUCTION
Combination antiretroviral therapies (cART) prolong the lives of HIV-1-infected subjects but are concomitantly limited to virus-based inhibitors that primarily block various steps, including reverse transcription and integration of proviral DNA, or prevent the formation of functional viral particles. Despite the success of cART in slowing disease progression and decreasing the incidence of HIV-1-associated dementia (HAD), the percentage of infected patients suffering from HIV-1-associated neurocognitive disorders (HAND), which are characterized by cognitive, motor, and behavioral anomalies, remains unchanged (1–3). Consequently, epidemiological data suggest that cART provides partial protection of HIV-1-infected individuals from neurological damage (4, 5). A portion of the current research efforts focus on finding novel host-based therapies that can provide viral inhibition in tandem with protection of cells from apoptosis. Cytoprotective effects are desirable in order to protect already depleted cell populations and to counteract detrimental effects of viral proteins, such as Tat and gp120 (Env), which are normally generated during infection (6–11).
The HIV-1 transactivator Tat is an important regulatory protein that directs viral transcriptional elongation by association with the transactivation response (TAR) RNA element present at the transcriptional initiation site (positions −3 to +57) on the HIV-1 long terminal repeat (LTR). The Tat/TAR complex is able to recruit various critical host cell factors, such as the pTEF-b complex (Cdk9/Cyclin T1), to the RNA polymerase II complex that occupies the LTR (12–19). Due to its small size and stretch of basic residues, Tat can be secreted into the extracellular environment, where it exerts various functional effects on bystander cells (8, 20–26). The neurotoxic effects of HIV-1 are largely attributed to the Tat and gp120 proteins (27, 28). For example, Cheng et al. (27) demonstrated that exogenous Tat treatment could cause depolarization of human fetal neurons in culture, suggesting the Tat protein may directly contribute to neurotoxicity. Similarly, gp120 treatment induced injury to primary rat dopamine cells and reduced dopamine transport, suggesting a neuropathological consequence of gp120 (27), while this negative effect was prevented by gp120 antibody. Moreover, studies carried out over the last 2 decades link extracellular Tat to HIV-1 HAND, where Tat functions as a neuroexcitatory toxin that plays a role in virus-mediated neuronal dysfunction (29–33). Postulated mechanisms of Tat neurotoxicity include altered calcium homeostasis and calcium dependence in fetal neurons (27, 34, 35), increased oxidative stress resulting from direct injection of Tat into the intrastriatal space (36), increased gliosis and glial infiltration (36–38), stimulation of the glutamatergic system (39), increased nitric oxide production in microglial cultures (40), and increased apoptosis from cell damage and death following Tat exposure (34, 41). Moreover, the paracrine-like function of Tat wields its effects on neuronal cells and entails excitotoxic mechanisms possibly triggered in a receptor-dependent manner (42). Although extracellular secretion of HIV-1 Tat protein by infected T cells has been well documented (20, 24) and is largely considered to play a role in HIV-associated neuronal disease, secretion of Tat by infected primary macrophages and its contribution to neurotoxicity are not clear. Several Tat targets have been previously described (43, 44) and include N-methyl-d-aspartate (NMDA) receptors and GPCR activation (42), vascular endothelial growth factor 1 receptor (45), αv integrin subunit-containing receptors (43, 46, 47), low-density lipoprotein receptor-related protein (48), and amino acid excitatory receptors (29, 49, 50). Thus, Tat is an important neurotoxin in the HIV-1-infected brain and a novel therapeutic target that could be utilized in HIV-1 inhibition to counter the effects of Tat in the brain.
To date, multiple drug candidates that counteract host-based targets (51–59) or specifically target viral components (60–64) have been examined. Several HIV-1 transcriptional inhibitors, including K-12 and Ro24-7429, have undergone clinical trials and have been determined not to be clinically efficacious (65–68). More recent findings, however, indicate that novel virus- and host-based inhibitors can inhibit HIV-1 transcription without affecting normal cellular functions. Such compounds include WP631, temacrazine, and various cyclin-dependent kinase (Cdk) inhibitors (69–75). In the last 10 years, host-based therapies have shed light on potential targets that had previously not been fully recognized. For instance, robust glycogen synthase kinase-3β (GSK-3β) inhibitors, such as lithium and valproic acid, have been shown to protect against Tat- and gp120-mediated neurotoxicity (59, 76, 77). Recently, small chemical molecules have taken the spotlight due to their capacity for conferring both potent Tat-dependent transcriptional inhibition and cytoprotection from Tat-induced neurotoxicity through mechanisms that remain to be determined (33, 78).
In this study, we present data on a small-molecule inhibitor that both achieves Tat-dependent transcription inhibition and protects cells from Tat-induced neurotoxicity. The small chemical molecule inhibits GSK-3β, an enzyme that has been associated with HAND. We have characterized 18BIOder as an effective noncytotoxic HIV-1 Tat-dependent transcriptional inhibitor. In addition, we have described GSK-3β upregulation in HIV-1-infected cells and the formation of new protein complexes during infection. We demonstrate that our small-molecule compound inhibits HIV-1 transcription by targeting this novel GSK-3β complex present only in infected cells. Therefore, targeting the host GSK-3β pathway can serve as an attractive strategy for the development of novel host-based therapeutics that target both viral transcription and neurotoxic side effects of HIV-1 infection.
MATERIALS AND METHODS
Cell culture and reagents.
The human fetal microglial cell line CHME5 (a generous gift from Pierre Talbot, Laboratory of Neuroimmunovirology, INRS-Institute, Armand-Frappier, Canada) were grown and cultured to confluence in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% l-glutamine, and 1% streptomycin/penicillin (Gibco/BRL, Gaithersburg, MD, USA). CHME5 cells have been well characterized (79–81) and were obtained from embryonic fetal human microglia through transformation with simian virus 40 (SV-40) T antigen (79, 80). TZM-bl and HLM-1 cells were obtained from the AIDS Research and Reference Reagents Program (NIH, Bethesda, MD, USA). TZM-bl cells are engineered HeLa cells that express CD4, CCR5, and CXCR4 and contain integrated reporter genes for firefly luciferase and β-galactosidase under the control of an HIV-1 long terminal repeat (82). HLM-1 cells are HeLa-T4+ cells containing one integrated copy of the HIV-1 genome with a Tat-defective mutation at the first AUG of the Tat gene. In the absence of stimulation, HLM-1 is completely negative for virus particle production, and viral transcripts are completely absent (83). Suspensions of J1.1 and ACH2 cells and their uninfected counterparts, Jurkat and CEM cells, as well as the promonocytic U1 cell line and the corresponding uninfected U937 cell line, were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated FBS, 1% l-glutamine, and 1% streptomycin/penicillin. The J1.1 cell line is a Jurkat E6.1 derivative chronically infected with HIV-1 (strain LAI) (84). ACH2 and J1.1 cells each contain a single integrated copy of the HIV-1 genome (85), whereas U1 cells harbor two copies (one wild type and one mutant) of the viral genome (strain LAV-1) in parental U937 cells (86). All cells were incubated at 37°C and 5% CO2. AP-7 SV-40 T-antigen-transformed olfactory neuronal cells (87) (a generous gift from Diane Griffin, Johns Hopkins University, Baltimore, MD, USA) were grown in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS, 1% l-glutamine, and 1% streptomycin/penicillin at 33°C in 7% CO2. Differentiated AP-7 (dAP-7) cells were derived from the AP-7 cells in the cycling state by placing them in differentiation medium composed of DMEM supplemented with insulin (2 μg/ml), 40 μM dopamine, and 200 μM ascorbic acid. The cells were allowed to differentiate at 39°C in 5% CO2 for 6 days prior to use in experiments. The HIV-1 Tat protein was prepared as described previously (88). Briefly, the Tat protein was expressed in Escherichia coli. Bacterial cells were sonicated and clarified by centrifugation before being chromatographed on a Sephacryl S-200 column and then, finally, purified on a reverse-phase high-performance liquid chromatography (HPLC) column. Full-length Tat protein (amino acids [aa] 1 to 86) was incubated with neuronal (dAP-7) cells in the presence or absence of 18BIOder. The effects of 18BIOder on Tat-mediated cell viability and cell death were analyzed by CellTiter-Glo and DNA fragmentation assays, respectively. Transcriptionally inactive Tat peptide containing a basic domain (aa 36 to 72) was used as a positive control for Tat-mediated neurotoxicity (32).
Transfections and luciferase assay.
TZM-bl cells were transfected with pc-Tat (0.5 μg) using the Attractene reagent (Qiagen, Chatsworth, CA, USA) according to the manufacturer's instructions. The next day, the cells were treated with dimethyl sulfoxide (DMSO) or the indicated compound at 1.0 μM. Forty-eight hours post-drug treatment, firefly luciferase activity was measured with the BrightGlo Luciferase Assay (Promega, Madison, WI, USA), and luminescence was read from a 96-well plate on an EG&G Berthold luminometer (Berthold Technologies, Oak Ridge, TN, USA).
Cell viability assay.
Fifty thousand cells per well were plated in a 96-well plate and treated the next day with 1.0 or 10 μM compound or DMSO. Forty-eight hours later, CellTiter-Glo (Promega, Madison, WI, USA) was used to measure viability following the manufacturer's recommendations. CellTiter-Glo is a luminescence assay used to measure cell viability by ATP level. The reagent was added to the wells (1:1 [reagent-medium]) and incubated at room temperature for 10 min protected from light. The luminescence was detected using the GloMax-Multi Detection System (Promega).
Small-molecule compounds.
The selected 2nd-generation 6-bromoindirubin-3′-oxime (6BIO) derivatives used in this study were 6:6-bromo-5-methyl-1H-indole-2,3-dione 3-[(6-bromo-5-methyl-2-oxo-1,2-dihydro-3H-indol-3-ylidene)hydrazone] (6BIOder), 10:5,7-dibromo-1H-indole-2,3-dione 3-oxime (10BIOder), 18:6-chloro-7-methyl-1H-indole-2,3-dione 3-oxime (18BIOder), and 19:4-chloro-7-methyl-1H-indole-2,3-dione 3-oxime (19BIOder). All inhibitors were obtained from Hit2Lead (ChemBridge Corporation, San Diego, CA, USA) and prepared in 10 mM stock solution dissolved in DMSO.
In silico docking of BIOders onto GSK-3β.
The small-molecule docking simulations on GSK-3β were performed using the AutoDock software package version 3.05 (Scripps Research Institute, La Jolla, CA, USA). For GSK-3β, the protein receptor, while held rigid, was taken from Protein Data Bank (PDB) file 1UV5, and the Kollman charges were added. PRODRG (89) was used to prepare the ligands, 6BIO, 6BIOder, 18BIOder, and 19BIOder. Mass-centered grid maps were generated with 0.15-Å spacing by the AutoGrid program only for the ATP pocket of GSK-3β, with default parameters. This grid was then used to dock the above-mentioned ligands locally to the ATP pocket. A Lamarckian genetic algorithm (LGA) scheme was chosen for conformational searching and optimization. For each receptor-ligand pair, 20 simulations with random initial ligand conformation were performed with LGA. The complex conformation with the lowest binding free energy was used to visualize the binding mode within the Chimera program (90), where only hydrogen bonds were highlighted with distance. The schematic diagram of a more detailed interaction network of the binding mode was generated by the LIGPLOT program (91).
Reverse-transcriptase activity and p24 enzyme-linked immunosorbent assays (ELISAs).
For reverse-transcriptase (RT) assays, supernatants from infected cells (10 μl) were incubated in a 96-well plate with RT reaction mixture containing 1× RT buffer (50 mM Tris-HCl, 1 mM dithiothreitol [DTT], 5 mM MgCl2, 20 mM KCl), 0.1% Triton, poly(A) (10 to 2 U), poly(dT) (2 to 10 U), and [3H]TTP. The mixture was incubated overnight at 37°C, and 5 μl of the reaction mixture was spotted on a DEAE filter mat paper (PerkinElmer, Shelton, CT, USA), washed four times with 5% Na2HPO4 and three times with water, and then dried completely. RT activity was measured in a Betaplate counter (Wallac, Gaithersburg, MD). Similarly, supernatants from infected cells were incubated in a 96-well plate precoated with HIV-1 anti-p24 antibody following the manufacturer's protocol (SAIC-NCI, Frederick, MD). Quantitative analysis of HIV-1 in the supernatant was performed based on a linear standard curve generated by the optical densities (ODs) of serial dilutions of known amounts of p24 antigen.
Protein extracts and immunoblotting.
Cells were collected, washed once with phosphate-buffered saline (PBS), and pelleted. The cells were lysed in a buffer containing Tris-HCl, pH 7.5, 120 mM NaCl, 5 mM EDTA, 0.5% NP-40, 50 mM NaF, 0.2 mM Na3VO4, 1 mM DTT, and one tablet of Complete protease inhibitor cocktail per 50 ml. Lysis was performed on ice, incubated on ice for 30 min, and spun at 4°C for 5 min at 14,000 rpm. The protein concentration for each preparation was determined with a Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA). Cell extracts were resolved by SDS-PAGE on a 4 to 20% Tris-glycine gel (Invitrogen, Carlsbad, CA, USA). Proteins were transferred to polyvinylidene difluoride microporous membranes using the iBlot dry-blotting system as described by the manufacturer (Invitrogen). The membranes were blocked with Dulbecco's PBS (0.1% Tween 20 plus 3% bovine serum albumin [BSA]). Primary antibody against the specified protein was incubated with the membrane in blocking solution overnight at 4°C. Antibodies against GSK-3β (1V001) and β-actin (C-11) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). HIV-IG, the anti-gp120 antibody (#3957), was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, and contributed by Jonathan Karn. Membranes were washed twice with PBS plus 0.1% Tween 20 and incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h in blocking solution. The presence of secondary antibody was detected by SuperSignal West Dura Extended Duration Substrate (Pierce, Rockford, IL, USA). Luminescence was visualized on a Kodak 1D image station (Carestream Health, Rochester, NY, USA).
Immunofluorescent staining.
HLM-1 or HeLa-T4+ cells were grown on glass slides for 3 days in the presence or absence of 2 mM sodium butyrate (NaB) or transfected with pc-Tat (5 μg) using the Attractene reagent (Qiagen). Cells were fixed for 1 h in 4% paraformaldehyde at room temperature. The cells were then permeabilized with 0.5% Triton X-100 in PBS for 20 min. Next, the cells were washed with PBS without Mg2+ and Ca2+. After the wash, the cells were incubated with RNase A at 10 μg/ml for 30 min at 37°C and washed again with PBS without Mg2+ and Ca2+. The cells were then blocked for 10 min at room temperature in PBS plus 3% BSA. Primary antibodies for anti-GSK-3β, GIT2 (sc-5416), and Nef (sc-17437) were obtained from Santa Cruz. The antibodies were incubated in fresh blocking buffer at 37°C for 1 h and washed 3 times for 3 min each time in 300 mM NaCl with 0.1% Triton X-100. Alexa Fluor 488 goat anti-rabbit (A11008; Invitrogen) and Texas Red goat anti-mouse (T862; Invitrogen) at a dilution of 1:200 were used as secondary antibodies and treated in the same manner as the primary antibody. DAPI (4′,6-diamidino-2-phenylindole) (dilution, 1:1,000) was used to visualize nuclei. Fluorescence microscopy was carried out using a Nikon Eclipse 90i microscope (Nikon Instruments Inc., Melville, NY, USA).
