

Abstract
Most avian influenza viruses do not replicate efficiently in human cells. This is partly due to the low activity of the RNA polymerase of avian influenza viruses in mammalian cells. Nevertheless, this impediment can be overcome through an E→K adaptive mutation at residue 627 of the PB2 subunit of the polymerase. Accordingly, viral ribonucleoprotein (RNP) reconstitution assays show that a viral polymerase containing PB2 627E has impaired activity in mammalian cells compared to a viral polymerase that contains PB2 627K, characteristic of mammalian-adapted influenza viruses. In contrast, purified viral polymerases containing either PB2 627E or PB2 627K show comparable levels of activity in transcription assays that require no RNP assembly. We sought to reconcile these conflicting observations by using an NP-independent cell-based transcription/replication assay to assess viral polymerase activity. We found that PB2 627E polymerase restriction in mammalian cells is independent of NP expression but is dependent on the length of the viral RNA template. In addition, restriction of PB2 627E polymerase was overcome by mutations specific to the viral RNA template promoter sequence. Consequently, we propose that PB2 627E affects recruitment of the viral RNA promoter by the viral polymerase in mammalian cells.
INTRODUCTION
A major obstacle blocking the efficient replication of avian influenza viruses in humans is the low activity of avian influenza virus polymerases in human cells (1). Accordingly, in order to function efficiently in human cells, avian-derived influenza virus polymerases are known to undergo adaptive changes. The viral polymerase, composed of three subunits, polymerase acidic (PA) and polymerase basic one and two (PB1 and PB2), catalyzes the replication and transcription of the viral genome, composed of eight single-stranded, negative-sense RNA segments. In order to accomplish this, the viral polymerase binds to the 5′ and 3′ ends of viral RNA (vRNA) segments that form the vRNA promoter (2, 3). The rest of the vRNA is bound by multiple copies of the viral nucleoprotein (NP) to form viral ribonucleoprotein complexes (vRNPs). vRNPs are transcribed by the viral polymerase, which produces mRNA and a positive-sense cRNA replication intermediate that is in turn used as a template for vRNA synthesis (4–6).
The archetypal adaptive mutation known to confer increased activity on viral polymerases of avian origin in mammalian cells occurs at residue 627 of the PB2 polymerase subunit (7). Avian viral isolates predominantly encode glutamic acid at PB2 627 (627E), whereas this residue is almost exclusively a lysine (627K) in influenza viruses isolated from humans, with the exception of the 2009 pandemic H1N1 viruses (8, 9). The presence of lysine at this position results in increased viral replication in mammalian models (10–13). In addition, the PB2 627 E→K mutation has been associated with lethality in humans infected with H5N1, H7N7, and H7N9 avian viruses (10, 14–16).
Several studies have sought to elucidate the molecular basis for the increase in viral polymerase activity in human cells associated with the PB2 627 E→K mutation. In this regard, it has been demonstrated that PB2 627K allows avian-derived viral polymerases to evade a cellular inhibitor in human cells (17). In contrast, a similar study proposed that human cells are deficient in an activating factor that acts selectively on polymerases containing PB2 627E (18). Moreover, it has been suggested that PB2 627E has decreased affinity for NP, thereby inhibiting the formation of transcriptionally active viral RNPs (17, 19–21). In this study, we use a recently developed cell-based replication assay to study the viral determinants of PB2 627E polymerase restriction. Using this approach, we demonstrate that restriction of viral polymerases containing PB2 627E occurs independently of the interaction between PB2 and NP. In addition, we show that PB2 627E polymerase activity can be rescued on short vRNA-like templates or by introducing mutations to the vRNA promoter sequence.
MATERIALS AND METHODS
Plasmids.
The plasmids expressing PB1, PB2, PA, and NP of influenza virus A/WSN/33, as well as plasmids expressing short (47- to 101-nucleotide [nt]) vRNA templates derived from segment 5, have been described previously (22, 23). Similar plasmids expressing short vRNA templates were constructed by PCR mutagenesis, producing internally deleted vRNA templates derived from segment 5 (NP37 [5′-1–17:CTAG:1549–1565-3′; the numbers indicate the nucleotide positions retained at the 5′ and 3′ ends, respectively, separated by a CTAG spacer]) and segment 6 (NA37 [5′-1–18:1390–1409-3′], NA47 [5′-1–25:1387–1409-3′], and NA76 [5′-1–37:1370–1409-3′]). Likewise, mutations to the PB2-coding sequence and vRNA promoter were introduced by PCR mutagenesis. For production of vRNA templates in avian cells, cDNA corresponding to viral segment 6 was cloned into pPRC249 (a kind gift from Nadia Naffakh, Pasteur Institute, Paris, France) (24), and mutagenesis was performed as described above.
