

Abstract
Epstein-Barr virus (EBV) oncogenes exert potent B cell proliferative effects. EBV infection gives rise to B cell lines that readily proliferate in culture. This ability of EBV represents a powerful tool to study cell proliferation. In efforts to delineate the contribution of signal transducer and activator of transcription 3 (STAT3) toward EBV-driven cell proliferation, we have discovered that B cells from patients with autosomal dominant hyper-IgE syndrome (AD-HIES) resist such EBV oncogene-driven outgrowth of cells. Patients with AD-HIES have a dominant negative mutation in their STAT3 gene which renders most of the protein nonfunctional. Exposure of healthy subject-derived B cells to EBV resulted in early activation of STAT3, rapidly followed by increased expression of its mRNA and protein. STAT3 upregulation preceded the expression of EBNA2, temporally one of the first viral oncogenes to be expressed. We found that STAT3 was necessary for subsequent survival and for proliferation of EBV-infected cells past the S phase of the cell cycle. Consequently, B cells from AD-HIES patients were prone to dying and accumulated in the S phase, thereby accounting for impaired cell outgrowth. Of importance, we have now identified a cohort of patients with a primary immunodeficiency disorder whose B cells oppose EBV-driven proliferative signals. These findings simultaneously reveal how EBV manipulates host STAT3 even before expression of viral oncogenes to facilitate cell survival and proliferation, processes fundamental to EBV lymphomagenesis.
INTRODUCTION
Epstein-Barr virus (EBV) is an oncovirus that infects B cells and epithelial cells (1–3). EBV establishes lifelong latency in memory B lymphocytes; periodic activation into the lytic cycle can lead to asymptomatic shedding of virus in saliva. Upon infection of primary B cells, EBV must first drive cell proliferation in order to establish latency (3). Latency contributes to viral persistence. Although most of mankind is persistently infected with EBV, only a small fraction develops EBV-related cancers of B and epithelial cells (3, 4). While this propensity for development of cancer, particularly posttransplant lymphoproliferative disorders, is prominently associated with loss of EBV-directed T cell immunity (3, 5, 6), the pathogenesis of other forms of EBV-cancers, such as endemic Burkitt lymphoma and nasopharyngeal cell carcinoma, is more complex. Not surprisingly, the contribution of host cellular proteins toward EBV-driven cell proliferation and potentially to EBV-related diseases is important (3, 7–9). Most of our understanding of the involvement of cellular proteins and mechanisms that may contribute to pathogenesis derives from investigations on downstream effects of EBV latent membrane proteins and the nuclear antigens (7, 8). Such studies have included new infection of B cells in culture, examination of EBV-derived B cell lines (lymphoblastoid cell lines [LCL]), and expression of individual viral proteins in culture. We are interested in understanding whether EBV can manipulate the host during the early stages of infection, possibly even before viral latency proteins are expressed.
Signal transducer and activator of transcription 3 (STAT3) is a transcription factor that is well known for its prosurvival and proproliferative functions (10–13). STAT3 is also constitutively active in many human cancers, including EBV-related cancers (11, 13–15). While STAT3 can be transcriptionally induced by the EBV oncoprotein LMP1 in already transformed B cells (16), whether STAT3 contributes to cell proliferation early after infection of primary B cells with EBV has not been investigated. Anecdotally, we have observed that B cells from patients with autosomal dominant hyper-IgE syndrome (AD-HIES or Job's syndrome) are difficult to transform with EBV. Patients with AD-HIES have a heterozygous dominant negative mutation in their STAT3 gene that renders the majority of cellular STAT3 nonfunctional despite normal levels of STAT3 protein (17). Such patients have a rare primary immunodeficiency characterized by eczema, skin and lung infections, extremely elevated serum IgE, and a variety of skeletal, connective tissue, and vascular abnormalities (18). In the setting of EBV infection in culture, we have observed that LCL from AD-HIES patients are slower to emerge than those from healthy subjects, but sometimes LCL cannot be generated even after repeated attempts, suggesting that there are inherent differences between B cells derived from AD-HIES patients and those from healthy subjects. Also of importance, studies have demonstrated that patients with AD-HIES have significant deficits in effector and memory T and B cell responses (19–23); however, the precise mechanisms underlying such defects remain unclear. These observations prompted us to examine the response of AD-HIES B cells to EBV infection and ask whether and when STAT3 contributes toward EBV-driven B cell proliferation.
We now demonstrate using primary B cells that cellular STAT3 is required for the initial phase of EBV-driven growth transformation. We also show that during EBV infection, STAT3 is exploited early by EBV to facilitate viral oncogene-driven cell proliferation. Notably, B cells derived from AD-HIES patients are impaired in their susceptibility to EBV-driven growth transformation because of a combination of factors that include early death of EBV-infected cells as well as accumulation of infected cells in the S phase of the cell cycle, described herein.
MATERIALS AND METHODS
Patient materials.
Blood was obtained from study subjects following informed consent. The study of human subjects was approved by the Institutional Review Boards at Stony Brook University and the NIAID. Healthy EBV-seronegative volunteers ranged from 18 to 28 years of age. Blood was obtained from a total of 11 AD-HIES patients followed at the NIAID (summarized in Table 1). Eight of these had a mutation in the SH2 domain (17); three had a mutation in the DNA binding domain (patients not previously reported). Of note, AD-HIES patients most commonly have a mutation in either the SH2 domain or the DNA binding domain of their STAT3 gene.
TABLE 1.
Characteristics of AD-HIES patients
| Patient ID | Age (yrs) | Gender | Mutation | STAT3 domain | Ability to generate LCL |
|---|---|---|---|---|---|
| J002 (daughter) | 30 | Female | 1865 C-T | SH2 | Yes |
| J002 (father) | 62 | Male | 1865 C-T | SH2 | Yes |
| J011 | 38 | Female | 1909 G-A | SH2 | Yes |
| J022 | 43 | Female | 1909 G-A | SH2 | Yes |
| J074 | 59 | Female | 1954 G-A | SH2 | No |
| J098 | 22 | Male | 1909 G-T | SH2 | Yes |
| J100 | 28 | Female | 1909 G-A | SH2 | Yes |
| A010 | 15 | Female | 1027 G-T | DBD | Yes |
| A011 | 10 | Male | 1294 G-A | DBD | Yes |
| A014 | 39 | Female | 1144 C-T | DBD | Yes |
| J015 | 43 | Female | 1861 T-G | SH2 | Yes |
Isolation of primary B lymphocytes.
