

Abstract
CspA of the Lyme disease spirochete Borrelia burgdorferi represents a key molecule in immune evasion, protecting borrelial cells from complement-mediated killing. As previous studies focused almost exclusively on CspA of B. burgdorferi, here we investigate the different binding capacities of CspA orthologs of Borrelia burgdorferi, B. afzelii, and B. spielmanii for complement regulator factor H and plasminogen and their ability to inhibit complement activation by either binding these host-derived plasma proteins or independently by direct interaction with components involved in formation of the lethal, pore-like terminal complement complex. To further examine their function in serum resistance in vivo, a serum-sensitive B. garinii strain was used to generate spirochetes, ectopically producing functional CspA orthologs. Irrespective of their species origin, all three CspA orthologs impart resistance to complement-mediated killing when produced in a serum-sensitive B. garinii surrogate strain. To analyze the inhibitory effect on complement activation and to assess the potential to inactivate C3b by binding of factor H and plasminogen, recombinant CspA orthologs were also investigated. All three CspA orthologs simultaneously bound factor H and plasminogen but differed in regard to their capacity to inactivate C3b via bound plasmin(ogen) and inhibit formation of the terminal complement complex. CspA of B. afzelii binds plasmin(ogen) and inhibits the terminal complement complex more efficiently than CspA of B. burgdorferi and B. spielmanii. Taken together, CspA orthologs of serum-resistant Lyme disease spirochetes act as multifunctional evasion molecules that inhibit complement on two central activation levels, C3b generation and assembly of the terminal complement complex.
INTRODUCTION
Lyme disease is the most commonly reported vector-borne disease in the United States and Europe. This multisystemic inflammatory disorder is caused by a variety of spirochetes belonging to the Borrelia burgdorferi sensu lato group (1). If left untreated, the infection may progress to more severe clinical manifestations, including neurological abnormalities, chronic arthritis, skin lesions, and cardiac complications (2).
In Europe, 11 distinct species have been detected so far, of which B. burgdorferi, B. afzelii, B. garinii, B. bavariensis, B. spielmanii, B. valaisiana, B. lusitaniae, B. bissettii, and B. finlandensis are endemic, and B. afzelii as well as B. garinii appear to be the dominant species (3, 4). The exclusive host-pathogen relationship of the dormice-associated B. spielmanii spirochetes, together with the specific adaptation of their reservoir host(s), results in a geographical distribution of this particular species that is more restricted than that of any other human-pathogenic Lyme disease spirochete (5). Of note, B. afzelii and B. spielmanii have frequently been detected in skin biopsy specimens obtained from patients with erythema migrans and acrodermatitis chronica atrophicans (6–10).
The ability of Lyme disease spirochetes to survive in diverse host environments necessitates strategies to overcome both adaptive and innate immune responses, in particular complement. Resistance of pathogenic B. burgdorferi, B. afzelii, and B. spielmanii to complement-mediated killing is accomplished, at least in part, by acquisition of diverse complement regulators of the factor H protein family via bacterial surface molecules of three distinct and genetically unrelated groups, collectively termed complement regulator-acquiring surface proteins (CRASPs) (11–16). Depending on the genetic composition, individual Borrelia species can produce up to five different binding proteins; hence, various combinations of these molecules exist on the borrelial outer surface. CRASPs differ not only in their binding capabilities for distinct complement regulators and other serum proteins but also in their protein sequences and structures, gene locations, gene regulatory mechanisms, and expression patterns during the tick-mammal infection cycle (11, 12, 17–30).
The complement system constitutes a first line of defense of the human innate immune system that collaboratively eliminates invading microorganisms through immediate activation of proteolytic-acting plasma proteins (31). Activation of complement via the alternative (AP), lectin (LP), and classical pathway (CP) converges in the formation and deposition of C3b, a key component of the complement cascade. C3b deposition on microbial cell surfaces leads to opsonization and marks the cell for phagocytosis. If activation progresses, the C5 convertase is generated and initiates the terminal pathway (TP) by cleavage of C5 into C5a and C5b. Once C5b attaches to the microbial surface, assembly of the terminal complement complex (TCC) takes place by subsequent binding of C6, C7, C8, and several molecules of C9, resulting in lysis of the invading pathogens. Normally, activation of complement effector molecules is tightly controlled on the surface of intact host cells by a number of fluid-phase and surface-attached regulators. The concerted action of those regulators ensures cell and tissue integrity (32–34). The 150-kDa factor H (CFH) protein is a key member of this fine-tuned regulatory network and plays a pivotal role in modulating, controlling, and regulating activation of the AP by acting as cofactor for factor I-mediated degradation of C3b, and it supports the dissociation (decay-accelerating activity) of the C3 convertase, C3bBb (33–35).
Dissemination of spirochetes involves interaction with host-derived plasminogen, which, upon activation to plasmin, degrades components of the host extracellular matrix, allowing the bacteria to traverse tissue barriers (36–39). Recently, it has been shown that plasmin also degrades the central complement component C3b, leading to inhibition of all three pathways of complement. Furthermore, the proteolytically inactive proenzyme plasminogen also enhances complement factor I-mediated inactivation of C3b in the presence of CFH (40).