Cell-based RNA replication assays.
HEK-293T or DF1 cells were transfected, as described previously (22), with plasmids encoding PA and PB1, as well as PB2 627K/E and NP where relevant, and a plasmid expressing a vRNA template. Cells were harvested 48 h after transfection, and the isolated RNA was analyzed by primer extension (22). Primers for detection of full-length viral segments 5 and 6, as well as short vRNA templates derived from segment 5, have been described previously (22, 25). To detect replication and transcription of segment 6-derived short vRNA templates, primers NA37+ (5′ GAGTTTTTAAACTCCTGCTTTCGC 3′) and NA37− (5′ GCGAAAGCAGGAGTTTAAAAACTC 3′) were used to detect mRNA and vRNA produced from the 37-nt template, while primers NA+ (5′ CATTTAAACTCCTGCTTTCGC 3′) and NA− (5′ GTTCAAAAAACTCCTTGTTTC 3′) were used for the 47- and 76-nt templates. Similarly, primers NP37+ (5′ GGTACTAGTCTACCCTGCTTTTGC 3′) and NP37− (5′ GCAAAAGCAGGGTAGACTAGTACC 3′) were used to detect mRNA and vRNA produced from the segment 5-derived 37-nt template. Quantification of viral RNA signals was performed by phosphorimage analysis using AIDA software (Raytest) and 5S rRNA as an internal control. The level of vRNA was used as a measure of viral polymerase replicative efficiency, as the cRNA signal was considered too weak to accurately quantify.
Recombinant polymerase transcription assay.
Recombinant viral polymerase was purified by IgG Sepharose chromatography from HEK-293T cells that had been cotransfected with plasmids expressing PA, PB1-TAP, and PB2 627K or 627E (26). Purified polymerase was analyzed by SDS-PAGE, and PB2 levels were assessed using AIDA software (Raytest). The purified polymerase was used in ApG or β-globin mRNA-primed in vitro transcription assays, as described previously (26, 27). Transcription products were analyzed by 20% PAGE containing 7 M urea in Tris-borate-EDTA (TBE) buffer and quantified by phosphorimage analysis using AIDA software (Raytest).
RESULTS AND DISCUSSION
In order to address the role of adaptive mutations associated with PB2 627, plasmids expressing PB2 genes derived from A/WSN/1933 (H1N1) with either 627K or 627E were transfected into human (HEK-293T) or avian (DF1) cells, along with plasmids encoding PA, PB1, and NP and expressing a segment 6 vRNA template. Transcription and replication of the vRNA template by reconstituted vRNPs were measured 48 h after transfection by primer extension analysis of steady-state viral RNA levels. In agreement with previous studies (17, 28), the PB2 627 K→E mutation resulted in a dramatic decrease in RNA replication and transcription by the viral polymerase in human cells but showed no effect when vRNP reconstitutions were conducted in avian cells (Fig. 1A). To determine whether the reduction in viral polymerase activity in human cells was due to a defect in viral polymerase assembly, polymerases containing either PB2 627K or PB2 627E were purified from HEK-293T cells using a TAP tag on the PB1 polymerase subunit (26). SDS-PAGE analysis of the purified polymerases indicated that all three viral polymerase subunits were expressed efficiently in human cells and copurified as a trimeric complex irrespective of the identity of PB2 627 (Fig. 1B). To determine if the purified viral polymerases were able to function outside the cellular context, both polymerases were incubated for 1 h at 37°C with a short double-stranded viral promoter and NTPs, including [α-32P]GTP. The PB2 627E viral polymerase, which exhibited restricted activity in the vRNP reconstitution assay, was able to catalyze ApG-primed and cap-dependent transcription as efficiently as the mammalian-adapted PB2 627K polymerase (Fig. 1C), in agreement with previous studies (29).
FIG 1.