Size exclusion chromatography.
Uninfected and infected log-phase Jurkat and J1.1 T cells were pelleted for analysis. The cell pellets were washed twice with PBS without Mg2+ and Ca2+ and resuspended in lysis buffer (50 mM Tris-HCl, pH 7.5, 120 mM NaCl, 5 M methylenediaminetetraacetic acid, 0.5% NP-40, 50 mM NaF, 0.2 mM Na3VO4, 1.0 mM DTT, and one Complete protease cocktail tablet per 50 ml) and incubated on ice for 20 min, with gentle vortexing every 5 min. The lysates were then centrifuged at 4°C at 10,000 rpm for 10 min. The supernatants were transferred to a fresh tube, and protein concentrations were determined using the Bradford protein assay (Bio-Rad, Hercules, CA, USA). For each cell line, 2.5 mg of protein was brought up to 1.0-ml total volume using chromatography running buffer (0.2 M Tris-HCl, pH 7.5, 0.5 M NaCl, and 5% glycerol). The lysates were run on a Superose 6 10/300 size exclusion chromatography column (GE Healthcare Bio-Sciences, Uppsala, Sweden) using the ÄKTA Purifier system (GE Healthcare Bio-Sciences, Piscataway, NJ, USA). A quarter-inch gap was introduced into the top of the Superose 6 column to better separate low-molecular-weight complexes from fractions eluting off the far-right side of the chromatogram. After sample injection (using a 1-ml loop), the running buffer was set at a flow rate of 0.3 ml/minute, and 0.5-ml fractions of the flowthrough were collected at 4°C for a total of approximately 60 fractions. Every 5th fraction was acetone precipitated (∼250 μl) using 4 volumes of ice-cold 100% acetone with incubation for 15 min on ice. The lysates were centrifuged at 4°C for 10 min at 14,000 rpm, the supernatants were removed, and the pellets were dried for about 10 min at 95°C. The pellets were resuspended in Laemmli buffer and analyzed by immunoblotting with GSK-3β and actin antibodies (Santa Cruz).
Mass spectrometry.
Fractions corresponding to the medium- and low-molecular-weight GSK-3β complexes were immunoprecipitated with an anti-GSK-3β antibody. The immunoprecipitated material was eluted from the A/G beads (Calbiochem). Identification of the peptides was performed with an LTQ tandem mass spectrometer (MS-MS) equipped with a reverse-phase liquid chromatography (LC) nanospray (Thermo Fisher Scientific, Huntsville, AL, USA). The reverse-phase column was slurry packed in house with 5-μm, 200-Å-pore-size C18 resin (Michrom Bioresources, Auburn, CA) in a 100-μm by 10-cm fused silica capillary (Polymicro Technologies, Lisle, IL) with a laser-pulled tip. After a sample injection, the column was washed for 5 min at 200 nl/minute with 0.1% formic acid; peptides were eluted using a 50-min linear gradient from 0 to 40% acetonitrile and an additional step of 80% acetonitrile (all in 0.1% formic acid) for 5 min. The LTQ MS was operated in a data-dependent mode in which each full MS scan was followed by five MS-MS scans where the five most abundant molecular ions were dynamically selected and fragmented by collision-induced dissociation using normalized collision energy of 35%. Tandem mass spectra were matched against the National Center for Biotechnology Information mouse database with SEQUEST BioWorks software (Thermo Fisher Scientific) with full tryptic cleavage constraints and static cysteine alkylation by iodoacetamide. For a peptide to be considered legitimately identified, it had to be the number one matched and had to achieve cross-correlation scores of 1.9 for [M+H]1+, 2.2 for [M+2H]2+, and 3.5 for [M+3H]3+ with a ΔCn value of >0.1 and a maximum probability of randomized identification of 0.01.
RNA extraction, RT-PCR, and qRT-PCR.
For mRNA analysis of Cdk9-related genes following drug treatments, total RNA was isolated from cells using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer's protocol. Five hundred nanograms of total RNA was used to generate cDNA with the iScript cDNA Synthesis kit (Bio-Rad) using oligo(dT) reverse primers. Cdk9-responsive gene primers have been previously described (92). RNA was analyzed by quantitative RT-PCR (qRT-PCR). qRT-PCR assays were performed using the ABI Prism 7000 and Invitrogen's RNA UltraSense one-step quantitative-RT-PCR system. The cycling conditions were as follows: 1 cycle at 50°C for 15 min, 1 cycle at 95°C for 2 min, and 40 cycles at 95°C for 15 s and 53°C for 30 s. The absolute quantification was calculated based on the threshold cycle (Ct) relative to the standard curve.
Immunoprecipitation and in vitro kinase assay.
For immunoprecipitation (IP), select fractions obtained from size exclusion chromatography were divided into equal parts and immunoprecipitated overnight at 4°C with GSK-3β or normal mouse IgG (Santa Cruz) antibody. The next day, the complexes were precipitated with A/G beads (Calbiochem, La Jolla, CA, USA) for 2 h at 4°C. IPs were washed twice with appropriate TNE buffer (0.5 ml/wash) and kinase buffer (0.5 ml/wash). Reaction mixtures (20 μl) contained TTK kinase buffer with 50 mM HEPES, pH 7.9, 10 mM MgCl2, 6 mM EGTA, 2.5 mM dithiothreitol, and γ-32P (0.2 mM; 1 μCi). Phosphorylation reactions were performed with IP material and 200 ng of glycogen synthase peptide 2 (12-241; Millipore, Temecula, CA, USA) as the substrate. Reaction mixtures were incubated at 37°C for 1 h, stopped by the addition of 1 volume of Laemmli sample buffer containing 5% β-mercaptoethanol, and analyzed by SDS-PAGE on a 4 to 20% gel. The gels were stained, destained, dried, and subjected to autoradiography. Quantitation was performed using Molecular Dynamics PhosphorImager software (Amersham Biosciences, Piscataway, NJ, USA).
Nanoparticle enrichment of gp120 from primary macrophage cell culture supernatants.
The nanoparticles NT082 (Cibacron Blue F3G-A) and NT084 (Acid Black 48) were generously provided by Ceres Nanosciences. The nanoparticles were synthesized as previously described (93, 94). Briefly, core shells were generated in the following manner: N-isopropylacrylamide (NIPAm), N′N-methylene bisacrylamide (BIS), and acrylic acid (AAc) or allylamine (AA) were polymerized via precipitation polymerization. The NIPAm-Bis-AAc core shell of NT084 was covalently functionalized with amino dyes by an amidation reaction in dimethylformamide in the case of Acid Black 48. The NIPAm–co-AA core shell of NT082 was functionalized with Cibacron Blue F3G-A in a mixture of sodium carbonate and water. To prepare cell culture supernatants for downstream Western blot analysis of gp120 and Tat, we generated a 30% slurry of both NT082 (Cibacron Blue F3G-A) and NT084 (Acid Black 48) in RPMI. We then added this slurry (35 μl) to HIV-1 89.6-infected primary macrophage culture supernatants (500 μl) pooled from three donors. After rotation for 30 min at 25°C, samples were centrifuged at 10,000 rpm for 5 min. The supernatants were aspirated, and samples were washed twice with RPMI. After a final centrifugation at 10,000 rpm for 5 min, Laemmli buffer (10 μl) was added to elute materials off the nanoparticles. The nanoparticles were incubated at −20°C for 10 min and then vortexed and heated at 95°C for 5 min. Next, the nanoparticles were centrifuged at 10,000 rpm for 5 min, and the entire volume of Laemmli buffer was loaded onto the gel.
Exosome isolation.
To collect and prepare exosomes for downstream cytokine array analysis, we purified the exosomes using ExoQuick reagent (EXOQ5A-1; System Biosciences, Mountain View, CA, USA) according to the manufacturer's instructions. Briefly, pooled primary cell culture supernatants (1.5 ml) were incubated with ExoQuick reagent (750 μl), and the two components were incubated overnight at 4°C. The next day, the exosomes were pelleted by centrifugation at 5,000 rpm and resuspended in PBS (25 μl) prior to use in cytokine assays.
Animal experiments.
All mice used in the study were maintained within the National Center for Biodefense and Infectious Disease's breeding colony (George Mason University, Manassas, VA, USA). All experiments were carried out in biosafety level 2 (BSL-2) facilities and in accordance with the Guide for the Care and Use of Laboratory Animals (Committee on Care And Use of Laboratory Animals of the Institute of Laboratory Animal Resources, National Research Council; NIH Publication no. 86-23, revised 1996). NOD.Cg-Rag1tm1MomIl2rgtm1Wjl/SzJ mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA; 007799) and were humanized as described previously (74). Briefly, neonatal animals were sublethally irradiated. The next day, the mice were intraperitoneally implanted (100 μl) with approximately 1 × 105 human cord blood-derived CD34+ hematopoietic stem cells (2C-101B; Lonza, Walkersville, MD, USA). The cells were allowed to differentiate for 2 to 3 months. Five groups of two animals each were injected subcutaneously with vehicle (DMSO) or 1.0 mg/kg of body weight and 10 mg/kg of two drugs (18- and 19BIOder) 1 day prior to infection. The animals were subsequently infected subcutaneously with the dual-tropic HIV-1 89.6 (100 μl) and then further injected subcutaneously with the respective doses of drugs. Blood samples were collected 14 days postinfection, and serum was separated using Sarstedt Microvette 200 Z-Gel (20.1291; Sarstedt AG & Co., Nümbrecht, Germany) for downstream assays. An additional two groups of two animals each were subcutaneously injected with DMSO or 1.0 mg/kg 18BIOder 1 day prior to infection, subsequently infected subcutaneously with HIV-1 89.6, and then further treated with 18BIOder (1.0 mg/kg) at days 2, 5, 8, and 22. Blood was collected at days 14, 18, 25, and 29, and serum was separated as described above for downstream assays.
RESULTS
18BIOder inhibits HIV-1 Tat-dependent transcription.
We previously identified the GSK-3β inhibitor 6BIO as a potent HIV-1 LTR transcriptional inhibitor by performing a high-throughput screen for Tat-dependent transcription inhibitors from Lopac-Sigma-Aldrich (1,280 compounds) and Spectrum-Microsource (2,000 compounds) (33). In order to identify more potent 6BIO analogs, a Hit2Lead compound query based on chemical structure yielded 38 commercially available derivatives. During HIV-1 transcriptional inhibition studies, two compounds of particular interest were identified as possible regulators of Tat-activated transcription. Both derivatives 18 and 19 are composed of half the core structure of the parent compound, 6BIO, differing only in the location of the chlorine R group. These analogs were tested at 1.0 μM in a luciferase reporter gene system, scoring for activity against Tat-dependent HIV-1 LTR transcription. TZM-bl cells were transfected with Tat, followed by treatment with the parental (6BIO) and second-generation (BIOder) compounds. The results in Fig. 1A show luciferase activity assays performed in TZM-bl cells 2 days posttreatment. The data indicated that derivative 18 inhibited HIV-1 LTR Tat-dependent transcription more efficiently than 6BIO (6.7-fold and 1.7-fold decreases, respectively), whereas derivative 19 did not show any inhibitory effect. We then compared the inhibitory effects of 6BIO and its derivatives against both HIV-1 and Venezuelan equine encephalitis virus (VEEV). We used VEEV, since the 6BIOder compound was previously shown by our group to significantly inhibit viral replication (95). Quantification of previously obtained data (33, 95) in Fig. 1B indicated that 18BIOder was effective in inhibiting Tat-dependent HIV-1 transcription (>75% inhibition) but ineffective in VEEV infection (no inhibition). Likewise, 19BIOder moderately inhibited VEEV replication in vitro (50 to 75% inhibition) but had minimal effect on HIV-1-activated transcription (<5% inhibition). These findings indicate that although BIOder may be an effective inhibitor of both viruses, only a few derivatives may be selective against each virus in vitro.
FIG 1.
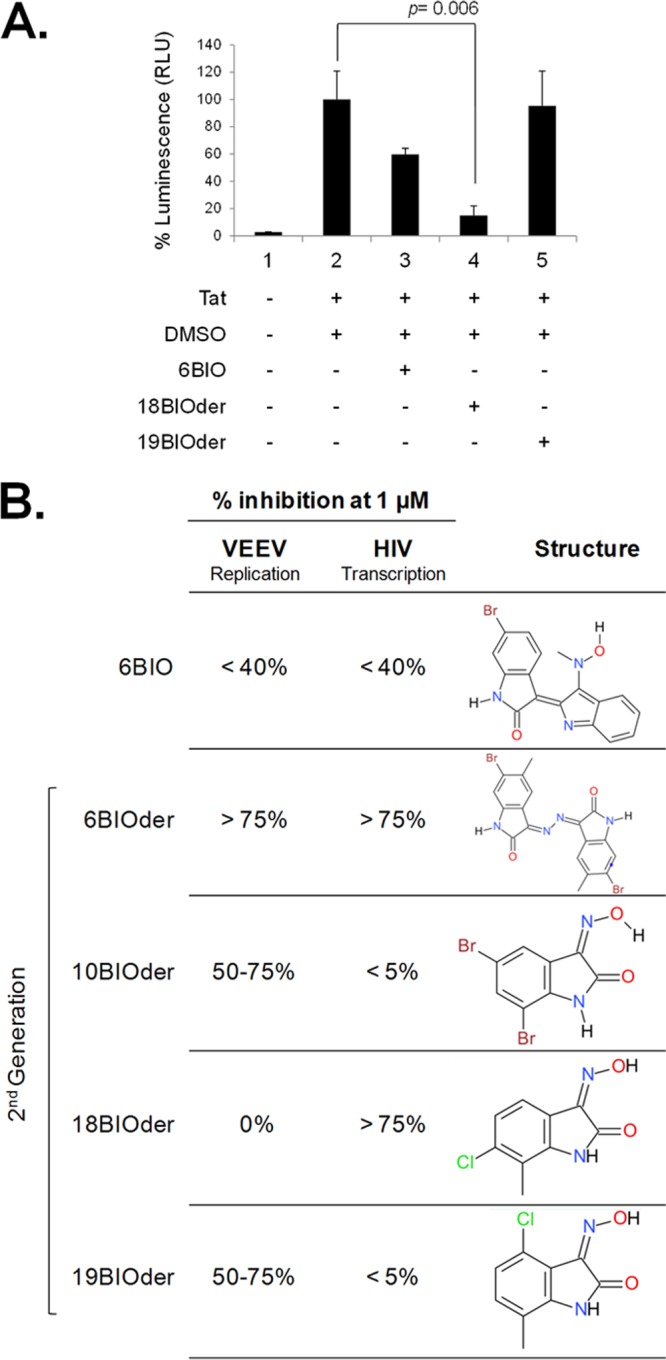
18BIOder inhibits HIV-1 Tat-dependent transcription. (A) TZM-bl cells were transfected with 0.5 μg of Tat plasmid and treated the next day with DMSO, 6BIO, and 18- and 19BIOder (1.0 μM). The cells were processed 48 h post-drug treatment for luciferase assays. The assays were performed in triplicate, and average values plus standard deviations are shown. Statistical significance was determined by Student's t test. RLU, relative light units. (B) Summary of virus-inhibitory properties of sample 6BIO second-generation derivatives against VEEV and HIV-1, and compound structures depicting the structural variance of 6BIOder, 6-bromo-5-methyl-1H-indole-2,3-dione 3-[(6-bromo-5-methyl-2-oxo-1,2-dihydro-3H-indol-3-ylidene)hydrazone]; 10BIOder, 5,7-dibromo-1H-indole-2,3-dione 3-oxime; 18BIOder, 6-chloro-7-methyl-1H-indole-2,3-dione 3-oxime; and 19BIOder, 4-chloro-7-methyl-1H-indole-2,3-dione 3-oxime. The percent inhibition of viral replication (VEEV) and transcription (HIV-1) was quantified and categorized based on previously published data (33, 95). Briefly, U87MG astrocytes were pretreated for 2 h with DMSO or BIO (5, 2.5, or 1.25 μM), infected with VEEV TC-83 at a multiplicity of infection (MOI) of 0.1, and posttreated with the compounds. Twenty-four hours postinfection, the viral supernatants were collected and assayed for viral replication by plaque assays. For HIV-1 studies, TZM-bl cells were processed as previously described (33). The data are representative of three technical triplicates and at least 2 biological replicates.