Peripheral blood mononuclear cells (PBMC) were harvested from blood, and B cells were isolated by negative selection using CD3 depletion (Stem Cell Technologies). Negative selection was performed to minimize nonspecific activation of B cells. Freshly isolated B cells were used in all experiments.
EBV preparation and infection of B cells.
EBV was isolated from the supernatant of EBV-infected B95-8 cells as described previously (24). Infectivity of virus preparations was assessed as described previously (25), and virus titer was calculated. Infections were performed using EBV as described previously (26) at a multiplicity of infection of 1 to 5. For UV inactivation of the EBV genome, 1 ml of virus suspension in a 100-mm dish was irradiated with UV (254 nm) at 10 mJ/cm2 using a Stratalinker 2400 (Stratagene). For mock infection, culture medium was added instead of EBV suspension.
Culture conditions.
Cells were cultured at 37°C under 5% CO2 in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin sodium, and 100 μg/ml streptomycin sulfate. For experiments using AG490 (25 μM; Cayman Chemical) and acyclovir (200 μM; Sigma), chemicals were added at time zero to cultures. For long-term outgrowth cultures, chemicals were supplemented at the initial concentration every fourth day. We had experimentally determined 25 μM AG490 to be minimally toxic to EBV-infected B cell lines. To measure the impact of STAT3 on long-term outgrowth of cells, freshly isolated CD3− primary B cells were infected with EBV and placed in culture. Cells were harvested at various times thereafter, and live cells were counted by Trypan blue staining. Total number of live cells was calculated for each time point to derive growth curves.
Antibodies.
The following primary antibodies (Ab) were used for immunologic applications: rabbit anti-human phosphoSTAT3 (Y705; catalog number 9145; Cell Signaling), rabbit anti-human STAT3 (catalog number SC482; Santa Cruz), mouse anti-human β-actin (catalog number A5316; Sigma), mouse anti-LMP1 (catalog number M897; Dako), allophycocyanin (APC)-conjugated mouse anti-human CD23 (catalog number 558690; BD), fluorescein isothiocyanate (FITC)-conjugated anti-human CD58 (catalog number MCA2126F; ABD Serotec), and rat anti-EBNA2 (clone R3) (27). Secondary antibodies included horseradish peroxidase (HRP)-conjugated anti-mouse Ab (catalog number A4416; Sigma), HRP-conjugated anti-rabbit Ab (catalog number A0545; Sigma), HRP-conjugated anti-rat Ab (catalog number A9037; Sigma), phycoerythrin (PE)-conjugated anti-mouse IgG1 (catalog number 550083; BD), PE-conjugated anti-mouse IgG (catalog number 550589; BD), FITC-conjugated anti-mouse IgG (catalog number F0257; Sigma), FITC-conjugated anti-rabbit IgG (catalog number A10526; Invitrogen), and Alexa 647-conjugated anti-rabbit IgG (catalog number A21245; Invitrogen). Isotype-matched control antibodies, including mouse IgG1 (catalog number M5284; Sigma) and normal rabbit sera (catalog number R9133; Sigma) were used as negative controls for fluorescence-activated cell sorting (FACS) staining. Apoptosis was detected using annexin V biotin (catalog number 556418; BD) followed by FITC-conjugated avidin (catalog number 43-4411; Invitrogen).
Flow cytometry.
Cells were fixed and permeabilized using Cytofix/Cytoperm (BD), incubated with saturating amounts of either directly conjugated antibody or primary antibody followed by fluorochrome-conjugated secondary antibody as previously described (28). For all flow cytometry experiments, matched isotype control-labeled cells were used to set cutoffs between cells negatively and positively stained with antibodies of interest during analyses. For assessment of cell cycle distribution, cells were stained with 50 μg/ml propidium iodide supplemented with 1 μg/ml RNase A. Samples were then acquired using FACSCalibur (BD), and data analyses were carried out using FlowJo software (Tree Star, Inc.), after gating on live cells based on forward scatter and side scatter values and exclusion of doublets. Statistical analyses were performed using Student t test.
Immunoblotting.
Total extracts from 1 × 106/ml cells were analyzed by immunoblotting as previously described (29). Briefly, cells were electrophoresed using 10% SDS-polyacrylamide gels and transferred to nitrocellulose membranes. Nonspecific binding sites were blocked by incubating membranes overnight in 0.05% Tween 20 (vol/vol in Tris-buffered saline [TBS]) supplemented with 5% nonfat powdered milk or bovine serum albumin (BSA). Following incubation with primary antibody, membranes were incubated with the appropriate HRP-conjugated secondary antibody. Signals were detected using extended chemiluminescence. The abundance of β-actin was monitored to ensure equal loading.
Quantitative RT-PCR.
RNA was isolated, and relative transcript levels were determined with gene-specific primers as previously described (29). Sequences of primers are as previously published: 18SrRNA, STAT3, LMP1 (30), Bcl-xL (31), and Bcl-2 (32). Primer sequences for IRF1 are as follows: forward, 5′AAAAGGAGCCAGATCCCAAGA; reverse, 5′CATCCGGTACACTCGCACAG3′. Relative expression levels were calculated using the threshold cycle (ΔΔCT) method by normalizing to 18S rRNA and comparing among uninfected, EBV-infected, vehicle-treated, or chemical-treated cells with StepOne software version 2.2 (Applied Biosystems). Individual samples were assayed in triplicate.
Statistical analyses.
P values were calculated by comparing the means of two groups of interest using an unpaired Student t test.
RESULTS
Exposure to EBV results in early phosphorylation and increased expression of STAT3.