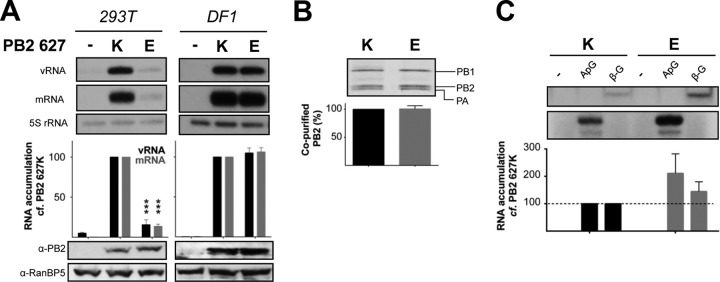
Effects of PB2 K627E mutation on polymerase assembly and activity in cultured cells and in vitro. (A) HEK-293T (human) or DF1 (chicken) cells were cotransfected with plasmids expressing PA, PB1, PB2 627K or 627E, NP, and a segment 6 vRNA. Accumulation of mRNA and vRNA was assessed by primer extension 48 h posttransfection (22). The RNA levels detected for the E627K polymerase were set to 100. The error bars indicate standard errors of the mean (SEM), and the asterisks indicate a significant difference from 100 (one-sample t test; ***, P < 0.001). Expression of PB2 was analyzed by Western blotting, with RanBP5 serving as a loading control. (B) HEK-293T cells were cotransfected with plasmids expressing PA, PB1-TAP, and PB2 627K or 627E, and polymerase was purified using IgG Sepharose chromatography 48 h posttransfection (26). The purified polymerase was analyzed by SDS-PAGE, and PB2 levels were assessed by using AIDA software (Raytest). PB2 627K levels were set to 100. The error bar indicates SEM of three experiments. (C) The activity of the purified polymerase from panel B was assessed in an in vitro transcription assay using ApG primer or β-globin mRNA as a cap donor (26, 27). The RNA levels detected for the E627K polymerase were set to 100. The error bars indicate SEM of three experiments.
In contrast to the vRNP reconstitution assay, in vitro transcription of a short vRNA promoter does not require the formation of a canonical vRNP or, indeed, the presence of NP. In agreement with this, reduced interaction between PB2 and NP has been proposed to account for the decreased activity associated with avian viral polymerases in mammalian cells (17, 19–21). To investigate the contribution of the interaction between PB2 and NP to the restricted phenotype of the PB2 627E polymerase, we made use of a recently developed NP-independent cell-based replication assay (22, 30). This system takes advantage of the observation that viral RNA transcription and replication can take place in the absence of NP on templates up to 76 nucleotides in length. Accordingly, replication assays were conducted in the presence and absence of NP on vRNA templates between 37 and 101 nucleotides in length (Fig. 2A and B, respectively). Omission of NP did not alter the accumulation of RNA products from templates that do not require NP for replication and transcription (37- to 76-nt templates) (compare Fig. 2A and B). This indicates that PB2 627E restricts polymerase activity in a manner that is independent of NP and consequently independent of canonical RNP formation. This is in agreement with the recent suggestion that the previously reported decrease in the affinity of PB2 627E for NP stems from a decrease in viral RNA replication (31). Surprisingly, on the shortest template assayed (37 nt), transcription and replication by the PB2 627E polymerase approach levels observed for the mammalian-adapted PB2 627K polymerase (Fig. 2). However, the level of RNA transcription and replication by the PB2 627E polymerase on longer templates (47 to 101 nt long) appears to decrease in a manner that is proportional to the template length (Fig. 2). The experiment was repeated using segment 6-derived short vRNA templates, producing comparable results (Fig. 3A). As expected, replication and transcription of the 37-nt vRNA template by PB2 627K and 627E polymerases were comparable in avian cells (Fig. 3B).
FIG 2.
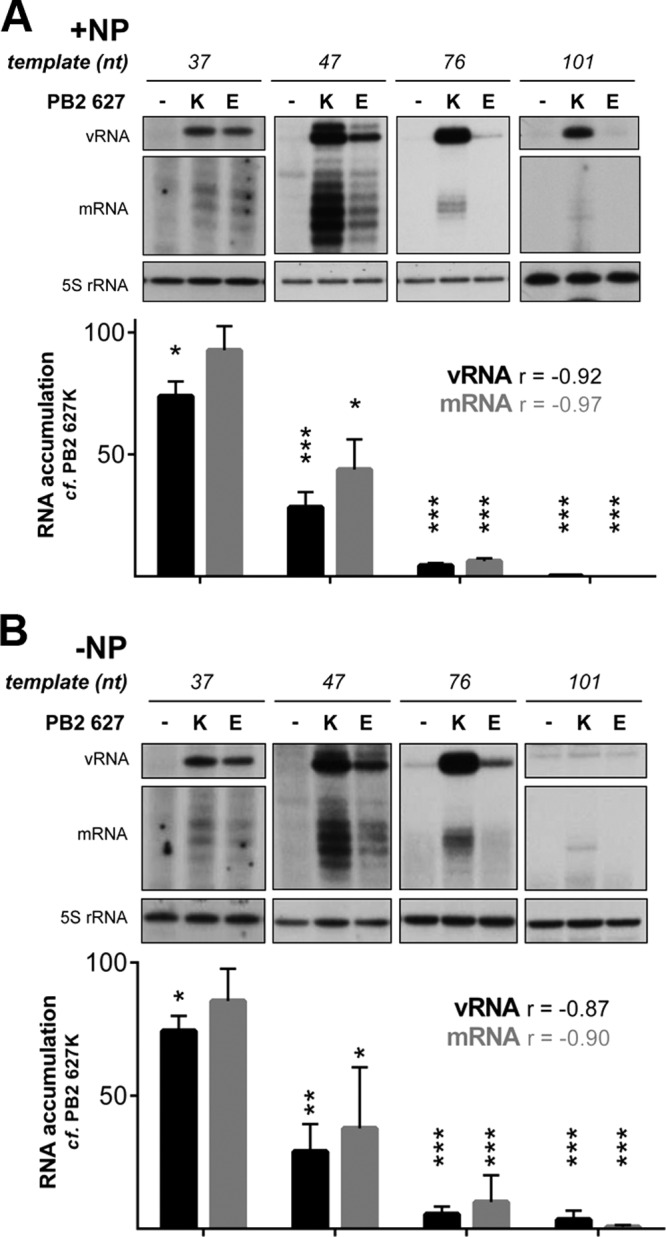