18BIOder is a cytoprotective small-molecule inhibitor.
GSK-3β inhibitors have been shown to have prosurvival activity and to regulate cell proliferation in various cell types (96–99). In order to exclude potential cytotoxicity of our compounds, we first assessed the effects of 18BIOder on cell viability in various cell lines, including uninfected (CEM and Jurkat) and HIV-1-infected T cells (ACH2 and J1.1) and monocytes (U937 and HIV-1-infected U1). Flavopiridol (flavo) (100 nM), a well-characterized pharmacological Cdk9 inhibitor, was used as a positive control for HIV-1 transcriptional inhibition in these assays (reference 100 and data not shown). The results in Fig. 2A indicate that the concentrations used in our transcriptional inhibition assays show minimal compromise in cell viability by 18BIOder in either uninfected or infected cell lines. Statistical significance was determined by Student's t test. Significant P values were observed only when DMSO (lane 2) was compared to18BIOder (lane 3) in the left panel (P = 0.013) and 18BIOder (lane 4) in the middle panel (P = 0.046). To further characterize the effect of 18BIOder on cell viability, we performed flow cytometry analysis of Jurkat cells treated with 1.0 and 10 μM 18- and 19BIOder. The cells were treated with each drug, maintained for 3 days, and collected for analysis. The results in Fig. 2B showed that there is no increase in the sub-G1 population with 18BIOder treatment. In contrast, there is a 10 to 25% increase with 19BIOder treatment, suggesting increased death in these cells. The cell cycle profile was not affected by drug treatment compared to the untreated control (data not shown).
FIG 2.

Lack of cellular toxicity when using 6BIO derivatives. (A) A panel of uninfected and infected cell pairs, including CEM and ACH2 (T cells), Jurkat and J1.1 (T cells), and U937 and U1 (monocytes), were treated with vehicle or 1.0 μM or 10 μM 18BIOder to determine possible compound toxicity in vitro. Cell viability was determined by CellTiter-Glo assay. Statistical significance was determined by Student's t test, and the significant P values are marked with asterisks. (B) Uninfected Jurkat cells were treated with 1.0 μM or 10 μM 18- or 19BIOder for 72 h. The cells were processed with PI stain, and the cell cycle was analyzed by flow cytometry. The percent sub-G1 (apoptosis) population is plotted to visualize the cell death detected. (C) Primary macrophages from three donors were infected with HIV-1 89.6 by spinoculation and treated with DMSO or 10 μM 18BIOder or 6BIOder on the day of infection, as well as every 2 days postinfection (total, 3 treatments). Cell viability was determined by CellTiter-Glo assay 6 days posttreatment. (D) The HIV-1 89.6-infected primary macrophages from the same donors were similarly treated with 10 μM 18BIOder (as shown in panel C) and then stained with PI and analyzed by flow cytometry, and the percent sub-G1 (apoptosis) population was plotted. The experiments for panels A to D were performed in triplicate. The error bars indicate standard deviations.
Next, we were interested in determining the effect of 18BIOder treatment in primary human cells. Primary macrophages were infected with HIV-1 89.6 and treated with 10 μM on the day of infection, as well as every 2 days postinfection (a total of 3 treatments). The results in Fig. 2C showed that after receiving three treatments of 18BIOder or 6BIOder, there was no significant loss of cell viability at day 6 posttreatment in cells from three different donors. The effect of 18BIOder on primary macrophages was further analyzed by staining these cells with propidium iodide (PI) and measuring apoptosis by flow cytometry. As shown in Fig. 2D, there was no increase in the sub-G1 cell population with 18BIOder treatment compared with DMSO. Collectively, these results indicate that 18BIOder can inhibit HIV-1 transcription without inducing cellular toxicity in either cell lines or primary cells.
18BIOder inhibits HIV-1 replication in a dose-dependent manner.
We next examined whether 18BIOder was effective in inhibiting HIV-1 replication in chronically infected cell lines. J1.1 cells were treated with vehicle (DMSO) or two concentrations (1.0 and 10 μM) of 18BIOder and 19BIOder. These cells are chronically infected and produce high viral titers in the supernatant without cell death. Cells were treated with each drug and maintained for 6 days, and the supernatants were collected for RT analysis. As shown in Fig. 3A, although not statistically significant (by the Student t test), multiple treatments of 10 μM 18BIOder inhibited virus replication by almost 50% (P = 0.12). Interestingly, there was no compromise in the viability of these cells when treated with 18BIOder as tested by CellTiter-Glo assays (Fig. 3B). While flavo treatment decreased cell viability to a moderate level, the viability of nucleoside RT inhibitor (NRTI)-treated cells was markedly reduced. To determine the levels of viral replication inhibition in primary cells, peripheral blood mononuclear cells (PBMCs) from 3 separate donors were activated with interleukin 2 (IL-2)-phytohemagglutinin (PHA) for 3 days prior to infection with HIV-1 89.6 and treated only once with 0.01, 0.1, and 1.0 μM 18BIOder. The results in Fig. 3C showed a dose-dependent decrease of viral replication in PBMCs from all 3 donors as measured by p24 ELISA at day 6 postinfection. The levels of 18BIOder inhibition were analyzed by comparing them with DMSO treatment using the Student t test. Statistically significant virus inhibition was achieved with both 0.1 and 1.0 μM concentrations of 18BIOder (P = 0.030 and 0.018, respectively). There was no significant loss in cell viability with 18BIOder treatment in infected PBMCs compared with DMSO treatment (Fig. 3D). Collectively, these data show suppression of viral replication in the absence of cytotoxicity.
FIG 3.

Effects of 18- and 19BIOder on HIV-1 replication. (A) Log-phase growing HIV-1-infected J1.1 T cells were treated once or every 2 days (total, 3 treatments) with 1.0 μM and 10 μM 18- or 19BIOder and vehicle. Flavopiridol (100 nM) was used as a positive control. The supernatants were processed for the presence of RT activity at day 6 posttreatment. (B) J1.1 cells were treated once or every 2 days (total, 3 treatments) with 1.0 μM and 10 μM 18- or 19BIOder or 1.0 μM 6Bioder. Flavopiridol (100 nM) and an NRTI cocktail (10 μM) were used as controls. Cell viability was determined by CellTiter-Glo assay 6 days posttreatment. (C) IL-2–PHA-activated PBMCs from 3 separate donors were pretreated once with 0.01, 0.1, or 1.0 μM 18BIOder for 24 h and then infected with HIV-1 89.6. The supernatants from infected PBMCs were collected for p24 ELISA 6 days postinfection. The statistical differences between 18BIOder inhibition and DMSO treatment were analyzed by Student t test, and the significant P values are marked with asterisks. (D) PBMCs from 3 donors were similarly treated and infected (as in panel C) with 18Bioder, and cell viability was determined by CellTiter-Glo assay 6 days postinfection. The error bars indicate standard deviations.
GSK-3β expression is upregulated in HIV-1-infected cells.
We have previously shown that 6BIOder inhibits GSK-3β both in vitro and in vivo (33, 95). We next examined the effects of GSK-3β in various uninfected and infected pairs of T cells and monocytes using Western blot analysis. They included the uninfected cell lines Jurkat, CEM, and U937 and the HIV-1-infected cell lines J1.1, ACH2, and U1. We further utilized monocytoid cells (U937 mΦ/U1 mΦ) by inducing monocyte pairs with phorbol myristate acetate (PMA) (101, 102). The results in Fig. 4A indicate that GSK-3β levels were generally higher (∼2- to 5-fold increase) in HIV-1 chronically infected cells compared to uninfected cells. Endogenous GSK-3β exists within both active and inactive complexes in the cytoplasm (103). We therefore asked whether GSK-3β localization was differently affected in HIV-1-infected cells. For that, we utilized a fractionation of cytoplasmic versus nuclear extracts and also performed localization assays using confocal microscopy. The results in Fig. 4B indicate that there was more GSK-3β in the cytoplasm (∼4-fold increase compared to uninfected cells) and to a lesser extent in the nucleus (∼2-fold increase compared to uninfected cells). Localization of lamin A (nuclear) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (cytoplasmic) served as positive controls. Finally, we utilized an HIV-1-infected silent cell line, HLM-1, which harbors an integrated provirus containing a mutated Tat gene with a triple termination codon (83). These cells can be induced to produce virus using histone deacetylase (HDAC) inhibitors, such as NaB. We therefore plated log-phase growing cells, induced virus production using NaB, and looked for localization of GSK-3β. The results in Fig. 4C show that GSK-3β was mainly dispersed throughout the nucleus and, to a lesser extent, the cytoplasm prior to induction of HLM-1 (Fig. 4C, I). However, after NaB treatment, the overall expression of GSK-3β was upregulated (Fig. 4C, II). The observed upregulation of GSK-3β in the cytoplasm was specific to virus activation, since treatment of HeLa-T4+ with NaB did not result in significantly altered GSK-3β expression (Fig. 4C, III and IV). Consistent with these results, we found increased GSK-3β levels in both the nucleus and cytoplasm of induced HLM-1 cells when counting more than 100 cells per condition (Fig. 4D). To further reinforce that the observed GSK-3β phenotype is specific to viral reactivation in HLM-1 cells, we performed confocal analysis of GSK-3β and Nef as viral markers in cells that had been treated with NaB or Tat (transfection). The GIT2 protein was used as a cytoplasmic positive-control marker. The results in Fig. 4E showed that cells exhibiting altered GSK-3β expression are positive for Nef expression, indicating active viral expression in these cells. As expected, the untreated HLM-1 cells did not show staining for GSK-3β, GIT2, or Nef (Fig. 4F). Taken together, these results are suggestive of altered GSK-3β dynamics during HIV-1 infection, as indicated by increased protein expression levels and intracellular distribution.
FIG 4.

GSK-3β expression is upregulated in HIV-1 infection. (A) To assess GSK-3β expression in a series of uninfected and infected cell pairs, 100 μg of whole-cell extract from Jurkat/J1.1, CEM/ACH2, U937/U1, and PMA-treated U937/U1 monocytoid cells (U937 mΦ/U1 mΦ) were run on 4 to 20% SDS-PAGE and immunoblotted against GSK-3β and β-actin as a loading control. Western blots were performed in duplicate. (B) Cytosolic and nuclear extracts were prepared from 5 × 106 Jurkat and J1.1 cells. The extracts were analyzed by Western blotting using anti-GSK-3β. Actin was utilized as a loading control. Lamin A and GAPDH were used as nuclear and cytoplasmic controls, respectively. Western blots were performed in duplicate. (C) HeLa-T4+ and HLM-1 cells were grown on coverslips in the presence or absence (mock) of NaB for 72 h. The cells were stained for GSK-3β and visualized with confocal microscopy. Cellular GSK-3β protein staining is represented in green in the cytoplasm, and nuclei were stained with DAPI (blue). Row I shows HLM-1 mock cells (control), row II shows HLM-1cells induced with NaB, row III shows HeLa-T4+ mock cells (control), and row IV shows HeLa-T4+ cells treated with NaB. Upregulated GSK-3β levels observed in induced HLM-1 (activated virus) are indicated with white arrows in row II (merge). (D) HLM-1 cells were treated with NaB for 72 h. GSK-3β localization was determined by confocal microscopy as in panel C. One hundred cells per condition were counted and scored for the presence of GSK-3β distribution and upregulation in HLM-1 and HeLa-T4+ cells. Data collection was performed in triplicate. (E) HLM-1 cells were grown on coverslips in the presence of NaB or transfected Tat for 72 h. The cells were stained for GSK-3β, Nef, and GIT2 and were visualized with confocal microscopy. Cellular GSK-3β protein staining is represented in green, Nef and GIT2 are red, and nuclei were stained with DAPI (blue). GIT2 served as a cytoplasmic protein control. (F) As background control, HLM-1 cells were also stained for Nef, GIT2, or GSK-3β in the absence of NaB treatment or Tat transfection. NS, nonspecific.
GSK-3β upregulation and complex redistribution in HIV-1-infected cells.