We addressed whether exposure of B cells to EBV results in activation of STAT3. Within 30 min of exposure of primary B cells from healthy EBV-naive individuals to EBV, we observed phosphorylation of STAT3 at Y705 (Fig. 1A), an event typically associated with activation of STAT3 via the Janus kinase (JAK)-STAT pathway (33). Phosphorylation continued to be observed thereafter, although the amount of phospho-STAT3 dropped off after 2 h of infection. Because activated STAT3 is known to drive STAT3 gene expression (15), we then examined levels of STAT3 protein after exposure to EBV. We found a modest but more sustained increase (2- to 3.4-fold) in STAT3 protein levels by 8 h after exposure to EBV compared to cells immediately after exposure to EBV (time zero; Fig. 1B). Uninfected cells harvested at 72 h had undetectable STAT3 protein consistent with loss of cell viability in culture. In determining the temporal relationship between increased expression of STAT3 and expression of EBNA2, one of the first EBV oncoproteins to be expressed, we found that EBNA2 was expressed by 24 h after EBV exposure (Fig. 1B). This timing was consistent with earlier observations on EBNA2 expression (34). Thus, exposure to EBV results in both activation and increased expression of STAT3, events that temporally precede expression of the EBV latency oncoprotein EBNA2. Similar to STAT3, early expression of other cellular proteins, such as CD23 and CD40, has also been observed within several hours of EBV infection (35, 36).
FIG 1.
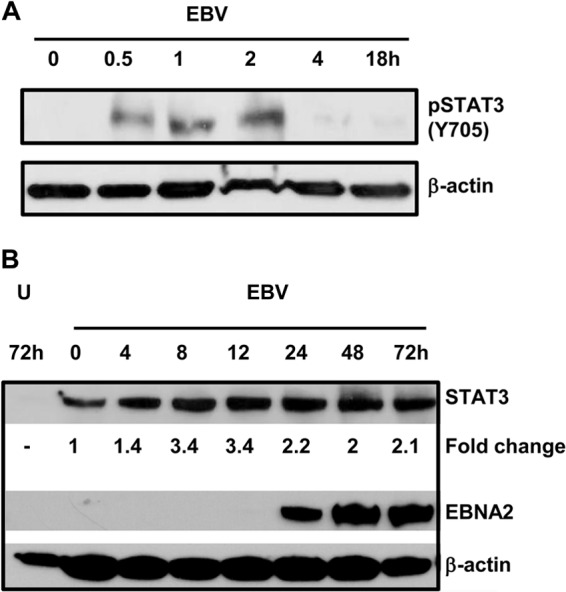
Exposure of primary B lymphocytes to EBV results in phosphorylation and increased levels of STAT3. Primary B cells from healthy EBV-seronegative subjects were exposed to EBV for the indicated times, and phosphorylation of STAT3 at Y705 (A) and total levels of STAT3 and EBV EBNA2 (B) were determined by immunoblotting. Fold change, STAT3 levels at time points postexposure compared to time zero after normalization to β-actin; U, uninfected cells harvested after 72 h in culture. These experiments were performed 3 times.
Early events following EBV exposure result in a transient increase in STAT3 transcript levels.
Because activated STAT3 transcriptionally activates its own expression (15), we examined the levels of STAT3 mRNA at various times after exposure to EBV. Figure 2A shows that there was an almost 70-fold increase in STAT3 transcript level by 1 h after exposure to EBV; this increase temporally followed STAT3 activation at 30 min (Fig. 1A). After the first hour, STAT3 continued to be expressed at much lower levels. The dramatic increase in STAT3 mRNA compared to a more modest increase in STAT3 protein (Fig. 1B), while surprising, has also been observed previously (37, 38). Almost instantaneous activation and increase in STAT3 expression, particularly before expression of EBNA2, suggested that very early events unrelated to viral gene expression were causal to the observed changes in STAT3. As shown in Fig. 2A to C, we observed an increase in phospho-STAT3, STAT3 mRNA, and total STAT3 despite UV inactivation of EBV. As expected, there was significant suppression of the EBNA2 transcript level consistent with loss of viral gene expression resulting from UV treatment (Fig. 2D). Since EBV lytic gene expression may contribute to EBV-driven cell proliferation and tumorigenesis (35, 39, 40), we tested the effect of acyclovir on STAT3 mRNA. Acyclovir selectively inhibits EBV DNA replication during the lytic cycle. The early increases in phospho-STAT3, STAT3 transcript level, and total STAT3 were unaffected by treatment with acyclovir (Fig. 2A to C). Since phosphorylation of STAT3 at Y705 is mediated by Janus kinases, we reasoned that AG490, a Janus kinase inhibitor that blocks activation of STAT3 (29, 41), should suppress STAT3 mRNA. Not surprisingly, we observed a near-total loss of STAT3 mRNA and absence of increases in phospho-STAT3 and total STAT3 following treatment with AG490 (Fig. 2A to C). These findings suggest that an early event such as virus binding or internalization triggers activation followed by increased expression of STAT3, likely via the JAK-STAT pathway. The lack of effect of acyclovir on STAT3 suggests that completion of the EBV lytic cycle is not necessary for the initial burst in STAT3 expression. Taken together, experiments shown in Fig. 1 and 2 suggest that EBV may be setting the stage for B cell proliferation even before expression of viral proproliferative oncoproteins.
FIG 2.

An early signaling event following exposure to EBV triggers an initial burst in STAT3 gene expression. (A to C) Primary B cells from healthy subjects were uninfected (U) or exposed to EBV (EBV), EBV and acyclovir (EBV+ACV), EBV and AG490 (EBV+AG490), or UV-inactivated EBV (uv-EBV). Cells were placed in culture and harvested at the indicated times for determination of relative amounts of STAT3 mRNA using qRT-PCR (transcript levels were compared to that at time zero) in panel A, phospho-STAT3 (Y705) in panel B, and total STAT3 in panel C. Fold changes in panel C were compared to time zero after normalization to β-actin. (D) Cells were harvested at 24 h for measurement of EBNA2 mRNA by qRT-PCR. Data from two experiments and three technical qRT-PCR replicates are incorporated for each data point. All error bars = standard errors of the means (SEM).
STAT3 is necessary for outgrowth of EBV-infected proliferating B cells.