Transcription and replication of short vRNA templates in the presence or absence of NP by influenza virus RNA polymerase containing PB2 627K or 627E in a cell-based replication assay. HEK-293T cells were cotransfected with plasmids expressing short segment 5-based vRNA-like templates (22) of the indicated lengths, PA, PB1, PB2 627K or 627E, and a plasmid expressing NP (A) or empty vector (B) (22). Accumulation of vRNA and mRNA was assessed by primer extension 48 h posttransfection (22). The level of vRNA template produced by cellular transcription was determined by the omission of the PB2 subunit of the viral polymerase and set to zero. The RNA levels detected for the 627K polymerase were set to 100 for each template length. The error bars indicate SEM of three experiments, and the asterisks indicate a significant difference from 100 (one-sample t test; *, P < 0.05; **, P < 0.01; ***, P < 0.001). Pearson's correlation coefficient was calculated for the PB2 627E polymerase activities, producing the r values shown.
FIG 3.
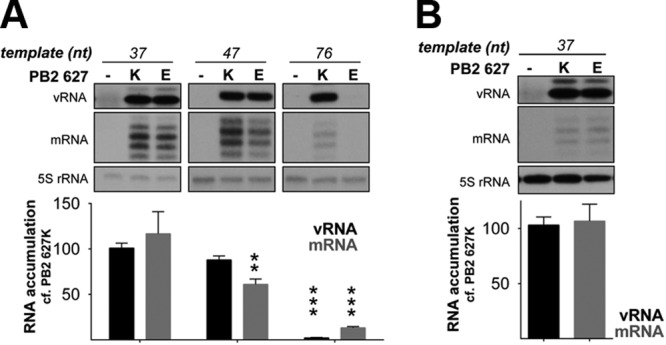
Transcription and replication of short NA segment vRNA templates by PB2 627K or 627E in human and avian cells. HEK-293T (A) or DF1 (B) cells were cotransfected with plasmids expressing short segment 6-based vRNA-like templates of the indicated lengths, as well as PA, PB1, and PB2 627K or 627E. Accumulation of vRNA and mRNA was assessed by primer extension 48 h posttransfection. The level of vRNA template produced by cellular transcription was determined by the omission of the PB2 subunit of the viral polymerase and set to zero. The RNA levels detected for the 627K polymerase were set to 100 for each template length. The error bars indicate SEM of three experiments, and the asterisks indicate a significant difference from 100 (one-sample t test; **, P < 0.01; ***, P < 0.001).
The intriguing finding that a restricted polymerase with PB2 627E is active on short vRNA templates in the context of a cell suggests that the restriction could occur at the level of template binding or elongation. In fact, the template or, more specifically, promoter stability has been implicated previously in the mechanism of restriction of avian influenza virus polymerases in mammalian cells (32). In order to test the effect of promoter stability on PB2 627E polymerase activity, a range of mutations were introduced into the promoter of a full-length noncoding segment 5 template. The mutations were designed not to disrupt intrastrand base pairing between nucleotides proposed to interact in the corkscrew model (2), but rather, to stabilize the formation of a panhandle structure. Compared to the activity of a vRNA template with a wild-type promoter, mutations in the 3′ promoter strand that are proposed to stabilize the panhandle structure led to increased replication and transcription by the PB2 627E polymerase (Fig. 4, i). Replication and transcription by a PB2 627E polymerase returned to the level observed for the wild-type promoter when the nucleotides stabilizing the panhandle conformation were substituted for nucleotides not capable of pairing with the opposing 5′ promoter strand (Fig. 4, ii). In contrast, panhandle-stabilizing mutations introduced into the 5′ strand of the vRNA promoter resulted in restricted activity of the PB2 627E polymerase (Fig. 4, iii). Surprisingly, this template could be replicated by the PB2 627K polymerase but could not be transcribed. However, destabilization of the panhandle by introducing mutations into the 3′ promoter strand resulted in increased transcription and replication by both PB2 627K and 627E polymerases relative to the wild-type promoter (Fig. 4, iv). When stimulatory mutations were introduced into the 3′ promoter strand of a segment 6 vRNA template, a comparable increase in PB2 627E polymerase activity was observed, confirming that this effect is not segment specific (Fig. 5A). In addition, viral polymerases containing PB2 627K and 627E retained similar activities when an identical experiment was conducted in avian cells (Fig. 5B).