It is widely accepted that viral infection causes profound changes in the dynamics of the host cell. For this reason, the elucidation of novel protein complexes in infected but not in uninfected cells is a great tool for the understanding of viral mechanisms and the development of new host-based therapies. Based on the observed GSK-3β upregulation and altered cellular distribution in infection, we next asked whether the virus could generate novel GSK-3β complexes that could serve as a target for 18BIOder. To this end, size exclusion chromatography serves as a tool to efficiently fractionate functional protein complexes of interest for further downstream analysis (95, 104, 105). In fact, in a recent study, this technique was utilized to successfully discern novel GSK-3β complex formation in VEEV-infected U87MG cells (95). In order to determine the elution profile of GSK-3β in HIV-1-infected cells, we performed size exclusion chromatography to separate protein complexes, followed by Western blot analysis. We prepared total protein extracts from matched parental uninfected Jurkat and infected J1.1 cell lines and loaded the samples on a Superose 6 size exclusion column in the presence of a stringent 500 mM salt concentration. The high salt concentration provides specificity for stable protein complexes. Samples were fractionated in water-soluble buffer, followed by precipitation to concentrate protein fractions and remove excess salt. Roughly every 5th fraction was assayed by Western blotting for the presence of GSK-3β and β-actin. The results in Fig. 5A show a dramatic differential GSK-3β signature in the lower-molecular-weight fractions between uninfected and infected cells. Uninfected cells showed a predominant band with a molecular mass of ∼300 kDa in fraction 35, most likely representing homo- or heterodimers alongside other bound proteins. However, when utilizing the HIV-1-infected cells, we observed a different distribution profile of GSK-3β. The infected J1.1 gel, lanes 7 to 11, showed significant presence of GSK-3β in fractions 40 to 55 compared to uninfected cells. Additionally, we observed increased expression and redistribution of actin in infected cells. This is in agreement with HIV-1 dependence on the actin cytoskeleton, where the virus utilizes actin to facilitate entry, reverse transcription, and nuclear migration for infection establishment (106). Interestingly, the levels of lamin A and gelsolin were similar in the two cell types; however, GAPDH levels slightly increased in the Jurkat cell uninfected fractions 45 to 55 (data not shown). We next examined the GSK-3β expression profile in a set of other cells, and the results in Fig. 5B indicate a similar pattern of GSK-3β redistribution observed in paired monocytic uninfected and infected cells. Similar to the T cell signature, GSK-3β protein is distributed to lower-molecular-weight fractions in U1 cells compared to the uninfected U937 cells. The low-molecular-weight GSK-3β protein complexes unique to the infected lysates were found in the fractions associated with the right-hand side of the A280 chromatogram (Fig. 5C). Of interest, the total protein lysate eluted as a bell curve (Fig. 5C, inset), with smaller amounts of proteins eluting in the high- and low-molecular-weight regions and the majority of proteins eluting around fraction 30 (data not shown). To further investigate protein distribution changes, we concentrated a small sample of neighboring relevant fractions of both Jurkat and J1.1 cell fractionations, resolved them by SDS-PAGE, and silver stained the gel. The results in Fig. 5D indicate that fractions 37 and 47 of uninfected and infected cells contain relatively similar amounts of total protein. To further characterize these complexes, we performed mass spectrometry analysis of J1.1 fractions 37 and 47. Briefly, GSK-3β was immunoprecipitated from these fractions and subjected to LC–MS-MS. Our data suggest that GSK-3β found in lower-molecular-weight fractions (fraction 47) is mostly present as free enzyme, while GSK-3β in larger complexes (fraction 37) interacts with putative binding partners. Identified binding proteins included activating signal cointegrator 1 (ASC-1), a complex known to enhance transactivation of NF-κB, SRF, and AP1 (107), all of which are factors linked to HIV-1 activation. To examine interaction between GSK-3β and ASC-1, we performed GSK-3β immunoprecipitation from Jurkat and J1.1 whole-cell lysates, in addition to J1.1 fractions 37 and 47. The results in Fig. 5E indicate that there is increased enrichment of ASC-1 in J1.1 cell lysates and fractions compared to Jurkat cells (compare lanes 3, 5, and 7 to 8), suggesting altered and novel binding dynamics between GSK-3β and ASC-1 in infection. Collectively, these results imply that novel GSK-3β complexes are formed during HIV-1 infection in both T cells and monocytic lines.
FIG 5.

Presence of novel GSK-3β complexes in HIV-1-infected cells. (A) Samples from uninfected Jurkat (top) and infected J1.1 (bottom) T cells were loaded in a size exclusion chromatography column and separated in the presence of 500 mM salt buffer. No detergent was used during fractionation. A sample (250 μl) of every 5th fraction from 10 to 55 was acetone precipitated and resuspended in 30 μl of Laemmli buffer. Fifteen microliters was run on a gel for Western blotting for the presence of GSK-3β and β-actin. Western blots were performed in duplicate. (B) Similar to panel A, uninfected U937 (top) and infected U1 (bottom) monocytic cells were loaded in a size exclusion chromatography column and separated. Western blots were performed in duplicate. (C) Two and a half milligrams of J1.1 WCE (lysis buffer described in Materials and Methods) was brought up to 500 μl in running buffer and processed through the Superose 6 column as described in Materials and Methods. The chromatogram shows a sample A280 trace of the processed WCE sample. The inset depicts the overall bell-shaped elution profile of the WCE as confirmed through Western blots. The red box indicates the tailing fractions in which the low-molecular-weight (LMW) complexes specific to the infected J1.1 WCE were contained. (D) Fifty microliters of J1.1 fractions 37 and 47 was acetone precipitated and processed as in panel A. Fifteen microliters was run on a gel and silver stained to visualize the total protein content in these relevant fractions as a measure of loading control. (E) GSK-3β was immunoprecipitated from J1.1 whole-cell lysates and fractions 37 and 47. Eluted samples were subjected to mass spectrometry analysis. ASC-1 was a novel candidate protein identified by LC-MS analysis. To verify this putative interaction, GSK-3β was immunoprecipitated from Jurkat and J1.1 whole-cell lysates and J1.1 fractions 37 and 47. IP material was run on a gel and Western blotted for ASC-1 and GSK-3β. Western blots were performed in duplicate. NS, nonspecific; WCE, whole-cell extract.
18BIOder does not inhibit cellular gene expression in the absence of Tat and is specific to GSK-3β.
It is well known that HIV-1 Tat-dependent transcription requires canonical cellular transcriptional machinery, such as p-TEFb (Cdk9/cyclin T1), for promoter activation and transcriptional elongation. We have shown that 18BIOder is able to inhibit HIV-1 transcription. In order to determine that this effect was not due to the general downregulation of cellular gene expression in drug-treated cells, we sought to determine if 18BIOder treatment was inhibitory to cellular genes that require p-TEFb. The results in Fig. 6A show RT-PCR amplifications of CAD, CIITA, cyclin D1, and PBX-1, a set of genes that require Cdk9/cyclin T1for their transcription (108–110). None of these genes showed a decrease in transcription in various cell types, including 293T cells, CHME5 human microglial cells, and Jurkat T cells. To verify these RT-PCR findings in a more quantitative manner, we performed a representative qRT-PCR on the same RNA used in Fig. 6A. The results in Fig. 6B showed that for both 293T and Jurkat cells there was no decrease in PBX-1 transcripts upon treatment with 10 μM 18BIOder. These results further reinforce the notion that the transcriptional inhibition caused by 18BIOder may be specific to the HIV-1 promoter.
FIG 6.

18BIOder does not inhibit cellular gene expression in the absence of Tat and is specific to GSK-3β. (A) Total RNA was isolated from cells treated with 18BIOder (1.0 and 10 μM for 293T cells and 1.0 μM for CHME5 and Jurkat cells) using RNeasy Mini Kit extraction. A total of 500 ng was used to generate cDNA with the iScript cDNA synthesis kit using oligo(dT) reverse primers. The primers for PCR were CAD, CIITA, cyclin D1, PBX-1, and GAPDH as a control. (B) A representative panel of intracellular RNA levels of PBX-1 were determined by qRT-PCR using total RNA extracted from cells shown in panel A. The error bars indicate standard deviations. (C) GSK-3β IPs from fractions 35 and 50 (250 μl each) were mixed with 5 μg of GSK-3β or IgG (control immunoprecipitation) antibodies overnight and washed the next day, first with RIPA buffer (1×) and then TNE and 0.1% NP-40 (2×), followed by kinase buffer (3×), prior to addition of substrate and [γ-32P]ATP. Samples were run on a gel, stained, destained (over 4 h), dried, and then exposed to a phosphorimager cassette. (D) IP/kinase reactions were treated with 18BIOder (10 nM) in vitro during the kinase reaction. Samples were run on a gel and exposed to a phosphorimager cassette. The kinase assay was quantified by densitometry and expressed as percent kinase activity compared to the immunoprecipitated Jurkat fraction 35 GSK-3β activity. (Bottom) Coomassie gels show p-Gly pep 2 substrate stains.
We next examined the functional effects of various GSK-3β complexes from both uninfected and infected cells. GSK-3β is known to be regulated by pathways that include phosphorylation of the enzyme, protein complex formation, and intracellular localization (111). Therefore, we asked whether GSK-3β found in these small and medium-size complexes were active and whether 18BIOder was capable of inhibiting these GSK-3β complexes in vitro. To that end, we performed GSK-3β immunoprecipitation from fractions 35 (medium size) and 50 (small) from both uninfected and infected cells and used the immunoprecipitated material in an in vitro kinase assay with glycogen synthase peptide 2 as the substrate. The results in Fig. 6C indicated that GSK-3β present in fractions 35 from both uninfected and infected cells was active. In addition, these results also confirm the absence of GSK-3β in Jurkat fraction 50 (∼6% activity) and increased GSK-3β present in J1.1 fraction 50 (32% activity). We next used 18BIOder in kinase assays and found that upon treatment of the immunoprecipitated material with 18BIOder (10 nM), there was little inhibition of kinase activity from Jurkat fraction 35 (∼33% activity) (Fig. 6D, compare lanes 1 and 2). Interestingly, treatment of J1.1 fractions 35 (lanes 3 and 4) and 50 (lanes 5 and 6) showed more sensitivity to 18BIOder, with 36% and 41% kinase inhibition, respectively. Taken together, these data support the notion that 18BIOder transcriptional inhibition is specific to the GSK-3β present in HIV-1-infected cells.
18BIOder protects neuronal cultures from HIV-1 proteins and cytokine toxicities.
Currently, cART does not fully protect against HAND. Recently, neuroimaging studies have shown the presence of virus in the brain even at early stages of infection (112). To date, there are very limited interventions available to prevent HIV-1-associated neurotoxicity or dementia. Although Nef, Rev, and Vpr have been linked to neurotoxicity, the neurotoxic effects of HIV-1 are largely attributed to gp120 and Tat proteins (7, 9, 10, 27, 28, 113–115). Current cART treatment does not block production of early viral proteins, such as Tat, even during successful virus suppression, and thus, extracellular Tat released into the extracellular space can exert neurotoxic effects on bystander cells (116, 117). However, the pivotal release of Tat by infected cells and the consequent uptake by neurons remained undefined until recently. Quantified neuronal uptake of circulating Tat was achieved by using high-performance capillary electrophoresis and was measured to ∼2 pg/ml in both a neuronal cell line and primary cultures (23), highlighting the involvement of Tat in HAND. Several GSK-3β inhibitors, such as cesium chloride (118), lithium chloride (119), valproate (120), AR-A014418/B6B30 (78), and 6BIOder (33), have been shown to confer neuroprotection from Tat-induced neurotoxicity, though efforts to fully halt Tat-mediated neurodegeneration both in vitro and in vivo have failed (23). In order to further explore the role of 18BIOder in a more relevant system, we obtained primary macrophages from three independent donors. The macrophages were spinoculated with HIV-1 89.6 and subjected to treatment with 10 μM 18BIOder or 6BIOder at days 1, 3, and 5 postinfection, and cells and supernatants were harvested at day 6 postinfection. The results shown in Fig. 7A indicate that both 18- and 6BIOder dramatically reduced HIV-1 replication in macrophages from two of three donors (donors 1 and 3) as Env RNA copies measured by qRT-PCR. Unexpectedly, 6BIOder was more effective in reducing virus replication in the macrophages from donor 2. To examine the potential neurotoxin contents of these infected macrophages, we performed 2 separate assays. First, we pooled supernatants from new donors, 1 and 3. The supernatants were enriched for gp120 and Tat using the NT082 (Cibacron Blue F3G-A) and NT084 (Acid Black 48) nanoparticles (Ceres Nanosciences, Manassas, VA, USA), which we have shown to specifically bind and trap HIV-1 gp120 (data not shown) and Tat (121) from complex and dilute solutions. A 30% slurry of nanoparticles was added to each indicated fraction at equal volumes. Samples were incubated with rotation for 30 min at room temperature, spun down, and aspirated. Nanoparticles were resuspended in an equal volume of Laemmli buffer. Figure 7B shows the presence of gp120 and Tat, as detected by Western blotting in infected donor samples, with percent densitometry counts for gp120 displayed above the capture. 18BIOder-treated macrophages (Fig. 7B, lane 4) showed a 28% decrease in gp120 abundance compared to untreated cells (Fig. 7B, lane 2); however, there was no decrease in Tat levels, and therefore, the inhibition may be occurring at the level of Tat-activated transcription, resulting in lower levels of singly spliced messages, such as Env. Media from uninfected cells were used in nanotrap experiments as a negative control (Fig. 7B, lane 1).
FIG 7.

18BIOder protects microglial and neuronal cultures from the HIV-1 Tat protein. (A) Primary macrophages from three donors were infected with HIV-1 89.6 by spinoculation. After overnight recovery, the cells were treated with DMSO or 10 μM 18BIOder or 6BIOder, and thereafter every 2 days postinfection (total, 3 treatments). The supernatants were collected, and virus production was measured by analyzing Env RNA copies using qRT-PCR at day 6 postinfection. Triplicate samples from donors 1 and 3 and a single sample from donor 2 were analyzed. Insufficient material was recovered from donor 2 for triplicate analysis. (B) Supernatants collected from the infected macrophages at day 6 postinfection as in panel A were pooled and subjected to nanoparticle capture of gp120 and Tat by using NT082 (Cibacron Blue F3G-A) and NT084 (Acid Black 48). Nanoparticle contents were resuspended in 10 μl Laemmli buffer and run on a gel. Membranes were immunoblotted with HIV-1-infected human immune serum (for gp120) and α-Tat mouse monoclonal antibody. Normal human serum and mouse IgG were used as isotype controls. Densitometry counts were performed for α-gp120 blotting, and the effects of 18Bioder were compared. Western blotting was performed in duplicate. (C) Supernatants from panel A were subjected to exosome purification using Exoquick reagent following the manufacturer's instructions. Exosome material obtained from the infected macrophage supernatants were analyzed for cytokine expression using a custom human cytokine array (RayBio Human Cytokine Array 1) following the manufacturer's instructions. (D) dAP-7 neuronal cells were differentiated for 6 days and treated with 18- or 6BIOder (0.1 μM) for 1 h at 39°C prior to 18 h of exposure to 500 nM Tat1-86. Cell viability was tested using a CellTiter-Glo assay to determine the cytoprotection effect of BIOders. The quantifications are based on triplicate experiments, and statistical analyses were performed between DMSO and Tat1-86 treatments and also between Tat1-86 and BIOder treatments using Student's t test, and a significant P value is marked by an asterisk. Transcriptionally inactive Tat peptide containing the essential basic domain (positive control) and BSA (negative control) were used. (E) dAP-7 cells were similarly treated as in panel D and analyzed for cell death (apoptosis) by measuring DNA fragmentation at 405-nm absorbance in cell lysates. The error bars indicate standard deviations.