To assess the contribution of STAT3 toward outgrowth of EBV-infected cells, we used complementary approaches. First, AG490 was included during EBV infection to pharmacologically impair STAT3. Second, to safeguard against potential off-target effects of AG490 and to evaluate their response to EBV, B cells from patients with AD-HIES were used. Using LMP1, a critical latency oncoprotein, to mark individual EBV-infected cells, we found that approximately 7-fold-fewer LMP1+ cells resulted 4 days after EBV infection when AG490 was added (Fig. 3A and B). Similarly, there was a 2.3-fold reduction in LMP1+ cells when AD-HIES B cells were infected compared to when healthy B cells were infected. We then asked if the significant fall in LMP1+ cells upon impairment of STAT3 had an effect on the outgrowth of LCL. Figure 4 shows that outgrowth of transformed B cell lines (LCL) either failed, in the case of AG490, or was delayed, for AD-HIES B cells; the latter is consistent with a dose effect due to a functional “STAT3 knockdown” in AD-HIES cells. While we were able to generate LCL from 10 of 11 patients tested (Table 1), these were slow to grow out. Thus, STAT3, which is upregulated before viral oncogene expression, is required for emergence of LMP1+ cells and for the initial outgrowth of LCL.
FIG 3.
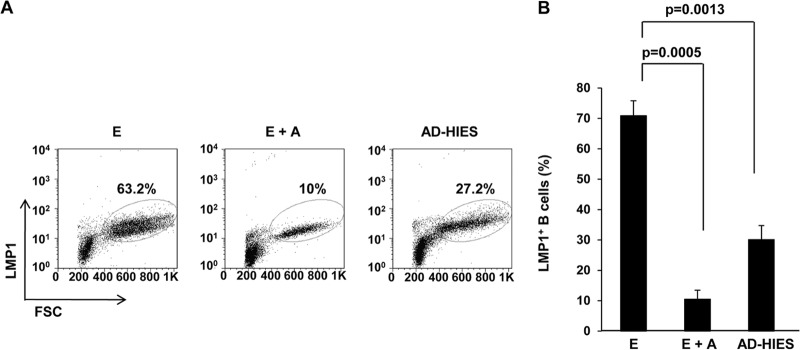
STAT3 is necessary for emergence of LMP1+ B cells after infection with EBV. Primary B cells from healthy subjects were infected with EBV in the absence (E) or presence (E+A) of AG490; B cells from AD-HIES patients were similarly infected with EBV (AD-HIES). Cells were harvested on day 4, and percentage of cells expressing EBV oncoprotein LMP1 was enumerated by flow cytometry (% within dot plots). Representative data are shown in panel A, and aggregate data from six healthy and six AD-HIES patients are shown in panel B. Error bars = SEM.
FIG 4.
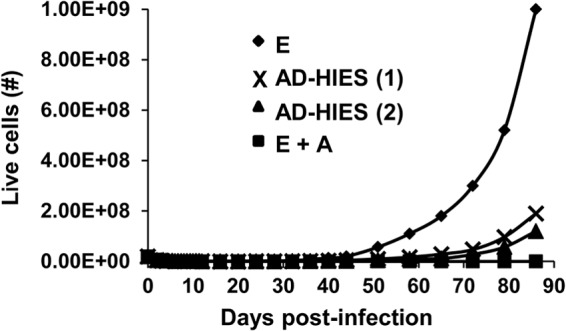
STAT3 is necessary for outgrowth of proliferating B cells following EBV infection. Primary B cells from a healthy subject were infected with EBV in the absence (E) or presence (E+A) of AG490; primary B cells from two patients with AD-HIES [AD-HIES (1) and AD-HIES (2), with different mutations in the SH2 domain] were also infected with EBV. Cells were harvested at various times after EBV infection, and live cells were counted by Trypan blue staining. Representative growth curves are shown. Experiments were performed more than three times.
Suppression of STAT3 causes death of EBV-infected B cells.
Fewer LMP1+ cells derived from AD-HIES patients or in the presence of AG490 suggested prosurvival effects of STAT3. This led us to examine the effect of STAT3 on cell survival during EBV infection. Figure 5A shows that as early as 2 days after EBV infection, there were approximately 7-fold more LMP1+ cells but not LMP1− cells undergoing apoptosis by annexin V staining in the presence of AG490. There was also an increase in LMP1+ cells undergoing apoptosis when derived from AD-HIES patients. This change, though not significant compared to LMP1+ healthy B cells, taken together with increase in death compared to that for AD-HIES-derived LMP1− cells, supports the observation of increased cell death in the presence of AG490. To understand if STAT3-mediated suppression of LMP1 gene expression contributes to death of and therefore fewer LMP1+ cells, we examined LMP1 transcript levels. As levels of LMP1 message were not significantly suppressed in the presence of AG490 (Fig. 5B), impaired LMP1 transcription did not significantly contribute to the reduction in LMP1+ cells observed upon interference with STAT3 function. We have shown earlier that suppression of STAT3 in LCL increases lytic activation (29), raising the possibility that cell death in AG490-treated cells could be a consequence of lytic activation in newly infected cells. We were unable to detect BZLF1 (the earliest lytic gene) expression in the presence of AG490 (data not shown), suggesting that lytic activation in newly infected cells does not contribute significantly to cell death when STAT3 is impaired.
FIG 5.

Impairment of STAT3 function causes death of EBV-infected cells and lower transcript levels of STAT3 targets but not of LMP1. (A) B cells from healthy EBV-seronegative subjects were infected with EBV in the absence (black bars) or presence (open bars) of AG490; B cells from AD-HIES patients were also infected with EBV (gray bars). Cells were harvested on day 2 and evaluated by flow cytometry for expression of LMP1 and staining with annexin V. Percent annexin V+ cells within LMP1+ and LMP1− populations was calculated, and aggregate data from six healthy and six AD-HIES patients are shown. (B and C) Cells were harvested 24 h after infection of healthy B cells, and relative levels of LMP1 mRNA (B) and mRNA from genes that are known transcriptional targets of STAT3 (Bcl-xL and Bcl-2) and STAT1 (IRF1) (C) were determined by qRT-PCR. Solid bars, EBV; open bars, EBV+AG490. Data from three experiments and three technical qRT-PCR replicates are incorporated for each data point. All error bars = SEM; N.S., not significant. (D) Infections were performed as described for panels B and C. Twenty-four hours after infection, intracellular levels of STAT3 were determined by flow cytometry. Solid line, EBV; dashed line, EBV+AG490.