FIG 4.
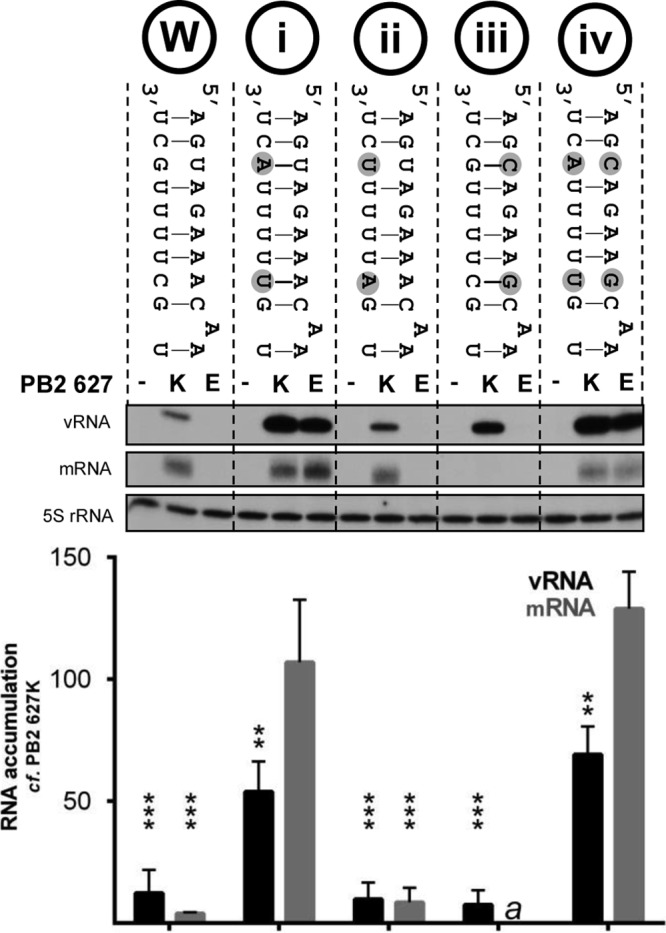
Effects of vRNA promoter mutations on transcription and replication by viral polymerase with PB2 627K or 627E. Shown is a schematic representation of wild-type (W) and mutant (i to iv) influenza virus vRNA promoter structures according to the panhandle model. Mutated residues are indicated by circles. HEK-293T cells were cotransfected with plasmids expressing PA, PB1, PB2 627K or 627E, NP, and segment 5 vRNA with the indicated promoter mutations and mutations to prevent expression of NP (22). Accumulation of mRNA and vRNA was assessed by primer extension 48 h posttransfection (22). The level of vRNA template produced by cellular transcription was determined by the omission of the PB2 subunit of the viral polymerase and set to zero. The RNA levels detected for the 627K polymerase were set to 100. The error bars indicate SEM of three experiments, and the asterisks indicate a significant difference from 100 (one-sample t test; ***, P < 0.001). a, no mRNA signal was detected above background levels for either PB2 627K or PB2 627E polymerases when template iii was used.
FIG 5.