In addition to virus-derived neurotoxins, cellular factors, such as inflammatory chemokines and cytokines released by infiltrating macrophages and T cells and infected astrocytes and microglia, have been linked to neurotoxicity (122). Thus, we were interested in assaying the cytokine contents of these supernatants. To this end, circulating exosomes were isolated by ExoQuick precipitation. Exoquick-purified exosomes have been previously shown to contain RNA and protein of higher purity and in greater quantity than other routinely used isolation methods (123). RayBio cytokine arrays were used to screen 23 immunoregulatory molecules in pooled supernatants from infected primary macrophages subjected to 18BIOder treatment. Cytokine detection revealed changes between untreated infected and treated cells (Fig. 7C). There was a decrease in the majority of molecules screened, including granulocyte colony-stimulating factor (G-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF); GRO and GRO-α; IL-1α, -2, -3, -5, -6, -7, -8, -10, -13, and -15; gamma interferon (IFN-γ); monocyte chemoattractant protein 1 (MCP-1) and -2; MIG; RANTES; transforming growth factor β1 (TGF-β1); and tumor necrosis factor alpha (TNF-α). However, MCP-3 and TNF-β levels were not changed compared to the DMSO control. Of interest, MCP-3 is a CCR5 antagonist and, as such, could provide endogenous protection by inhibiting HIV-1 entry (124).
Consequently, we were interested in determining whether 18BIOder would also display comparable cytoprotection of neuronal cultures from HIV-1 Tat-induced cell death (125). dAP-7 cells were preincubated with 18- or 6BIOder (0.1 μM) for 1 h prior to exposure to the Tat protein. Both cell viability and cell death (apoptosis) were analyzed 18 h following Tat exposure by CellTiter-Glo and DNA fragmentation measurement assays, respectively. The results in Fig. 7D showed that treatment with functionally active full-length Tat protein (Tat1-86) significantly reduced cell viability compared to negative-control BSA treatment using the Student t test (P = 0.02). Transcriptionally nonfunctional Tat peptide, which contains the basic domain and has been shown to be neurotoxic (32), served as a positive control. Of importance, 18BIOder was protective against Tat-mediated cell death. Interestingly 18BIOder also showed marked, although not statistically significant, reduction of apoptosis caused by Tat1-86 treatment when DNA fragmentation was measured (Fig. 7E). Collectively, these results indicate that 18BIOder may be able to confer cytoprotection from Tat-induced cell death on neuronal cultures.
DISCUSSION
In the current study, we focused on defining unique cellular targets that may be used to inhibit HIV-1, decrease viral transcription at a low 50% inhibitory concentration (IC50), and avoid interference with native cellular transcription. We have expanded the characterization of potent HIV-1 Tat-dependent transcription inhibitors derived from a screen of over 3,000 compounds that initially identified the parental 6BIO compound (33) as a potent inhibitor of GSK-3 (126).
Originally described as a key regulatory enzyme in the process of glycogen synthesis, mammalian GSK-3 is a serine/threonine kinase that harbors two highly homologous isoforms, GSK-3α and GSK-3β (127). GSK-3β is a constitutively active endogenous enzyme that has a variety of multifaceted roles in cellular signaling that include regulation of neuronal plasticity, gene expression, and cell survival. GSK-3β is currently believed to be a key component of certain neurodegenerative and psychiatric diseases (111). Identification of GSK-3β inhibition by lithium at an IC50 of approximately 2 mM (128) provided an extraordinarily valuable tool that expanded the repertoire of inhibitors, along with SB-216763 (IC50, 34 nM) and SB-415286 (IC50, 78 nM) (129). The parental compound in this study, 6BIO, is a synthetic compound derived from its natural 6-bromoindirubin product, which displays a low IC50 of 5 nM and specific inhibition of GSK-3 both in vitro and in vivo in Xenopus embryos (126). Further characterized by the same group via cocrystallization experiments, 6BIO was demonstrated to bind the ATP pocket of GSK-3β. In addition, 6BIO has been shown to inhibit the cyclin-dependent kinase complexes Cdk1/cyclin B, Cdk2/cyclin A, and Cdk5/p35 at higher IC50s of 0.32, 0.30, and 0.08 μM, respectively (126). Also, the previously characterized 6BIO derivative 6BIOder has been shown to exert specific GSK-3β inhibition and to confer neuronal protection with decreased cytotoxicity compared to the parental 6BIO compound (33). Of interest, 6BIOder also exhibited potent viral suppression and cytoprotection in neurodegenerative infection by VEEV (95). 6BIOder was shown to decrease VEEV-induced cell death and replication at an IC50 of approximately 0.5 μM.
Our studies demonstrate the ability of a 6BIO derivative, 18BIOder, to inhibit Tat-dependent transcription. This small-molecule inhibitor has a low IC50 in transcriptional inhibitory assays and is composed of half the structure of 6BIO, making it an easier and more cost-effective compound to synthesize (Fig. 1A and B). To better define the mechanistic structure of these inhibitors, we docked these compounds to GSK-3β to probe their binding modes using a known crystal structure of the GSK-3β complex with a related small-molecule ligand, 6BIO (PDB ID 1UV5). We verified the accuracy of our docking simulations by closely reproducing the experimentally known binding mode of 6BIO (data not shown). Indeed, a very small root mean square deviation (RMSD) of 0.79 Å between the simulated and experimental binding conformations of 6BIO was obtained. The binding modes of the other three BIOder compounds, based on our docking simulations, were very distinct from each other.
From our simulation results, 6BIO and 6BIOder presented rather similar binding modes, forming hydrogen bonds with residues D133 and V135 (data not shown). Hydrogen bonding with the backbone portion of these two key residues, D133 and V135, was also observed among many other GSK-3β inhibitors (130, 131). Gentile et al. (130), using affinity-based screening of large-scale DNA-encoded chemical libraries, were able to select GSK-3β inhibitors of the desired specificity that formed one additional, albeit weak, hydrogen bond with the side chain of K85. While it remains unknown why interactions with K85 might enhance specificity, K85 could play a very important role, since it is well known that its equivalent, K33, in CDK2 with a flexible side chain was able to form salt bridges with the phosphate groups of ATP upon cyclin binding to activate CDK2 (132, 133). It is thus interesting to note that 18BIOder formed a very strong hydrogen bond with K85 (data not shown). However, 18BIOder could no longer maintain hydrogen bonding with D133 and V135 like 6BIO/6BIOder. It remains to be seen if it is desirable to design a BIOder that merges 6BIOder and 18BIOder to form strong hydrogen bonds with all three residues, D133, V135, and K85. Finally, 19BIOder moved to a surprisingly different location in its binding mode to GSK-3β from that used by 6BIOder and 18BIOder.
Similar to its potent inhibitory parent compound, 6BIOder, 18BIOder demonstrates its ability to inhibit Tat-dependent HIV-1 transcription without inducing significant cellular toxicity (Fig. 2). 18BIOder was also found to significantly inhibit viral replication in chronically infected T cells and PBMCs (Fig. 3A and C, respectively). Previously, it was shown that 6BIOder can potently inhibit viral replication in infected PBMCs at lower concentrations, though this small-molecule inhibitor was not tested in chronically infected high-viral-load-producing cells, as was done in this study (33). Interestingly, we have observed different levels of GSK-3β in the fractions containing lower-molecular-weight complexes present only in infected T cells or monocytoid cells (Fig. 5A and B, compare lanes 8 through 11), showing that protein levels or availability of GSK-3β present in these lower-molecular-weight fractions may play a role in the 18BIOder mechanism of inhibition.
Here, we also describe the general expression signature of GSK-3β in HIV-1 infection (Fig. 4A to C and 5A to E). We found that endogenous GSK-3β is overexpressed during infection in a variety of cells, including T cells, monocytes, and PMA-treated monocytoid cells. Furthermore, though modest, we have identified a slightly increased redistribution of cytoplasmic GSK-3β in cells that have undergone viral activation, suggesting that GSK-3β may be modulating signal transduction pathways in a way that benefits viral mechanisms of survival and infectivity. GSK-3β is characterized by regulation of transcription factors and cofactors that include β-catenin, c-Jun, c-Myc, C/EBPα/β, NFATc, RelA, and CREB (134, 135). Moreover, most of these regulatory targets have been implicated in Tat-mediated transcription (136). Members of the lymphoid enhancer factor-1/T-cell factor (LEF-1/TCF) family of transcription factors interact with β-catenin and play a role as nuclear effectors of Wnt signaling, which includes gene targets that impact cell differentiation, communication, apoptosis and survival, and proliferation (137, 138). A possibility of increased cytoplasmic GSK-3β in the presence of activated HIV-1 involves the β-catenin signaling pathway, which has been shown to function as an intrinsic molecular pathway restricting HIV-1 replication in PBMCs (137). GSK-3β forms part of a repressive multiprotein cytoplasmic complex, in addition to adenomatous polyposis coli (APC) and axin proteins, which target β-catenin for degradation. The TCF-4/β-catenin interaction is linked to HIV-1 replication inhibition in multiple cell types, including astrocytes and lymphocytes. Recently, TCF-4 and β-catenin have been shown to have multiple binding sites relative to the transcriptional initiation site at the 5′ HIV-1 LTR (139). In particular, TCF-4 and β-catenin at position −143 associate with the nuclear matrix binding protein SMAR1, which may tether the HIV-1 DNA segment into the nuclear matrix and away from transcriptional machinery, resulting in repression of basal HIV-1 LTR transcription. Thus, increased cytoplasmic GSK-3β may be acting as a transcriptional activating complex by repressing the β-catenin HIV-1 restrictive signaling pathway.
HIV-1 selectively targets the basal ganglion region of the brain, which results in loss of dopaminergic and nigrostriatal neurons (140, 141). Overexpression of exogenous GSK-3 in neurons impairs neurite growth (142), and this impairment can be overcome when neurons overexpressing exogenous active enzyme are treated with lithium. Likewise, Tat has been shown to induce GSK-3β activity, which can be abrogated by lithium treatment (143). 18BIOder is able to confer neuronal protection with less toxicity than 6BIO and exhibits potent inhibition of HIV-1 Tat-dependent transcription. In addition to viral transcriptional inhibition and thus decrease in Tat production and release, 18BIOder confers microglial and neuronal cytoprotection (Fig. 7). Although the mechanism of neuroprotection is not known, modulation of the GSK-3β signaling pathways and GSK-3β-mediated transcriptional regulation of neurotrophic growth factors may play an important role. The complete role of GSK-3β in HAND remains unclear; however, its actions have been well characterized in various prevailing diseases, such as neurocognitive and mood disorders, Alzheimer's disease, diabetes, and cancer (144). Recently, GSK-3β has been found to play a vital role in the inflammation process, providing a common link between the enzyme and the various diseases it influences, possibly including HAND (145). Given that indirect neurodegeneration in HAND involves soluble factors released by macrophages and microglia as part of the host inflammatory response to HIV-1 infection (some of which have been shown to act as nonviral neurotoxins), GSK-3-mediated modulation of the inflammatory response may add a layer of complexity to its role in HAND (146–148).
Currently, the possibility that these compounds exert their inhibitory function through other, unidentified single or collective kinases, which would result in synergistic HIV-1 transcriptional inhibition, cannot be ruled out. However, we have presented novel data indicating the exclusive presence of new GSK-3β complexes in infected cells (Fig. 5A to E) and that they are more susceptible to 18BIOder kinase inhibition without altering normal cellular transcription (Fig. 6A to C). In uninfected cells, GSK-3β was present in a complex with a molecular mass of ∼300 kDa, which is likely to be composed of either homo- or heterodimers of GSK-3β in conjunction with possible chaperone proteins or other bound proteins. However, HIV-1-infected cells displayed an extended set of lower-molecular-weight complexes in addition to the dominant ∼300-kDa complex. We hypothesize that the larger complex is formed by multimers of GSK-3β and possibly unknown proteins, while the smaller complexes may be constituted of monomers only. These elution profiles of GSK-3β are novel and reveal a redistribution of GSK-3β during infection that is not observed in uninfected cells. Interestingly, this elution profile was confirmed in two chronically HIV-1-infected cell line models. Relevant to this study, size fractionation of mock- and VEEV-infected cells revealed a similar distribution of GSK-3β during infection (95). Though only speculative at this point, it is possible that infection alters cytoplasmic complexes that are normally masking the bipartite nuclear localization signal (NLS) present in GSK-3β, which spans residues 85 to 103 (103). Inhibitory cytoplasmic complexes may allow protein localization regulation predominantly in the cytoplasm while permitting nuclear accumulation in response to demand, in this case viral infection. The NLS was found to be both sufficient and required for nuclear localization. Current proteomics experiments from lower fraction extracts from HIV-1-infected versus VEEV-infected cells may uncover stoichiometry of GSK-3β and its possible partners, defining the mechanism of 18BIOder inhibition in HIV-1-infected cells. This may also explain the inherent specificity of 18BIOder for HIV-1 transcription (nuclear event) versus 19BIOder inhibition of VEEV transcription (cytoplasmic event).
Lastly, we have demonstrated the efficacy of 18BIOder in HIV-1 replication inhibition in vivo (data not shown). Treatment with both 1.0 mg/kg and 10 mg/kg of 18BIOder resulted in viral suppression, as measured through reverse transcriptase activity from treated animals. Longitudinal measurements over a span of 29 days showed effective viral suppression at 1.0 mg/kg 18BIOder shortly after treatment. When left untreated, the virus would resume replication, a response comparable to that of cART, which requires daily dosage for effective plasma control of the viral load. Of interest, we observed effective inhibition after treatment interruption. This is significant, since it reinforces the notion that generating host-based therapies may offer new opportunities to overcome viral adaptation and resistance to virus-based inhibitors. A possible in vivo model is depicted in Fig. 8, where HIV-1 neurotoxicity may be derived from cytokines and soluble viral proteins, gp120 and Tat, released from infected astrocytes and microglia and 18BIOder may exert its effects by altering these neurotoxin releases.
FIG 8.
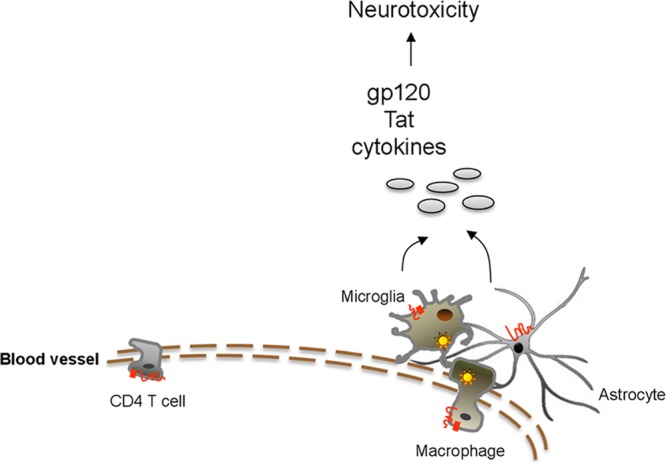
Model depicting mechanisms contributing to the development of HAND. Infiltration of peripheral macrophages generates latent reservoirs in astrocytes and microglial cells. HIV-1 neurotoxicity is derived from soluble viral (gp120 and Tat) and host (cytokines and chemokines) proteins that are released from infected astrocytes and microglia. 18BIOder may exert its effects by altering neurotoxin release.