In addressing the effect of STAT3 on transcription of prosurvival genes and to confirm that AG490 interfered with STAT3 level and its functions, we found that AG490 suppressed the level of STAT3 protein (Fig. 5D). Consistent with death of LMP1+ cells, AG490 also suppressed mRNA levels of antiapoptotic genes Bcl-xL and Bcl-2, both transcriptional targets of STAT3 (Fig. 5C). In contrast, the transcript level of IRF1, a STAT1-responsive gene, was not affected. These results support the concept that infection with EBV causes early activation and expression of host STAT3, which promotes cell survival contributing toward EBV-mediated B cell transformation.
Suppression of STAT3 results in impaired cell proliferation due to delay in S phase.
Because outgrowth of LCL is likely to be a net result of cells undergoing proliferation and death, and because STAT3 is well known for its proproliferative effects, we asked if STAT3 contributes toward transformation also by facilitating cell proliferation. We and others have shown that only a small fraction of EBV-infected cells undergo proliferation (9, 36, 42). In our earlier efforts to predict which EBV-infected cells undergo proliferation, we discovered a cell surface marker pattern specific to proliferating cells. These cells, identified by the marker profile CD23hi CD58+, expressed LMP1, were detectable as early as 3 days after EBV infection, and subsequently underwent proliferation (36). Figures 6A and B show that in the presence of AG490, there was a 10-fold reduction in CD23hi CD58+ cells compared to untreated EBV-infected cells. Fewer CD23hi CD58+ cells could be due to death of EBV-infected cells, a defect in proliferation of EBV-infected cells, or both. We therefore examined cell cycle profiles after EBV infection of B cells derived from healthy subjects and AD-HIES patients. We observed more LMP1+ cells in S phase, both in the presence of AG490 and when AD-HIES cells were infected, compared to infected healthy B cells. An increased number of cells in S phase could be the result of increased transit of cells from G1 to S phase and/or decreased transit of cells from S to G2 phase. Compared to cells with intact STAT3 (Fig. 6C, “E”), when AG490 was used to suppress STAT3 expression (Fig. 6C, “E+A”), there was a 13% reduction in the fraction of G1 cells compared to a 26% increase in the fraction of S phase cells. This reduction did not account for the remaining 13% increase (26% − 13%) observed in S phase in the presence of AG490; however, this increase correlated with a concomitant 12% fall in the fraction of cells in G2 phase in E+A cells compared to E cells. Therefore, while increased transit of cells from G1 to S could partially contribute to an increase in cells in S phase, it is unlikely that loss of STAT3, a proproliferative protein, which causes loss of cell outgrowth (Fig. 4), would cause a paradoxical increase in cell progress from G1 to S phase. Therefore, these observations are most consistent with delay in transit of cells from S to G2 phase upon impairment of STAT3. To quantitate this delay in S phase, we calculated the fraction of cells with >2 N nuclear DNA that was in G2 [G2/(S + G2)]. Suppression of STAT3 resulted in a smaller fraction than in cells with intact STAT3 (0.2% for E+A and 9.7% and 9.6% for AD-HIES patients, compared to 27.7% for E). As expected, uninfected cells in culture did not proliferate (Fig. 6A and B) and neither did LMP1− cells (data not shown). These data demonstrate that following infection with EBV, early upregulation of STAT3 is necessary for promoting cell proliferation beyond the S phase and that its impairment results in arrest/delay in S phase.
FIG 6.

Impairment of STAT3 results in fewer proliferating cells and delay in S phase. Primary B cells from healthy EBV-seronegative subjects were infected with EBV alone (E) or in the presence of AG490 (E+A) in panels A to C; B cells from two AD-HIES patients [AD-HIES (1) and (2)] were also infected with EBV in panel C. Cells were harvested on day 4. (A and B) CD23hi CD58+ proliferating cells were enumerated by flow cytometry. U, uninfected cells in culture for 4 days. Representative data (percent CD23hi CD58+ cells depicted in boxes) are shown in panel A, and aggregate data from 6 experiments are shown in panel B. (C) LMP1+ cells were evaluated for cell cycle profile by flow cytometry, and percent cells in G1 phase (open bars), S phase (black bars), and G2/M phase (gray bars) are shown. U, total uninfected B cells on day 0. Percentages represent G2/(S + G2). Experiment shown in panel C was performed twice.
DISCUSSION
This study functionally links STAT3 to EBV oncogene-driven B cell proliferation by showing that STAT3 is necessary for cell survival and for progress of cells past the S phase to facilitate cell proliferation. EBV targets STAT3 almost immediately after exposure of B cells to virus. The kinetics of STAT3 phosphorylation, of expression of its gene products, and of expression of EBNA2, taken together with infection in the presence of inhibitors and with UV-inactivated virus, indicate that early events, such as virus binding or internalization, lead to activation of STAT3, possibly by the JAK-STAT signaling pathway. Using B cells from rare patients with AD-HIES adds to the biological and clinical relevance of these findings while simultaneously providing much-needed insights into the ability of B cells from AD-HIES patients to proliferate.
We have now identified a cohort of individuals who are at least partially resistant to EBV-mediated cell proliferation; to our knowledge, this is a first for EBV. This identification now also provides a powerful tool to further understand the contribution of STAT3 toward susceptibility to EBV-driven cell proliferation, establishment of latency, and lytic activation from latency. While it is practically impossible to exclude all other causes, we believe the underlying cause for this resistance to EBV-driven cell proliferation to be functional knockdown of STAT3 function. This contention is supported by similar responses to EBV infection by cells from several AD-HIES patients, despite the presence of a distinct and unique mutation in the STAT3 gene in each patient. It is further supported by similar outcomes in the presence of AG490. Whether patients with AD-HIES are less susceptible to EBV infection is unknown because of very low disease frequency. Whether AD-HIES patients are less susceptible to EBV cancers is also not known; available data would support death of EBV-infected cells due to increased lytic activation on the one hand (22, 29), with impaired clearance of EBV-infected cells because of poor effector and memory T cell responses on the other (20–22). On rare occasion, EBV lymphomas have been described in AD-HIES patients. However, in a recent cohort of 60 AD-HIES patients, 7% developed lymphomas, none associated with EBV (43). Whether deficiency of STAT3 in these patients may impact clinical outcomes of EBV infection also remains a question.