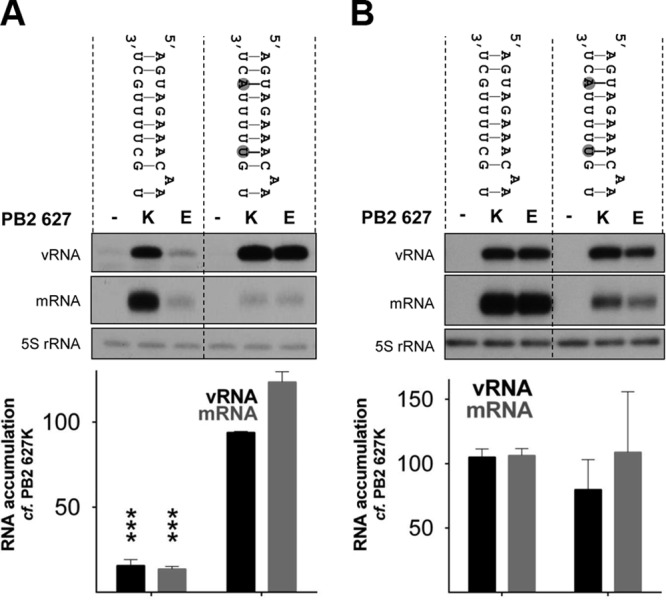
Effects of vRNA promoter mutations on PB2 627K or 627E polymerase activity in human and avian cells. HEK-293T (A) or DF1 (B) cells were cotransfected with plasmids expressing full-length segment 6 containing the indicated mutations, as well as PA, PB1, and PB2 627K or 627E. Accumulation of vRNA and mRNA was assessed by primer extension 48 h posttransfection. The level of vRNA template produced by cellular transcription was determined by the omission of the PB2 subunit of the viral polymerase and set to zero. The RNA levels detected for the 627K polymerase were set to 100 for each template length. The error bars indicate SEM of three experiments, and the asterisks indicate a significant difference from 100 (one-sample t test; ***, P < 0.001).
Cumulatively, our data suggest that stimulation of PB2 627E polymerase activity by mutations in the vRNA promoter (Fig. 4, i and iv) is not dependent on the stability of the promoter conformation but is specific to the sequence of the 3′ promoter strand. We speculate that a host factor specific for the 3′-end promoter sequence might be responsible for inhibiting the activity of a polymerase with PB2 627E. However, this restriction can be overcome by mutations in the 3′ end or by shortening the template, thereby possibly precluding the binding of cellular factors and allowing the viral promoter to be more efficiently recognized by a PB2 627E viral polymerase (and, indeed, by the PB2 627K polymerase [Fig. 4, i and iv]). In contrast, a PB2 627K polymerase might efficiently compete with such a host factor for promoter binding, as suggested by the increased RNA-binding affinity of an isolated 627K domain compared to 627E (33). Hence, our data suggest that the C terminus of PB2 might modulate viral promoter binding, in agreement with previous observations that PB2 can be cross-linked to promoter RNA (3, 34, 35). Moreover, our data show that PB2 627E polymerase is not restricted at the level of elongation, as full-length templates with mutated 3′ ends can be efficiently replicated and transcribed.
Infection of humans by avian influenza viruses requires that the viral polymerase overcome restricted activity in human cells. However, the molecular determinants of avian viral polymerase restriction remain elusive. Using a cell-based replication and transcription assay that does not rely on the formation of canonical RNPs, we show here that restriction of PB2 627E polymerase occurs independently of NP. In addition, we observed that a viral polymerase containing PB2 627E shows unrestricted activity in human cells when provided with a 37-nt vRNA template containing a wild-type promoter. This activity, however, declined steeply as the template length increased. Finally, we observed that mutations to the 3′ strand of the vRNA promoter were able to increase the activity of a PB2 627E polymerase. These observations indicate that restriction of avian viral polymerases is likely to occur at the level of promoter binding by the viral polymerase.
ACKNOWLEDGMENTS
We thank Nadia Naffakh for providing the pPRC249 plasmid.
D.P. is supported by a Polonsky Foundation Scholarship and Lincoln College; A.J.W.T.V. is supported by Netherlands Organization for Scientific Research (NWO) grant 825.11.029, Wellcome Trust grant 098721/Z/12/Z, and a Kemp postdoctoral fellowship from Lincoln College; F.T.V. is supported by Medical Research Council (MRC) grant G1100138; and E.F. is supported by MRC grants G0700848 and MR/K000241/1.