Future studies will focus more on identification of candidate factors that could regulate both LTR and the host. Using reverse-phase protein microarray (RPMA) technology, we have determined in preliminary experiments that GSK-3β affects protein nodes that control apoptosis and survival, especially in 18BIOder-treated cells. Validation of these nodes will require future studies in primary cells.
ACKNOWLEDGMENTS
We thank Diane Griffin (professor and chair in Molecular Microbiology and Immunology at Johns Hopkins Bloomberg School of Public Health) and Pierre Talbot (Directeur du Laboratoire de Neuroimmunovirologie, Université du Québec, Institut National de Recherche Scientifique Health) for generously providing reagents for this study, including AP-7 neuronal and CHME5 cells, respectively.
This work was supported by NIH grant AI070740 to F.K.
The content is solely our responsibility and does not necessarily represent the official views of the National Institutes of Health.
We declare no conflict of interest.
Footnotes
Published ahead of print 13 November 2013
REFERENCES
- 1.Ances BM, Ellis RJ. 2007. Dementia and neurocognitive disorders due to HIV-1 infection. Semin. Neurol. 27:86–92. 10.1055/s-2006-956759 [DOI] [PubMed] [Google Scholar]
- 2.Crews L, Lentz MR, Gonzalez RG, Fox HS, Masliah E. 2008. Neuronal injury in simian immunodeficiency virus and other animal models of neuroAIDS. J. Neurovirol. 14:327–339. 10.1080/13550280802132840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, Collier AC, Marra CM, Morgello S, Mindt MR, Taylor MJ, Marcotte TD, Atkinson JH, Wolfson T, Gelman BB, McArthur JC, Simpson DM, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I. 2011. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J. Neurovirol. 17:3–16. 10.1007/s13365-010-0006-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McArthur JC, Brew BJ, Nath A. 2005. Neurological complications of HIV infection. Lancet Neurol. 4:543–555. 10.1016/S1474-4422(05)70165-4 [DOI] [PubMed] [Google Scholar]
- 5.McArthur JC, Brew BJ. 2010. HIV-associated neurocognitive disorders: is there a hidden epidemic? AIDS 24:1367–1370. 10.1097/QAD.0b013e3283391d56 [DOI] [PubMed] [Google Scholar]
- 6.Zou S, El-Hage N, Podhaizer EM, Knapp PE, Hauser KF. 2011. PTEN gene silencing prevents HIV-1 gp120(IIIB)-induced degeneration of striatal neurons. J. Neurovirol. 17:41–49. 10.1007/s13365-010-0016-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ju SM, Goh AR, Kwon DJ, Youn GS, Kwon HJ, Bae YS, Choi SY, Park J. 2012. Extracellular HIV-1 Tat induces human beta-defensin-2 production via NF-kappaB/AP-1 dependent pathways in human B cells. Mol. Cells 33:335–341. 10.1007/s10059-012-2287-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thakur BK, Chandra A, Dittrich T, Welte K, Chandra P. 2012. Inhibition of SIRT1 by HIV-1 viral protein Tat results in activation of p53 pathway. Biochem. Biophys. Res. Commun. 424:245–250. 10.1016/j.bbrc.2012.06.084 [DOI] [PubMed] [Google Scholar]
- 9.Kim SE, Lee EO, Yang JH, Kang JH, Suh YH, Chong YH. 2012. 15-Deoxy-delta(1)(2),(1)(4)-prostaglandin J(2) inhibits human immunodeficiency virus-1 Tat-induced monocyte chemoattractant protein-1/CCL2 production by blocking the extracellular signal-regulated kinase-1/2 signaling pathway independently of peroxisome proliferator-activated receptor-gamma and heme oxygenase-1 in rat hippocampal slices. J. Neurosci. Res. 90:1732–1742. 10.1002/jnr.23051 [DOI] [PubMed] [Google Scholar]
- 10.Bachis A, Avdoshina V, Zecca L, Parsadanian M, Mocchetti I. 2012. Human immunodeficiency virus type 1 alters brain-derived neurotrophic factor processing in neurons. J. Neurosci. 32:9477–9484. 10.1523/JNEUROSCI.0865-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reddy PV, Gandhi N, Samikkannu T, Saiyed Z, Agudelo M, Yndart A, Khatavkar P, Nair MP. 2012. HIV-1 gp120 induces antioxidant response element-mediated expression in primary astrocytes: role in HIV associated neurocognitive disorder. Neurochem Int. 61:807–814. 10.1016/j.neuint.2011.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujinaga K, Cujec TP, Peng J, Garriga J, Price DH, Grana X, Peterlin BM. 1998. The ability of positive transcription elongation factor B to transactivate human immunodeficiency virus transcription depends on a functional kinase domain, cyclin T1, and Tat. J. Virol. 72:7154–7159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herrmann CH, Rice AP. 1995. Lentivirus Tat proteins specifically associate with a cellular protein kinase, TAK, that hyperphosphorylates the carboxyl-terminal domain of the large subunit of RNA polymerase II: candidate for a Tat cofactor. J. Virol. 69:1612–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laspia MF, Rice AP, Mathews MB. 1989. HIV-1 Tat protein increases transcriptional initiation and stabilizes elongation. Cell 59:283–292. 10.1016/0092-8674(89)90290-0 [DOI] [PubMed] [Google Scholar]
- 15.Nabel G, Baltimore D. 1987. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 326:711–713. 10.1038/326711a0 [DOI] [PubMed] [Google Scholar]
- 16.Peng J, Zhu Y, Milton JT, Price DH. 1998. Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 12:755–762. 10.1101/gad.12.5.755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rice AP, Mathews MB. 1988. Transcriptional but not translational regulation of HIV-1 by the tat gene product. Nature 332:551–553. 10.1038/332551a0 [DOI] [PubMed] [Google Scholar]
- 18.Sobhian B, Laguette N, Yatim A, Nakamura M, Levy Y, Kiernan R, Benkirane M. 2010. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol. Cell 38:439–451. 10.1016/j.molcel.2010.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. 1998. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 92:451–462. 10.1016/S0092-8674(00)80939-3 [DOI] [PubMed] [Google Scholar]
- 20.Ensoli B, Buonaguro L, Barillari G, Fiorelli V, Gendelman R, Morgan RA, Wingfield P, Gallo RC. 1993. Release, uptake, and effects of extracellular human immunodeficiency virus type 1 Tat protein on cell growth and viral transactivation. J. Virol. 67:277–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chihara T, Hashimoto M, Osman A, Hiyoshi-Yoshidomi Y, Suzu I, Chutiwitoonchai N, Hiyoshi M, Okada S, Suzu S. 2012. HIV-1 proteins preferentially activate anti-inflammatory M2-type macrophages. J. Immunol. 188:3620–3627. 10.4049/jimmunol.1101593 [DOI] [PubMed] [Google Scholar]
- 22.Pocernich CB, Boyd-Kimball D, Poon HF, Thongboonkerd V, Lynn BC, Klein JB, Calebrese V, Nath A, Butterfield DA. 2005. Proteomics analysis of human astrocytes expressing the HIV protein Tat. Brain Res. Mol. Brain Res. 133:307–316. 10.1016/j.molbrainres.2004.10.023 [DOI] [PubMed] [Google Scholar]
- 23.Deshmane SL, Mukerjee R, Fan S, Sawaya BE. 2011. High-performance capillary electrophoresis for determining HIV-1 Tat protein in neurons. PLoS One 6:e16148. 10.1371/journal.pone.0016148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rayne F, Debaisieux S, Yezid H, Lin YL, Mettling C, Konate K, Chazal N, Arold ST, Pugniere M, Sanchez F, Bonhoure A, Briant L, Loret E, Roy C, Beaumelle B. 2010. Phosphatidylinositol-(4,5)-bisphosphate enables efficient secretion of HIV-1 Tat by infected T-cells. EMBO J. 29:1348–1362. 10.1038/emboj.2010.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao H, Neuveut C, Tiffany HL, Benkirane M, Rich EA, Murphy PM, Jeang KT. 2000. Selective CXCR4 antagonism by Tat: implications for in vivo expansion of coreceptor use by HIV-1. Proc. Natl. Acad. Sci. U. S. A. 97:11466–11471. 10.1073/pnas.97.21.11466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson TP, Patel K, Johnson KR, Maric D, Calabresi PA, Hasbun R, Nath A. 2013. Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc. Natl. Acad. Sci. U. S. A. 110:13588–13593. 10.1073/pnas.1308673110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng J, Nath A, Knudsen B, Hochman S, Geiger JD, Ma M, Magnuson DS. 1998. Neuronal excitatory properties of human immunodeficiency virus type 1 Tat protein. Neuroscience 82:97–106 [DOI] [PubMed] [Google Scholar]
- 28.Bennett BA, Rusyniak DE, Hollingsworth CK. 1995. HIV-1 gp120-induced neurotoxicity to midbrain dopamine cultures. Brain Res. 705:168–176. 10.1016/0006-8993(95)01166-8 [DOI] [PubMed] [Google Scholar]
- 29.Magnuson DS, Knudsen BE, Geiger JD, Brownstone RM, Nath A. 1995. Human immunodeficiency virus type 1 tat activates non-N-methyl-D-aspartate excitatory amino acid receptors and causes neurotoxicity. Ann. Neurol. 37:373–380. 10.1002/ana.410370314 [DOI] [PubMed] [Google Scholar]
- 30.Maragos WF, Tillman P, Jones M, Bruce-Keller AJ, Roth S, Bell JE, Nath A. 2003. Neuronal injury in hippocampus with human immunodeficiency virus transactivating protein, Tat. Neuroscience 117:43–53. 10.1016/S0306-4522(02)00713-3 [DOI] [PubMed] [Google Scholar]
- 31.Sabatier JM, Vives E, Mabrouk K, Benjouad A, Rochat H, Duval A, Hue B, Bahraoui E. 1991. Evidence for neurotoxic activity of tat from human immunodeficiency virus type 1. J. Virol. 65:961–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weeks BS, Lieberman DM, Johnson B, Roque E, Green M, Loewenstein P, Oldfield EH, Kleinman HK. 1995. Neurotoxicity of the human immunodeficiency virus type 1 tat transactivator to PC12 cells requires the Tat amino acid 49–58 basic domain. J. Neurosci. Res. 42:34–40. 10.1002/jnr.490420105 [DOI] [PubMed] [Google Scholar]
- 33.Kehn-Hall K, Guendel I, Carpio L, Skaltsounis L, Meijer L, Al-Harthi L, Steiner JP, Nath A, Kutsch O, Kashanchi F. 2011. Inhibition of Tat-mediated HIV-1 replication and neurotoxicity by novel GSK3-beta inhibitors. Virology 415:56–68. 10.1016/j.virol.2011.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kruman II, Nath A, Mattson MP. 1998. HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp. Neurol. 154:276–288. 10.1006/exnr.1998.6958 [DOI] [PubMed] [Google Scholar]
- 35.Haughey NJ, Holden CP, Nath A, Geiger JD. 1999. Involvement of inositol 1,4,5-trisphosphate-regulated stores of intracellular calcium in calcium dysregulation and neuron cell death caused by HIV-1 protein tat. J. Neurochem. 73:1363–1374 [DOI] [PubMed] [Google Scholar]
- 36.Aksenov MY, Hasselrot U, Bansal AK, Wu G, Nath A, Anderson C, Mactutus CF, Booze RM. 2001. Oxidative damage induced by the injection of HIV-1 Tat protein in the rat striatum. Neurosci. Lett. 305:5–8. 10.1016/S0304-3940(01)01786-4 [DOI] [PubMed] [Google Scholar]
- 37.Aksenov MY, Hasselrot U, Wu G, Nath A, Anderson C, Mactutus CF, Booze RM. 2003. Temporal relationships between HIV-1 Tat-induced neuronal degeneration, OX-42 immunoreactivity, reactive astrocytosis, and protein oxidation in the rat striatum. Brain Res. 987:1–9. 10.1016/S0006-8993(03)03194-9 [DOI] [PubMed] [Google Scholar]
- 38.Bansal AK, Mactutus CF, Nath A, Maragos W, Hauser KF, Booze RM. 2000. Neurotoxicity of HIV-1 proteins gp120 and Tat in the rat striatum. Brain Res. 879:42–49. 10.1016/S0006-8993(00)02725-6 [DOI] [PubMed] [Google Scholar]
- 39.Nath A, Haughey NJ, Jones M, Anderson C, Bell JE, Geiger JD. 2000. Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: protection by memantine. Ann. Neurol. 47:186–194. [DOI] [PubMed] [Google Scholar]
- 40.Polazzi E, Levi G, Minghetti L. 1999. Human immunodeficiency virus type 1 Tat protein stimulates inducible nitric oxide synthase expression and nitric oxide production in microglial cultures. J. Neuropathol. Exp. Neurol. 58:825–831. 10.1097/00005072-199908000-00005 [DOI] [PubMed] [Google Scholar]
- 41.Gavriil ES, Cooney R, Weeks BS. 2000. Tat mediates apoptosis in vivo in the rat central nervous system. Biochem. Biophys. Res. Commun. 267:252–256. 10.1006/bbrc.1999.1894 [DOI] [PubMed] [Google Scholar]
- 42.Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD. 2001. HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J. Neurochem. 78:457–467. 10.1046/j.1471-4159.2001.00396.x [DOI] [PubMed] [Google Scholar]
- 43.Noonan D, Albini A. 2000. From the outside in: extracellular activities of HIV Tat. Adv. Pharmacol. 48:229–250. 10.1016/S1054-3589(00)48008-7 [DOI] [PubMed] [Google Scholar]
- 44.Rusnati M, Presta M. 2002. HIV-1 Tat protein and endothelium: from protein/cell interaction to AIDS-associated pathologies. Angiogenesis 5:141–151. 10.1023/A:1023892223074 [DOI] [PubMed] [Google Scholar]
- 45.Krum JM, Rosenstein JM. 1998. VEGF mRNA and its receptor flt-1 are expressed in reactive astrocytes following neural grafting and tumor cell implantation in the adult CNS. Exp. Neurol. 154:57–65. 10.1006/exnr.1998.6930 [DOI] [PubMed] [Google Scholar]
- 46.Barillari G, Gendelman R, Gallo RC, Ensoli B. 1993. The Tat protein of human immunodeficiency virus type 1, a growth factor for AIDS Kaposi sarcoma and cytokine-activated vascular cells, induces adhesion of the same cell types by using integrin receptors recognizing the RGD amino acid sequence. Proc. Natl. Acad. Sci. U. S. A. 90:7941–7945. 10.1073/pnas.90.17.7941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Etienne-Manneville S, Hall A. 2001. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell 106:489–498. 10.1016/S0092-8674(01)00471-8 [DOI] [PubMed] [Google Scholar]