The contribution of STAT3 toward the life cycle of other herpesviruses has been described for Kaposi's sarcoma-associated herpesvirus (KSHV) and varicella zoster virus (VZV) (44–46). In the case of infection of endothelial cells with KSHV, an oncogenic gamma-herpesvirus-like EBV, STAT3 is activated in a biphasic manner. The early phase of activation is attributed to virus binding and entry leading to signaling via the JAK-STAT pathway (45). More recently, the late phase of activation has been shown to be associated with phosphorylation of STAT3 at S727, mediated by the KSHV latency protein kaposin B (44). Although currently the status of p(S727)STAT3 is unexplored for EBV, lack of substantial p(Y705)STAT3 beyond 2 to 4 h following exposure to EBV in our experiments could be consistent with ongoing activation of STAT3 via phosphorylation at only S727, similar to that observed for KSHV, especially since high levels of STAT3 protein continue to persist.
The contribution of EBV latency proteins as well as cellular targets downstream of these proteins to cell proliferation has been extensively studied. EBV latency proteins LMP1, LMP2a, EBNA2, EBNA3A, EBNA3B, EBNA3C, and EBNA-LP intersect with and manipulate several cell signaling pathways which ultimately contribute toward successful EBV-mediated B cell proliferation. These activities are aided further by epigenetic regulation of cellular gene expression via some of the above-mentioned proteins (7, 8). STAT3 activation described here and its downstream effects are set in motion before the oncoprotein EBNA2 and therefore before EBNA3 proteins, EBNA-LP, or LMP1 are expressed. However, this does not exclude the possibility that at later times, these and other viral proteins may contribute toward or modulate STAT3-mediated functions; indeed, LMP1 has been shown to transcriptionally activate STAT3 in LCL (16). Conversely, STAT3 may affect viral gene expression, including that of LMP1, whose promoter contains a STAT3 binding site (47). In our experiments, while there was a suggestion that STAT3 might regulate LMP1 gene expression (Fig. 5B), the ultimate effect on LMP1 protein levels (Fig. 3A, intensity of staining on y axis) was not convincing, a finding consistent with that of Zhang et al. (16).
STAT3 is a well-studied transcription factor whose overexpression or activation is linked to many cancers (11, 13–15). As such, STAT3 has been recognized as a target for anticancer drug development (13). Our earlier discovery that impairment of STAT3 increases lytic susceptibility in EBV cancer and EBV-transformed cells (29), combined with our present finding that suppression of STAT3 limits the ability of EBV to drive B cell proliferation, now provides experimental evidence that pharmacologic manipulation of STAT3 is likely to target EBV-infected B cells by at least two independent mechanisms. Certainly, these findings warrant further investigations into the mechanisms by which STAT3 contributes toward EBV-driven cell proliferation and lytic activation, both critical to development of EBV-related diseases.
ACKNOWLEDGMENTS
This study was supported by K08 AI062732, K12 HD001401, 1UL1RR024139-02, and funds from The Research Foundation for The State University of New York to S.B.-M.
We thank Steven Holland at the NIAID for providing access to AD-HIES patient samples and AD-HIES patients as well as healthy blood donors at the NIH Primary Immune Deficiency Clinic for their participation.
S.K. and S.B.-M. designed the study. S.K. and A.D.L.P. carried out the experiments, A.F.F. helped to obtain materials from patients, S.K. and S.B.-M. analyzed data and interpreted the findings, and S.B.-M. wrote the manuscript.
The authors declare no competing financial interests.
Footnotes
Published ahead of print 30 October 2013
REFERENCES
- 1.Shannon-Lowe C, Adland E, Bell AI, Delecluse HJ, Rickinson AB, Rowe M. 2009. Features distinguishing Epstein-Barr virus infections of epithelial cells and B cells: viral genome expression, genome maintenance, and genome amplification. J. Virol. 83:7749–7760. 10.1128/JVI.00108-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shannon-Lowe C, Rowe M. 2011. Epstein-Barr virus infection of polarized epithelial cells via the basolateral surface by memory B cell-mediated transfer infection. PLoS Pathog. 7:e1001338. 10.1371/journal.ppat.1001338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thorley-Lawson DA, Gross A. 2004. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N. Engl. J. Med. 350:1328–1337. 10.1056/NEJMra032015 [DOI] [PubMed] [Google Scholar]
- 4.Young LS, Rickinson AB. 2004. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 4:757–768. 10.1038/nrc1452 [DOI] [PubMed] [Google Scholar]
- 5.Heslop HE, Brenner MK, Rooney CM. 1994. Donor T cells to treat EBV-associated lymphoma. N. Engl. J. Med. 331:679–680. 10.1056/NEJM199409083311017 [DOI] [PubMed] [Google Scholar]
- 6.Papadopoulos EB, Ladanyi M, Emanuel D, Mackinnon S, Boulad F, Carabasi MH, Castro-Malaspina H, Childs BH, Gillio AP, Small TN, et al. 1994. Infusions of donor leukocytes to treat Epstein-Barr virus-associated lymphoproliferative disorders after allogeneic bone marrow transplantation. N. Engl. J. Med. 330:1185–1191. 10.1056/NEJM199404283301703 [DOI] [PubMed] [Google Scholar]