Footnotes
Published ahead of print 23 October 2013
REFERENCES
- 1.Sorrell E, Schrauwen E, Linster M, De Graaf M, Herfst S, Fouchier R. 2011. Predicting “airborne” influenza viruses: (trans-) mission impossible? Curr. Opin. Virol. 1:635–642. 10.1016/j.coviro.2011.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flick R, Neumann G, Hoffmann E, Neumeier E, Hobom G. 1996. Promoter elements in the influenza vRNA terminal structure. RNA 2:1046–1057 [PMC free article] [PubMed] [Google Scholar]
- 3.Fodor E, Pritlove DC, Brownlee GG. 1994. The influenza virus panhandle is involved in the initiation of transcription. J. Virol. 68:4092–4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fodor E. 2013. The RNA polymerase of influenza A virus: mechanisms of viral transcription and replication. Acta Virol. 57:113–122. 10.4149/av_2013_02_113 [DOI] [PubMed] [Google Scholar]
- 5.Krug R, Fodor E. 2013. The virus genome and its replication, p 57–66 In Webster RG, Monto AS, Braciale TJ, Lamb RA. (ed), Textbook of influenza, 2nd ed. John Wiley & Sons, Ltd., New York, NY [Google Scholar]
- 6.Resa-Infante P, Jorba N, Coloma R, Ortin J. 2011. The influenza virus RNA synthesis machine: advances in its structure and function. RNA Biol. 8:207–215. 10.4161/rna.8.2.14513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Subbarao EK, London W, Murphy BR. 1993. A single amino acid in the PB2 gene of influenza A virus is a determinant of host range. J. Virol. 67:1761–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen G-W, Chang S-C, Mok C-K, Lo Y-L, Kung Y-N, Huang J-H, Shih Y-H, Wang J-Y, Chiang C, Chen C-J, Shih S-R. 2006. Genomic signatures of human versus avian influenza A viruses. Emerg. Infect. Dis. 12:1353–1360. 10.3201/eid1209.060276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mehle A, Doudna JA. 2009. Adaptive strategies of the influenza virus polymerase for replication in humans. Proc. Natl. Acad. Sci. U. S. A. 106:21312–21316. 10.1073/pnas.0911915106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hatta M, Gao P, Halfmann P, Kawaoka Y. 2001. Molecular basis for high virulence of Hong Kong H5N1 influenza A viruses. Science 293:1840–1842. 10.1126/science.1062882 [DOI] [PubMed] [Google Scholar]
- 11.Salomon R, Franks J, Govorkova EA, Ilyushina NA, Yen H-L, Hulse-Post DJ, Humberd J, Trichet M, Rehg JE, Webby RJ, Webster RG, Hoffmann E. 2006. The polymerase complex genes contribute to the high virulence of the human H5N1 influenza virus isolate A/Vietnam/1203/04. J. Exp. Med. 203:689–697. 10.1084/jem.20051938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steel J, Lowen AC, Mubareka S, Palese P. 2009. Transmission of influenza virus in a mammalian host is increased by PB2 amino acids 627K or 627E/701N. PLoS Pathog. 5:e1000252. 10.1371/journal.ppat.1000252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herfst S, Schrauwen EJA, Linster M, Chutinimitkul S, de Wit E, Munster VJ, Sorrell EM, Bestebroer TM, Burke DF, Smith DJ, Rimmelzwaan GF, Osterhaus ADME, Fouchier RAM. 2012. Airborne transmission of influenza A/H5N1 virus between ferrets. Science 336:1534–1541. 10.1126/science.1213362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fouchier RAM, Schneeberger PM, Rozendaal FW, Broekman JM, Kemink SAG, Munster V, Kuiken T, Rimmelzwaan GF, Schutten M, van Doornum GJJ, Koch G, Bosman A, Koopmans M, Osterhaus ADME. 2004. Avian influenza A virus (H7N7) associated with human conjunctivitis and a fatal case of acute respiratory distress syndrome. Proc. Natl. Acad. Sci. U. S. A. 101:1356–1361. 10.1073/pnas.0308352100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao P, Watanabe S, Ito T, Goto H, Wells K, McGregor M, Cooley AJ, Kawaoka Y. 1999. Biological heterogeneity, including systemic replication in mice, of H5N1 influenza A virus isolates from humans in Hong Kong. J. Virol. 73:3184–3189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kageyama T, Fujisaki S, Takashita E, Xu H, Yamada S, Uchida Y, Neumann G, Saito T, Kawaoka Y, Tashiro M. 2013. Genetic analysis of novel avian A(H7N9) influenza viruses isolated from patients in China, February to April 2013. Euro Surveill. 18(15):pii=20453 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20453 [PMC free article] [PubMed] [Google Scholar]
- 17.Mehle A, Doudna JA. 2008. An inhibitory activity in human cells restricts the function of an avian-like influenza virus polymerase. Cell Host Microbe 4:111–122. 10.1016/j.chom.2008.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moncorgé O, Mura M, Barclay WS. 2010. Evidence for avian and human host cell factors that affect the activity of influenza virus polymerase. J. Virol. 84:9978–9986. 10.1128/JVI.01134-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Labadie K, Dos Santos Afonso E, Rameix-Welti M-A, van der Werf S, Naffakh N. 2007. Host-range determinants on the PB2 protein of influenza A viruses control the interaction between the viral polymerase and nucleoprotein in human cells. Virology 362:271–282. 10.1016/j.virol.2006.12.027 [DOI] [PubMed] [Google Scholar]