- 48.Liu Y, Jones M, Hingtgen CM, Bu G, Laribee N, Tanzi RE, Moir RD, Nath A, He JJ. 2000. Uptake of HIV-1 tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat. Med. 6:1380–1387. 10.1038/82199 [DOI] [PubMed] [Google Scholar]
- 49.Haughey NJ, Mattson MP. 2002. Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins Tat and gp120. J. Acquir. Immune Defic. Syndr 31(Suppl 2):S55–S61. 10.1097/00126334-200210012-00005 [DOI] [PubMed] [Google Scholar]
- 50.Nath A, Psooy K, Martin C, Knudsen B, Magnuson DS, Haughey N, Geiger JD. 1996. Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. J. Virol. 70:1475–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Toggas SM, Masliah E, Mucke L. 1996. Prevention of HIV-1 gp120-induced neuronal damage in the central nervous system of transgenic mice by the NMDA receptor antagonist memantine. Brain Res. 706:303–307. 10.1016/0006-8993(95)01197-8 [DOI] [PubMed] [Google Scholar]
- 52.Wu P, Price P, Du B, Hatch WC, Terwilliger EF. 1996. Direct cytotoxicity of HIV-1 envelope protein gp120 on human NT neurons. Neuroreport 7:1045–1049. 10.1097/00001756-199604100-00018 [DOI] [PubMed] [Google Scholar]
- 53.Holden CP, Haughey NJ, Nath A, Geiger JD. 1999. Role of Na+/H+ exchangers, excitatory amino acid receptors and voltage-operated Ca2+ channels in human immunodeficiency virus type 1 gp120-mediated increases in intracellular Ca2+ in human neurons and astrocytes. Neuroscience 91:1369–1378. 10.1016/S0306-4522(98)00714-3 [DOI] [PubMed] [Google Scholar]
- 54.Khurdayan VK, Buch S, El-Hage N, Lutz SE, Goebel SM, Singh IN, Knapp PE, Turchan-Cholewo J, Nath A, Hauser KF. 2004. Preferential vulnerability of astroglia and glial precursors to combined opioid and HIV-1 Tat exposure in vitro. Eur. J. Neurosci. 19:3171–3182. 10.1111/j.0953-816X.2004.03461.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Muller WE, Pergande G, Ushijima H, Schleger C, Kelve M, Perovic S. 1996. Neurotoxicity in rat cortical cells caused by N-methyl-D-aspartate (NMDA) and gp120 of HIV-1: induction and pharmacological intervention. Prog. Mol. Subcell. Biol. 16:44–57. 10.1007/978-3-642-79850-4_3 [DOI] [PubMed] [Google Scholar]
- 56.Chao CC, Hu S, Gekker G, Lokensgard JR, Heyes MP, Peterson PK. 2000. U50,488 protection against HIV-1-related neurotoxicity: involvement of quinolinic acid suppression. Neuropharmacology 39:150–160. 10.1016/S0028-3908(99)00063-5 [DOI] [PubMed] [Google Scholar]
- 57.Wallace DR, Dodson SL, Nath A, Booze RM. 2006. Delta opioid agonists attenuate TAT(1-72)-induced oxidative stress in SK-N-SH cells. Neurotoxicology 27:101–107. 10.1016/j.neuro.2005.07.008 [DOI] [PubMed] [Google Scholar]
- 58.Pulliam L, Irwin I, Kusdra L, Rempel H, Flitter WD, Garland WA. 2001. CPI-1189 attenuates effects of suspected neurotoxins associated with AIDS dementia: a possible role for ERK activation. Brain Res. 893:95–103. 10.1016/S0006-8993(00)03293-5 [DOI] [PubMed] [Google Scholar]
- 59.Everall IP, Bell C, Mallory M, Langford D, Adame A, Rockestein E, Masliah E. 2002. Lithium ameliorates HIV-gp120-mediated neurotoxicity. Mol. Cell Neurosci. 21:493–501. 10.1006/mcne.2002.1196 [DOI] [PubMed] [Google Scholar]
- 60.Hamy F, Brondani V, Florsheimer A, Stark W, Blommers MJ, Klimkait T. 1998. A new class of HIV-1 Tat antagonist acting through Tat-TAR inhibition. Biochemistry 37:5086–5095. 10.1021/bi972947s [DOI] [PubMed] [Google Scholar]
- 61.Bailly C, Colson P, Houssier C, Hamy F. 1996. The binding mode of drugs to the TAR RNA of HIV-1 studied by electric linear dichroism. Nucleic Acids Res. 24:1460–1464. 10.1093/nar/24.8.1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dassonneville L, Hamy F, Colson P, Houssier C, Bailly C. 1997. Binding of Hoechst 33258 to the TAR RNA of HIV-1. Recognition of a pyrimidine bulge-dependent structure. Nucleic Acids Res. 25:4487–4492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Edwards TE, Sigurdsson ST. 2002. Electron paramagnetic resonance dynamic signatures of TAR RNA-small molecule complexes provide insight into RNA structure and recognition. Biochemistry 41:14843–14847. 10.1021/bi026299a [DOI] [PubMed] [Google Scholar]
- 64.Mestre B, Arzumanov A, Singh M, Boulme F, Litvak S, Gait MJ. 1999. Oligonucleotide inhibition of the interaction of HIV-1 Tat protein with the trans-activation responsive region (TAR) of HIV RNA. Biochim. Biophys. Acta 1445:86–98. 10.1016/S0167-4781(99)00019-6 [DOI] [PubMed] [Google Scholar]
- 65.Baba M, Okamoto M, Makino M, Kimura Y, Ikeuchi T, Sakaguchi T, Okamoto T. 1997. Potent and selective inhibition of human immunodeficiency virus type 1 transcription by piperazinyloxoquinoline derivatives. Antimicrob. Agents Chemother. 41:1250–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baba M, Okamoto M, Kawamura M, Makino M, Higashida T, Takashi T, Kimura Y, Ikeuchi T, Tetsuka T, Okamoto T. 1998. Inhibition of human immunodeficiency virus type 1 replication and cytokine production by fluoroquinoline derivatives. Mol. Pharmacol. 53:1097–1103 [PubMed] [Google Scholar]
- 67.Hsu MC, Dhingra U, Earley JV, Holly M, Keith D, Nalin CM, Richou AR, Schutt AD, Tam SY, Potash MJ. 1993. Inhibition of type 1 human immunodeficiency virus replication by a tat antagonist to which the virus remains sensitive after prolonged exposure in vitro. Proc. Natl. Acad. Sci. U. S. A. 90:6395–6399. 10.1073/pnas.90.14.6395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Haubrich RH, Flexner C, Lederman MM, Hirsch M, Pettinelli CP, Ginsberg R, Lietman P, Hamzeh FM, Spector SA, Richman DD. 1995. A randomized trial of the activity and safety of Ro 24-7429 (Tat antagonist) versus nucleoside for human immunodeficiency virus infection. The AIDS Clinical Trials Group 213 Team. J. Infect. Dis. 172:1246–1252 [DOI] [PubMed] [Google Scholar]
- 69.Agbottah E, de La Fuente C, Nekhai S, Barnett A, Gianella-Borradori A, Pumfery A, Kashanchi F. 2005. Antiviral activity of CYC202 in HIV-1-infected cells. J. Biol. Chem. 280:3029–3042. 10.1074/jbc.M406435200 [DOI] [PubMed] [Google Scholar]
- 70.Galons H, Oumata N, Meijer L. 2010. Cyclin-dependent kinase inhibitors: a survey of recent patent literature. Expert Opin. Ther. Pat. 20:377–404. 10.1517/13543770903524284 [DOI] [PubMed] [Google Scholar]
- 71.Kashanchi F, Kehn-Hall K. 2009. Cyclin dependent kinases as attractive targets to prevent transcription from viral genomes. Curr. Pharm. Des. 15:2520–2532. 10.2174/138161209788682280 [DOI] [PubMed] [Google Scholar]
- 72.Guendel I, Agbottah ET, Kehn-Hall K, Kashanchi F. 2010. Inhibition of human immunodeficiency virus type-1 by cdk inhibitors. AIDS Res. Ther. 7:7. 10.1186/1742-6405-7-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Malumbres M, Pevarello P, Barbacid M, Bischoff JR. 2008. CDK inhibitors in cancer therapy: what is next? Trends Pharmacol. Sci. 29:16–21. 10.1016/j.tips.2007.10.012 [DOI] [PubMed] [Google Scholar]
- 74.Van Duyne R, Cardenas J, Easley R, Wu W, Kehn-Hall K, Klase Z, Mendez S, Zeng C, Chen H, Saifuddin M, Kashanchi F. 2008. Effect of transcription peptide inhibitors on HIV-1 replication. Virology 376:308–322. 10.1016/j.virol.2008.02.036 [DOI] [PubMed] [Google Scholar]
- 75.Narayanan A, Sampey G, Van Duyne R, Guendel I, Kehn-Hall K, Roman J, Currer R, Galons H, Oumata N, Joseph B, Meijer L, Caputi M, Nekhai S, Kashanchi F. 2012. Use of ATP analogs to inhibit HIV-1 transcription. Virology 432:219–231. 10.1016/j.virol.2012.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dou H, Birusingh K, Faraci J, Gorantla S, Poluektova LY, Maggirwar SB, Dewhurst S, Gelbard HA, Gendelman HE. 2003. Neuroprotective activities of sodium valproate in a murine model of human immunodeficiency virus-1 encephalitis. J. Neurosci. 23:9162–9170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dou H, Ellison B, Bradley J, Kasiyanov A, Poluektova LY, Xiong H, Maggirwar S, Dewhurst S, Gelbard HA, Gendelman HE. 2005. Neuroprotective mechanisms of lithium in murine human immunodeficiency virus-1 encephalitis. J. Neurosci. 25:8375–8385. 10.1523/JNEUROSCI.2164-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nguyen TB, Lucero GR, Chana G, Hult BJ, Tatro ET, Masliah E, Grant I, Achim CL, Everall IP. 2009. Glycogen synthase kinase-3beta (GSK-3beta) inhibitors AR-A014418 and B6B3O prevent human immunodeficiency virus-mediated neurotoxicity in primary human neurons. J. Neurovirol. 15:434–438. 10.3109/13550280903168131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Atanassov CL, Muller CD, Dumont S, Rebel G, Poindron P, Seiler N. 1995. Effect of ammonia on endocytosis and cytokine production by immortalized human microglia and astroglia cells. Neurochem Int. 27:417–424. 10.1016/0197-0186(95)00023-2 [DOI] [PubMed] [Google Scholar]
- 80.Peudenier S, Hery C, Montagnier L, Tardieu M. 1991. Human microglial cells: characterization in cerebral tissue and in primary culture, and study of their susceptibility to HIV-1 infection. Ann. Neurol. 29:152–161. 10.1002/ana.410290207 [DOI] [PubMed] [Google Scholar]
- 81.de Gannes FM, Merle M, Canioni P, Voisin PJ. 1998. Metabolic and cellular characterization of immortalized human microglial cells under heat stress. Neurochem Int. 33:61–73. 10.1016/S0197-0186(05)80010-5 [DOI] [PubMed] [Google Scholar]
- 82.Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O'Brien WA, Ratner L, Kappes JC, Shaw GM, Hunter E. 2000. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J. Virol. 74:8358–8367. 10.1128/JVI.74.18.8358-8367.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Berro R, Kehn K, de la Fuente C, Pumfery A, Adair R, Wade J, Colberg-Poley AM, Hiscott J, Kashanchi F. 2006. Acetylated Tat regulates human immunodeficiency virus type 1 splicing through its interaction with the splicing regulator p32. J. Virol. 80:3189–3204. 10.1128/JVI.80.7.3189-3204.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Perez VL, Rowe T, Justement JS, Butera ST, June CH, Folks TM. 1991. An HIV-1-infected T cell clone defective in IL-2 production and Ca2+ mobilization after CD3 stimulation. J. Immunol. 147:3145–3148 [PubMed] [Google Scholar]
- 85.Clouse KA, Powell D, Washington I, Poli G, Strebel K, Farrar W, Barstad P, Kovacs J, Fauci AS, Folks TM. 1989. Monokine regulation of human immunodeficiency virus-1 expression in a chronically infected human T cell clone. J. Immunol. 142:431–438 [PubMed] [Google Scholar]
- 86.Folks TM, Justement J, Kinter A, Dinarello CA, Fauci AS. 1987. Cytokine-induced expression of HIV-1 in a chronically infected promonocyte cell line. Science 238:800–802. 10.1126/science.3313729 [DOI] [PubMed] [Google Scholar]
- 87.Murrell JR, Hunter DD. 1999. An olfactory sensory neuron line, odora, properly targets olfactory proteins and responds to odorants. J. Neurosci. 19:8260–8270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kashanchi F, Duvall JF, Brady JN. 1992. Electroporation of viral transactivator proteins into lymphocyte suspension cells. Nucleic Acids Res. 20:4673–4674. 10.1093/nar/20.17.4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schuttelkopf AW, van Aalten DM. 2004. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60:1355–1363. 10.1107/S0907444904011679 [DOI] [PubMed] [Google Scholar]
- 90.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612. 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 91.Wallace AC, Laskowski RA, Thornton JM. 1995. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8:127–134. 10.1093/protein/8.2.127 [DOI] [PubMed] [Google Scholar]
- 92.Carpio L, Klase Z, Coley W, Guendel I, Choi S, Van Duyne R, Narayanan A, Kehn-Hall K, Meijer L, Kashanchi F. 2010. microRNA machinery is an integral component of drug-induced transcription inhibition in HIV-1 infection. J. RNAi Gene Silencing 6:386–400 [PMC free article] [PubMed] [Google Scholar]
- 93.Tamburro D, Fredolini C, Espina V, Douglas TA, Ranganathan A, Ilag L, Zhou W, Russo P, Espina BH, Muto G, Petricoin EF, III, Liotta LA, Luchini A. 2011. Multifunctional core-shell nanoparticles: discovery of previously invisible biomarkers. J. Am. Chem. Soc. 133:19178–19188. 10.1021/ja207515j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Luchini A, Geho DH, Bishop B, Tran D, Xia C, Dufour RL, Jones CD, Espina V, Patanarut A, Zhou W, Ross MM, Tessitore A, Petricoin EF, III, Liotta LA. 2008. Smart hydrogel particles: biomarker harvesting: one-step affinity purification, size exclusion, and protection against degradation. Nano Lett. 8:350–361. 10.1021/nl072174l [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kehn-Hall K, Narayanan A, Lundberg L, Sampey G, Pinkham C, Guendel I, Van Duyne R, Senina S, Schultz KL, Stavale E, Aman MJ, Bailey C, Kashanchi F. 2012. Modulation of GSK-3beta activity in Venezuelan equine encephalitis virus infection. PLoS One 7:e34761. 10.1371/journal.pone.0034761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Spokoini R, Kfir-Erenfeld S, Yefenof E, Sionov RV. 2010. Glycogen synthase kinase-3 plays a central role in mediating glucocorticoid-induced apoptosis. Mol. Endocrinol. 24:1136–1150. 10.1210/me.2009-0466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li M, Wang X, Meintzer MK, Laessig T, Birnbaum MJ, Heidenreich KA. 2000. Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase 3beta. Mol. Cell. Biol. 20:9356–9363. 10.1128/MCB.20.24.9356-9363.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chin PC, Majdzadeh N, D'Mello SR. 2005. Inhibition of GSK3beta is a common event in neuroprotection by different survival factors. Brain Res. Mol. Brain Res. 137:193–201. 10.1016/j.molbrainres.2005.03.004 [DOI] [PubMed] [Google Scholar]