- 7.Saha A, Kaul R, Murakami M, Robertson ES. 2010. Tumor viruses and cancer biology: modulating signaling pathways for therapeutic intervention. Cancer Biol. Ther. 10:961–978. 10.4161/cbt.10.10.13923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sompallae R, Callegari S, Kamranvar SA, Masucci MG. 2010. Transcription profiling of Epstein-Barr virus nuclear antigen (EBNA)-1 expressing cells suggests targeting of chromatin remodeling complexes. PLoS One 5:e12052. 10.1371/journal.pone.0012052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thorley-Lawson DA, Mann KP. 1985. Early events in Epstein-Barr virus infection provide a model for B cell activation. J. Exp. Med. 162:45–59. 10.1084/jem.162.1.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Darnell JE., Jr 1997. STATs and gene regulation. Science 277:1630–1635. 10.1126/science.277.5332.1630 [DOI] [PubMed] [Google Scholar]
- 11.Darnell JE., Jr 2002. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2:740–749. 10.1038/nrc906 [DOI] [PubMed] [Google Scholar]
- 12.Yang J, Stark GR. 2008. Roles of unphosphorylated STATs in signaling. Cell Res. 18:443–451. 10.1038/cr.2008.41 [DOI] [PubMed] [Google Scholar]
- 13.Yu H, Jove R. 2004. The STATs of cancer—new molecular targets come of age. Nat. Rev. Cancer 4:97–105. 10.1038/nrc1275 [DOI] [PubMed] [Google Scholar]
- 14.Nepomuceno RR, Snow AL, Robert Beatty P, Krams SM, Martinez OM. 2002. Constitutive activation of Jak/STAT proteins in Epstein-Barr virus-infected B-cell lines from patients with posttransplant lymphoproliferative disorder. Transplantation 74:396–402. 10.1097/00007890-200208150-00017 [DOI] [PubMed] [Google Scholar]
- 15.Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, Stark GR. 2005. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 65:939–947 http://cancerres.aacrjournals.org/content/65/3/939.long [PubMed] [Google Scholar]
- 16.Zhang L, Hong K, Zhang J, Pagano JS. 2004. Multiple signal transducers and activators of transcription are induced by EBV LMP-1. Virology 323:141–152. 10.1016/j.virol.2004.03.007 [DOI] [PubMed] [Google Scholar]
- 17.Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, Freeman AF, Demidowich A, Davis J, Turner ML, Anderson VL, Darnell DN, Welch PA, Kuhns DB, Frucht DM, Malech HL, Gallin JI, Kobayashi SD, Whitney AR, Voyich JM, Musser JM, Woellner C, Schaffer AA, Puck JM, Grimbacher B. 2007. STAT3 mutations in the hyper-IgE syndrome. N. Engl. J. Med. 357:1608–1619. 10.1056/NEJMoa073687 [DOI] [PubMed] [Google Scholar]
- 18.Freeman AF, Holland SM. 2009. Clinical manifestations, etiology, and pathogenesis of the hyper-IgE syndromes. Pediatr. Res. 65:32R–37R. 10.1203/PDR.0b013e31819dc8c5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Avery DT, Deenick EK, Ma CS, Suryani S, Simpson N, Chew GY, Chan TD, Palendira U, Bustamante J, Boisson-Dupuis S, Choo S, Bleasel KE, Peake J, King C, French MA, Engelhard D, Al-Hajjar S, Al-Muhsen S, Magdorf K, Roesler J, Arkwright PD, Hissaria P, Riminton DS, Wong M, Brink R, Fulcher DA, Casanova JL, Cook MC, Tangye SG. 2010. B cell-intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J. Exp. Med. 207:155–171. 10.1084/jem.20091706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, Fulcher DA, Tangye SG, Cook MC. 2008. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J. Exp. Med. 205:1551–1557. 10.1084/jem.20080218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, Davis J, Hsu A, Asher AI, O'Shea J, Holland SM, Paul WE, Douek DC. 2008. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452:773–776. 10.1038/nature06764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siegel AM, Heimall J, Freeman AF, Hsu AP, Brittain E, Brenchley JM, Douek DC, Fahle GH, Cohen JI, Holland SM, Milner JD. 2011. A critical role for STAT3 transcription factor signaling in the development and maintenance of human T cell memory. Immunity 35:806–818. 10.1016/j.immuni.2011.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tangye SG, Cook MC, Fulcher DA. 2009. Insights into the role of STAT3 in human lymphocyte differentiation as revealed by the hyper-IgE syndrome. J. Immunol. 182:21–28 http://www.jimmunol.org/content/182/1/21.full [DOI] [PubMed] [Google Scholar]
- 24.Oh HM, Oh JM, Choi SC, Kim SW, Han WC, Kim TH, Park DS, Jun CD. 2003. An efficient method for the rapid establishment of Epstein-Barr virus immortalization of human B lymphocytes. Cell Prolif. 36:191–197. 10.1046/j.1365-2184.2003.00276.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henle W, Henle GE, Horwitz CA. 1974. Epstein-Barr virus specific diagnostic tests in infectious mononucleosis. Hum. Pathol. 5:551–565. 10.1016/S0046-8177(74)80006-7 [DOI] [PubMed] [Google Scholar]
- 26.Hui-Yuen J, McAllister S, Koganti S, Hill E, Bhaduri-McIntosh S. 2011. Establishment of Epstein-Barr virus growth-transformed lymphoblastoid cell lines. J. Exp. 10.3791/3321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kremmer E, Kranz BR, Hille A, Klein K, Eulitz M, Hoffmann-Fezer G, Feiden W, Herrmann K, Delecluse HJ, Delsol G, Bornkamm GW, Mueller-Lantzsch N, Grassert FA. 1995. Rat monoclonal antibodies differentiating between the Epstein-Barr virus nuclear antigens 2A (EBNA2A) and 2B (EBNA2B). Virology 208:336–342. 10.1006/viro.1995.1157 [DOI] [PubMed] [Google Scholar]