- 20.Ng AK-L, Chan W-H, Choi S-T, Lam MK-H, Lau K-F, Chan PK-S, Au SW-N, Fodor E, Shaw P-C. 2012. Influenza polymerase activity correlates with the strength of interaction between nucleoprotein and PB2 through the host-specific residue K/E627. PLoS One 7:e36415. 10.1371/journal.pone.0036415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rameix-Welti M-A, Tomoiu A, Afonso EDS, van der Werf S, Naffakh N. 2009. Avian influenza A virus polymerase association with nucleoprotein, but not polymerase assembly, is impaired in human cells during the course of infection. J. Virol. 83:1320–1331. 10.1128/JVI.00977-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turrell L, Lyall JW, Tiley LS, Fodor E, Vreede FT. 2013. The role and assembly mechanism of nucleoprotein in influenza A virus ribonucleoprotein complexes. Nat. Commun. 4:1591. 10.1038/ncomms2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fodor E, Crow M, Mingay LJ, Deng T, Sharps J, Fechter P, Brownlee GG. 2002. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J. Virol. 76:8989–9001. 10.1128/JVI.76.18.8989-9001.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Massin P, Rodrigues P, Marasescu M, van der Werf S, Naffakh N. 2005. Cloning of the chicken RNA polymerase I promoter and use for reverse genetics of influenza A viruses in avian cells. J. Virol. 79:13811–13816. 10.1128/JVI.79.21.13811-13816.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robb NC, Smith M, Vreede FT, Fodor E. 2009. NS2/NEP protein regulates transcription and replication of the influenza virus RNA genome. J. Gen. Virol. 90:1398–1407. 10.1099/vir.0.009639-0 [DOI] [PubMed] [Google Scholar]
- 26.Deng T, Sharps J, Fodor E, Brownlee GG. 2005. In vitro assembly of PB2 with a PB1-PA dimer supports a new model of assembly of influenza A virus polymerase subunits into a functional trimeric complex. J. Virol. 79:8669–8674. 10.1128/JVI.79.13.8669-8674.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hara K, Schmidt FI, Crow M, Brownlee GG. 2006. Amino acid residues in the N-terminal region of the PA subunit of influenza A virus RNA polymerase play a critical role in protein stability, endonuclease activity, cap binding, and virion RNA promoter binding. J. Virol. 80:7789–7798. 10.1128/JVI.00600-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Massin P, van der Werf S, Naffakh N. 2001. Residue 627 of PB2 is a determinant of cold sensitivity in RNA replication of avian influenza viruses. J. Virol. 75:5398–5404. 10.1128/JVI.75.11.5398-5404.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aggarwal S, Dewhurst S, Takimoto T, Kim B. 2011. Biochemical impact of the host adaptation-associated PB2 E627K mutation on the temperature-dependent RNA synthesis kinetics of influenza A virus polymerase complex. J. Biol. Chem. 286:34504–34513. 10.1074/jbc.M111.262048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Resa-Infante P, Recuero-Checa MÁ, Zamarreño N, Llorca Ó, Ortín J. 2010. Structural and functional characterization of an influenza virus RNA polymerase-genomic RNA complex. J. Virol. 84:10477–10487. 10.1128/JVI.01115-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cauldwell AV, Moncorgé O, Barclay WS. 2013. Unstable polymerase-NP interaction is not responsible for avian influenza polymerase restriction in human cells. J. Virol. 87:1278–1284. 10.1128/JVI.02597-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crescenzo-Chaigne B, van der Werf S, Naffakh N. 2002. Differential effect of nucleotide substitutions in the 3′ arm of the influenza A virus vRNA promoter on transcription/replication by avian and human polymerase complexes is related to the nature of PB2 amino acid 627. Virology 303:240–252. 10.1006/viro.2002.1637 [DOI] [PubMed] [Google Scholar]
- 33.Kuzuhara T, Kise D, Yoshida H, Horita T, Murazaki Y, Nishimura A, Echigo N, Utsunomiya H, Tsuge H. 2009. Structural basis of the influenza A virus RNA polymerase PB2 RNA-binding domain containing the pathogenicity-determinant lysine 627 residue. J. Biol. Chem. 284:6855–6860. 10.1074/jbc.C800224200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fodor E, Seong BL, Brownlee GG. 1993. Photochemical cross-linking of influenza A polymerase to its virion RNA promoter defines a polymerase binding site at residues 9 to 12 of the promoter. J. Gen. Virol. 74:1327–1333. 10.1099/0022-1317-74-7-1327 [DOI] [PubMed] [Google Scholar]
- 35.Pritlove DC, Fodor E, Seong BL, Brownlee GG. 1995. In vitro transcription and polymerase binding studies of the termini of influenza A virus cRNA: evidence for a cRNA panhandle. J. Gen. Virol. 76:2205–2213. 10.1099/0022-1317-76-9-2205 [DOI] [PubMed] [Google Scholar]