- 99.Cao H, Chu Y, Lv X, Qiu P, Liu C, Zhang H, Li D, Peng S, Dou Z, Hua J. 2012. GSK3 inhibitor-BIO regulates proliferation of immortalized pancreatic mesenchymal stem cells (iPMSCs). PLoS One 7:e31502. 10.1371/journal.pone.0031502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chao SH, Fujinaga K, Marion JE, Taube R, Sausville EA, Senderowicz AM, Peterlin BM, Price DH. 2000. Flavopiridol inhibits P-TEFb and blocks HIV-1 replication. J. Biol. Chem. 275:28345–28348. 10.1074/jbc.C000446200 [DOI] [PubMed] [Google Scholar]
- 101.Coley W, Van Duyne R, Carpio L, Guendel I, Kehn-Hall K, Chevalier S, Narayanan A, Luu T, Lee N, Klase Z, Kashanchi F. 2010. Absence of DICER in monocytes and its regulation by HIV-1. J. Biol. Chem. 285:31930–31943. 10.1074/jbc.M110.101709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Del Bufalo A, Bernad J, Dardenne C, Verda D, Meunier JR, Rousset F, Martinozzi-Teissier S, Pipy B. 2011. Contact sensitizers modulate the arachidonic acid metabolism of PMA-differentiated U-937 monocytic cells activated by LPS. Toxicol. Appl. Pharmacol. 256:35–43. 10.1016/j.taap.2011.06.025 [DOI] [PubMed] [Google Scholar]
- 103.Meares GP, Jope RS. 2007. Resolution of the nuclear localization mechanism of glycogen synthase kinase-3: functional effects in apoptosis. J. Biol. Chem. 282:16989–17001. 10.1074/jbc.M700610200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kim YK, Mbonye U, Hokello J, Karn J. 2011. T-cell receptor signaling enhances transcriptional elongation from latent HIV proviruses by activating P-TEFb through an ERK-dependent pathway. J. Mol. Biol. 410:896–916. 10.1016/j.jmb.2011.03.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Van Duyne R, Guendel I, Narayanan A, Gregg E, Shafagati N, Tyagi M, Easley R, Klase Z, Nekhai S, Kehn-Hall K, Kashanchi F. 2011. Varying modulation of HIV-1 LTR activity by Baf complexes. J. Mol. Biol. 411:581–596. 10.1016/j.jmb.2011.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Spear M, Guo J, Wu Y. 2012. The trinity of the cortical actin in the initiation of HIV-1 infection. Retrovirology. 9:45. 10.1186/1742-4690-9-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jung DJ, Sung HS, Goo YW, Lee HM, Park OK, Jung SY, Lim J, Kim HJ, Lee SK, Kim TS, Lee JW, Lee YC. 2002. Novel transcription coactivator complex containing activating signal cointegrator 1. Mol. Cell. Biol. 22:5203–5211. 10.1128/MCB.22.14.5203-5211.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Carlson B, Lahusen T, Singh S, Loaiza-Perez A, Worland PJ, Pestell R, Albanese C, Sausville EA, Senderowicz AM. 1999. Down-regulation of cyclin D1 by transcriptional repression in MCF-7 human breast carcinoma cells induced by flavopiridol. Cancer Res. 59:4634–4641 [PubMed] [Google Scholar]
- 109.Kanazawa S, Okamoto T, Peterlin BM. 2000. Tat competes with CIITA for the binding to P-TEFb and blocks the expression of MHC class II genes in HIV infection. Immunity 12:61–70. 10.1016/S1074-7613(00)80159-4 [DOI] [PubMed] [Google Scholar]
- 110.Chao SH, Walker JR, Chanda SK, Gray NS, Caldwell JS. 2003. Identification of homeodomain proteins, PBX1 and PREP1, involved in the transcription of murine leukemia virus. Mol. Cell. Biol. 23:831–841. 10.1128/MCB.23.3.831-841.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Grimes CA, Jope RS. 2001. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog. Neurobiol. 65:391–426. 10.1016/S0301-0082(01)00011-9 [DOI] [PubMed] [Google Scholar]
- 112.Boska MD, Mosley RL, Nawab M, Nelson JA, Zelivyanskaya M, Poluektova L, Uberti M, Dou H, Lewis TB, Gendelman HE. 2004. Advances in neuroimaging for HIV-1 associated neurological dysfunction: clues to the diagnosis, pathogenesis and therapeutic monitoring. Curr. HIV Res. 2:61–78. 10.2174/1570162043485095 [DOI] [PubMed] [Google Scholar]
- 113.Nath A, Geiger J. 1998. Neurobiological aspects of human immunodeficiency virus infection: neurotoxic mechanisms. Prog. Neurobiol. 54:19–33. 10.1016/S0301-0082(97)00053-1 [DOI] [PubMed] [Google Scholar]
- 114.Piller SC, Jans P, Gage PW, Jans DA. 1998. Extracellular HIV-1 virus protein R causes a large inward current and cell death in cultured hippocampal neurons: implications for AIDS pathology. Proc. Natl. Acad. Sci. U. S. A. 95:4595–4600. 10.1073/pnas.95.8.4595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Reddy PV, Gandhi N, Samikkannu T, Saiyed Z, Agudelo M, Yndart A, Khatavkar P, Nair MP. 2012. HIV-1 gp120 induces antioxidant response element-mediated expression in primary astrocytes: role in HIV associated neurocognitive disorder. Neurochem Int. 61:807–814. 10.1016/j.neuint.2011.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Li W, Li G, Steiner J, Nath A. 2009. Role of Tat protein in HIV neuropathogenesis. Neurotox. Res. 16:205–220. 10.1007/s12640-009-9047-8 [DOI] [PubMed] [Google Scholar]
- 117.Chauhan A, Turchan J, Pocernich C, Bruce-Keller A, Roth S, Butterfield DA, Major EO, Nath A. 2003. Intracellular human immunodeficiency virus Tat expression in astrocytes promotes astrocyte survival but induces potent neurotoxicity at distant sites via axonal transport. J. Biol. Chem. 278:13512–13519. 10.1074/jbc.M209381200 [DOI] [PubMed] [Google Scholar]
- 118.Zhong J, Yao W, Lee W. 2007. Cesium chloride protects cerebellar granule neurons from apoptosis induced by low potassium. Int. J. Dev. Neurosci. 25:359–365. 10.1016/j.ijdevneu.2007.07.003 [DOI] [PubMed] [Google Scholar]
- 119.Cross DA, Culbert AA, Chalmers KA, Facci L, Skaper SD, Reith AD. 2001. Selective small-molecule inhibitors of glycogen synthase kinase-3 activity protect primary neurones from death. J. Neurochem. 77:94–102. 10.1046/j.1471-4159.2001.t01-1-00251.x [DOI] [PubMed] [Google Scholar]
- 120.Villalba M, Bockaert J, Journot L. 1997. Pituitary adenylate cyclase-activating polypeptide (PACAP-38) protects cerebellar granule neurons from apoptosis by activating the mitogen-activated protein kinase (MAP kinase) pathway. J. Neurosci. 17:83–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Van Duyne R, Guendel I, Jaworski E, Sampey G, Klase Z, Chen H, Zeng C, Kovalskyy D, El Kouni MH, Lepene B, Patanarut A, Nekhai S, Price DH, Kashanchi F. 2013. Effect of mimetic CDK9 inhibitors on HIV-1-activated transcription. J. Mol. Biol. 425:812–829. 10.1016/j.jmb.2012.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jaeger LB, Nath A. 2012. Modeling HIV-associated neurocognitive disorders in mice: new approaches in the changing face of HIV neuropathogenesis. Dis. Model Mech. 5:313–322. 10.1242/dmm.008763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Taylor DD, Zacharias W, Gercel-Taylor C. 2011. Exosome isolation for proteomic analyses and RNA profiling. Methods Mol. Biol. 728:235–246. 10.1007/978-1-61779-068-3_15 [DOI] [PubMed] [Google Scholar]
- 124.Blanpain C, Libert F, Vassart G, Parmentier M. 2002. CCR5 and HIV infection. Receptors Channels 8:19–31. 10.1080/10606820212135 [DOI] [PubMed] [Google Scholar]
- 125.King JE, Eugenin EA, Buckner CM, Berman JW. 2006. HIV tat and neurotoxicity. Microbes Infect. 8:1347–1357. 10.1016/j.micinf.2005.11.014 [DOI] [PubMed] [Google Scholar]
- 126.Meijer L, Skaltsounis AL, Magiatis P, Polychronopoulos P, Knockaert M, Leost M, Ryan XP, Vonica CA, Brivanlou A, Dajani R, Crovace C, Tarricone C, Musacchio A, Roe SM, Pearl L, Greengard P. 2003. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem. Biol. 10:1255–1266. 10.1016/j.chembiol.2003.11.010 [DOI] [PubMed] [Google Scholar]
- 127.Woodgett JR. 1990. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 9:2431–2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Klein PS, Melton DA. 1996. A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. U. S. A. 93:8455–8459. 10.1073/pnas.93.16.8455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Coghlan MP, Culbert AA, Cross DA, Corcoran SL, Yates JW, Pearce NJ, Rausch OL, Murphy GJ, Carter PS, Roxbee Cox L, Mills D, Brown MJ, Haigh D, Ward RW, Smith DG, Murray KJ, Reith AD, Holder JC. 2000. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 7:793–803. 10.1016/S1074-5521(00)00025-9 [DOI] [PubMed] [Google Scholar]
- 130.Gentile G, Merlo G, Pozzan A, Bernasconi G, Bax B, Bamborough P, Bridges A, Carter P, Neu M, Yao G, Brough C, Cutler G, Coffin A, Belyanskaya S. 2012. 5-Aryl-4-carboxamide-1,3-oxazoles: potent and selective GSK-3 inhibitors. Bioorg. Med. Chem. Lett. 22:1989–1994. 10.1016/j.bmcl.2012.01.034 [DOI] [PubMed] [Google Scholar]
- 131.Bhat R, Xue Y, Berg S, Hellberg S, Ormo M, Nilsson Y, Radesater AC, Jerning E, Markgren PO, Borgegard T, Nylof M, Gimenez-Cassina A, Hernandez F, Lucas JJ, Diaz-Nido J, Avila J. 2003. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J. Biol. Chem. 278:45937–45945. 10.1074/jbc.M306268200 [DOI] [PubMed] [Google Scholar]
- 132.De Bondt HL, Rosenblatt J, Jancarik J, Jones HD, Morgan DO, Kim SH. 1993. Crystal structure of cyclin-dependent kinase 2. Nature 363:595–602. 10.1038/363595a0 [DOI] [PubMed] [Google Scholar]
- 133.Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, Massague J, Pavletich NP. 1995. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 376:313–320. 10.1038/376313a0 [DOI] [PubMed] [Google Scholar]
- 134.Buss H, Dorrie A, Schmitz ML, Frank R, Livingstone M, Resch K, Kracht M. 2004. Phosphorylation of serine 468 by GSK-3beta negatively regulates basal p65 NF-kappaB activity. J. Biol. Chem. 279:49571–49574. 10.1074/jbc.C400442200 [DOI] [PubMed] [Google Scholar]
- 135.Frame S, Cohen P. 2001. GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 359:1–16. 10.1042/0264-6021:3590001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rohr O, Marban C, Aunis D, Schaeffer E. 2003. Regulation of HIV-1 gene transcription: from lymphocytes to microglial cells. J. Leukoc. Biol. 74:736–749. 10.1189/jlb.0403180 [DOI] [PubMed] [Google Scholar]
- 137.Kumar A, Zloza A, Moon RT, Watts J, Tenorio AR, Al-Harthi L. 2008. Active beta-catenin signaling is an inhibitory pathway for human immunodeficiency virus replication in peripheral blood mononuclear cells. J. Virol. 82:2813–2820. 10.1128/JVI.02498-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Eastman Q, Grosschedl R. 1999. Regulation of LEF-1/TCF transcription factors by Wnt and other signals. Curr. Opin. Cell Biol. 11:233–240. 10.1016/S0955-0674(99)80031-3 [DOI] [PubMed] [Google Scholar]
- 139.Henderson LJ, Narasipura SD, Adarichev V, Kashanchi F, Al-Harthi L. 2012. Identification of novel T cell factor 4 (TCF-4) binding sites on the HIV long terminal repeat which associate with TCF-4, beta-catenin, and SMAR1 to repress HIV transcription. J. Virol. 86:9495–9503. 10.1128/JVI.00486-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yao H, Peng F, Fan Y, Zhu X, Hu G, Buch SJ. 2009. TRPC channel-mediated neuroprotection by PDGF involves Pyk2/ERK/CREB pathway. Cell Death Differ. 16:1681–1693. 10.1038/cdd.2009.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zauli G, Secchiero P, Rodella L, Gibellini D, Mirandola P, Mazzoni M, Milani D, Dowd DR, Capitani S, Vitale M. 2000. HIV-1 Tat-mediated inhibition of the tyrosine hydroxylase gene expression in dopaminergic neuronal cells. J. Biol. Chem. 275:4159–4165. 10.1074/jbc.275.6.4159 [DOI] [PubMed] [Google Scholar]
- 142.Munoz-Montano JR, Lim F, Moreno FJ, Avila J, Diaz-Nido J. 1999. Glycogen synthase kinase-3 modulates neurite outgrowth in cultured neurons: possible implications for neurite pathology in Alzheimer's disease. J. Alzheimers Dis. 1:361–378 [DOI] [PubMed] [Google Scholar]
- 143.Maggirwar SB, Tong N, Ramirez S, Gelbard HA, Dewhurst S. 1999. HIV-1 Tat-mediated activation of glycogen synthase kinase-3beta contributes to Tat-mediated neurotoxicity. J. Neurochem. 73:578–586 [DOI] [PubMed] [Google Scholar]
- 144.Jope RS, Yuskaitis CJ, Beurel E. 2007. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem. Res. 32:577–595. 10.1007/s11064-006-9128-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Martin M, Rehani K, Jope RS, Michalek SM. 2005. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 6:777–784. 10.1038/ni1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kaul M, Lipton SA. 1999. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc. Natl. Acad. Sci. U. S. A. 96:8212–8216. 10.1073/pnas.96.14.8212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Klein RS, Williams KC, Alvarez-Hernandez X, Westmoreland S, Force T, Lackner AA, Luster AD. 1999. Chemokine receptor expression and signaling in macaque and human fetal neurons and astrocytes: implications for the neuropathogenesis of AIDS. J. Immunol. 163:1636–1646 [PubMed] [Google Scholar]
- 148.Gonzalez-Scarano F, Martin-Garcia J. 2005. The neuropathogenesis of AIDS. Nat. Rev. Immunol. 5:69–81. 10.1038/nri1527 [DOI] [PubMed] [Google Scholar]