- 28.Bhaduri-McIntosh S, Rotenberg MJ, Gardner B, Robert M, Miller G. 2008. Repertoire and frequency of immune cells reactive to Epstein-Barr virus-derived autologous lymphoblastoid cell lines. Blood 111:1334–1343. 10.1182/blood-2007-07-101907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hill ER, Koganti S, Zhi J, Megyola C, Freeman AF, Palendira U, Tangye SG, Farrell PJ, Bhaduri-McIntosh S. 2013. Signal transducer and activator of transcription 3 limits Epstein-Barr virus lytic activation in B lymphocytes. J. Virol. 87:11438–11446. 10.1128/JVI.01762-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daigle D, Megyola C, El-Guindy A, Gradoville L, Tuck D, Miller G, Bhaduri-McIntosh S. 2010. Upregulation of STAT3 marks Burkitt lymphoma cells refractory to Epstein-Barr virus lytic cycle induction by HDAC inhibitors. J. Virol. 84:993–1004. 10.1128/JVI.01745-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kung CP, Meckes DG, Jr, Raab-Traub N. 2011. Epstein-Barr virus LMP1 activates EGFR, STAT3, and ERK through effects on PKCdelta. J. Virol. 85:4399–4408. 10.1128/JVI.01703-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pigazzi M, Manara E, Baron E, Basso G. 2009. miR-34b targets cyclic AMP-responsive element binding protein in acute myeloid leukemia. Cancer Res. 69:2471–2478. 10.1158/0008-5472.CAN-08-3404 [DOI] [PubMed] [Google Scholar]
- 33.Wen Z, Zhong Z, Darnell JE., Jr 1995. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 82:241–250. 10.1016/0092-8674(95)90311-9 [DOI] [PubMed] [Google Scholar]
- 34.Allday MJ, Crawford DH, Griffin BE. 1989. Epstein-Barr virus latent gene expression during the initiation of B cell immortalization. J. Gen. Virol. 70(Part 7):1755–1764. 10.1099/0022-1317-70-7-1755 [DOI] [PubMed] [Google Scholar]
- 35.Halder S, Murakami M, Verma SC, Kumar P, Yi F, Robertson ES. 2009. Early events associated with infection of Epstein-Barr virus infection of primary B-cells. PLoS One 4:e7214. 10.1371/journal.pone.0007214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Megyola C, Ye J, Bhaduri-McIntosh S. 2011. Identification of a sub-population of B cells that proliferates after infection with Epstein-Barr virus. Virol. J. 8:84. 10.1186/1743-422X-8-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ichiba M, Nakajima K, Yamanaka Y, Kiuchi N, Hirano T. 1998. Autoregulation of the Stat3 gene through cooperation with a cAMP-responsive element-binding protein. J. Biol. Chem. 273:6132–6138. 10.1074/jbc.273.11.6132 [DOI] [PubMed] [Google Scholar]
- 38.Nakajima K, Yamanaka Y, Nakae K, Kojima H, Ichiba M, Kiuchi N, Kitaoka T, Fukada T, Hibi M, Hirano T. 1996. A central role for Stat3 in IL-6-induced regulation of growth and differentiation in M1 leukemia cells. EMBO J. 15:3651–3658 [PMC free article] [PubMed] [Google Scholar]
- 39.Kalla M, Schmeinck A, Bergbauer M, Pich D, Hammerschmidt W. 2010. AP-1 homolog BZLF1 of Epstein-Barr virus has two essential functions dependent on the epigenetic state of the viral genome. Proc. Natl. Acad. Sci. U. S. A. 107:850–855. 10.1073/pnas.0911948107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hong GK, Gulley ML, Feng WH, Delecluse HJ, Holley-Guthrie E, Kenney SC. 2005. Epstein-Barr virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. J. Virol. 79:13993–14003. 10.1128/JVI.79.22.13993-14003.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meydan N, Grunberger T, Dadi H, Shahar M, Arpaia E, Lapidot Z, Leeder JS, Freedman M, Cohen A, Gazit A, Levitzki A, Roifman CM. 1996. Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature 379:645–648. 10.1038/379645a0 [DOI] [PubMed] [Google Scholar]
- 42.Thorley-Lawson DA, Nadler LM, Bhan AK, Schooley RT. 1985. BLAST-2 [EBVCS], an early cell surface marker of human B cell activation, is superinduced by Epstein Barr virus. J. Immunol. 134:3007–3012 [PubMed] [Google Scholar]
- 43.Chandesris MO, Melki I, Natividad A, Puel A, Fieschi C, Yun L, Thumerelle C, Oksenhendler E, Boutboul D, Thomas C, Hoarau C, Lebranchu Y, Stephan JL, Cazorla C, Aladjidi N, Micheau M, Tron F, Baruchel A, Barlogis V, Palenzuela G, Mathey C, Dominique S, Body G, Munzer M, Fouyssac F, Jaussaud R, Bader-Meunier B, Mahlaoui N, Blanche S, Debre M, Le Bourgeois M, Gandemer V, Lambert N, Grandin V, Ndaga S, Jacques C, Harre C, Forveille M, Alyanakian MA, Durandy A, Bodemer C, Suarez F, Hermine O, Lortholary O, Casanova JL, Fischer A, Picard C. 2012. Autosomal dominant STAT3 deficiency and hyper-IgE syndrome: molecular, cellular, and clinical features from a French national survey. Medicine 91:e1–e19. 10.1097/MD.0b013e31825f95b9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.King CA. 2013. Kaposi's sarcoma-associated herpesvirus kaposin B induces unique monophosphorylation of STAT3 at serine 727 and MK2-mediated inactivation of the STAT3 transcriptional repressor TRIM28. J. Virol. 87:8779–8791. 10.1128/JVI.02976-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Punjabi AS, Carroll PA, Chen L, Lagunoff M. 2007. Persistent activation of STAT3 by latent Kaposi's sarcoma-associated herpesvirus infection of endothelial cells. J. Virol. 81:2449–2458. 10.1128/JVI.01769-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sen N, Che X, Rajamani J, Zerboni L, Sung P, Ptacek J, Arvin AM. 2012. Signal transducer and activator of transcription 3 (STAT3) and survivin induction by varicella-zoster virus promote replication and skin pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 109:600–605. 10.1073/pnas.1114232109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen H, Lee JM, Zong Y, Borowitz M, Ng MH, Ambinder RF, Hayward SD. 2001. Linkage between STAT regulation and Epstein-Barr virus gene expression in tumors. J. Virol. 75:2929–2937. 10.1128/JVI.75.6.2929-2937.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]