CspA (also referred to as complement regulator-acquiring surface protein 1) is the key CFH-binding protein of B. burgdorferi (29) and is produced during both tick-to-mammal and mammal-to-tick transmission stages (17, 23). Mutants lacking CspA are highly susceptible to complement-mediated killing, and complementation with the cspA gene of B. burgdorferi completely restores serum resistance (41, 42). In addition to the aspects already addressed above, CspA interacts with late complement components and thereby inhibits assembly of the pore-forming terminal complement complex (43). CspA has also been identified as a ligand for plasminogen, suggesting a dual role of CspA in immune evasion of B. burgdorferi (30).
While previous studies focused almost exclusively on CspA of B. burgdorferi, this work aims to include B. afzelii and B. spielmanii, the species more prevalent in Eurasia. In the present study, we sought to (i) investigate the impact of CspA orthologs of serum-resistant B. afzelii and B. spielmanii on serum resistance by generating CspA-producing spirochetes, (ii) examine whether CspA directly interacts with complement, (iii) analyze the ability of CspA orthologs to bind plasminogen, (iv) characterize the nature of the CspA-plasminogen interaction, and (v) elucidate whether CspA-bound plasmin(ogen) contributes to inactivation of central complement component C3b.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
B. burgdorferi LW2 (skin isolate; Germany), B. afzelii PKo (skin isolate, Germany), B. spielmanii A14S (skin isolate; The Netherlands), and B. garinii G1 (CSF isolate; Germany) as well as transformants G1/pKFSS1, G1/pCspA LW2, G1/pCspA PKo, and G1/pCspA A14S were grown at 33°C in Barbour-Stoenner-Kelly (BSK) medium (Bio & Sell, Feucht, Germany) or BSK supplemented with streptomycin as described previously (14). Escherichia coli JM109 and DH5α were grown at 37°C in yeast tryptone broth supplemented with the appropriate antibiotics.
Human sera, antibodies, and other reagents.
Nonimmune human serum (NHS) obtained from healthy blood donors was used as a source of CFH. The study and the respective consent documents were approved by the ethics committee at the Johann Wolfgang Goethe-University of Frankfurt (control number 160/10). All healthy blood donors provided written informed consent.
Complement components as well as polyclonal antibodies for detection of C5b-6, C7, C8, and C9 were purchased from Complement Technology (Tyler, TX). Vitronectin was purchased from Sigma-Aldrich (Taufkirchen, Germany). Polyclonal antisera used to detect CFH, C3, and C6 were obtained from Calbiochem. The monoclonal antibody IXF9 was used for detection of CFH (44). A monoclonal anti-C5b-9 antibody was purchased from Quidel (San Diego, CA), and the polyclonal anti-plasminogen antibody was from Acris Antibodies (Herford, Germany). Rabbit and sheep erythrocytes (RE and SE, respectively) in Alsevers were purchased from Rockland (Gilbertsville, PA), and amboceptor was purchased from Siemens Healthcare Diagnostics (Eschborn, Germany). The generation of MAb L41 1C11 for detection of FlaB is described elsewhere (45).
Generation and purification of CspA orthologs.
Specific primers were used to generate vectors pQE CspA LW2, pQE CspA PKo, and pQE CspA A14S for production of the respective His-tagged CspA orthologs (Table 1). Expression of recombinant proteins and purification by Ni-nitrilotriacetic (NTA) agarose (Qiagen, Hilden, Germany) affinity chromatography have been described previously (14).
TABLE 1.
Oligonucleotides used in this study
| Oligonucleotide | Sequencea (5′–3′) | Use in this work |
|---|---|---|
| Crasp-1-Bam | GCTAAAACTTCTCTTTTTTTTAGGATCCCAACCCAAATCC | Cloning of cspA LW2 in pKFSS1 |
| Crasp-1-Hind | GAAAGAAAAAAAATAAGCTTTTGCACTTGATATTTTTAAAAAG | Cloning of cspA LW2 in pKFSS1 |
| BaCRASP-1(+)Bam | GCCAGTGTGCTGGGATCCGCCCTTCATTGC | Cloning of cspA PKo and cspA A14S in pKFSS1 |
| BaCRASP-1(−)Hind | GATGGATATCTGCAAGCTTCGCCCTTGGT | Cloning of cspA PKo and cspA A14S in pKFSS1 and cloning of cspA PKo in pQE-30Xa |
| BBA68 Bam | ACCGGATCCGCACCTTTTAGC | Cloning of cspA LW2 in pQE-30 Xa |
| BBA68 Eco 2 | TTTCGAATTCTTAGTAAAAGGCAGGTTTTAAAG | Cloning of cspA LW2 in pQE-30 Xa |
| BsCRASP-1 Bam | CTTTAATTTGCATCGGATCCACGCCTATTAATAAC | Cloning of cspA PKo in pQE-30 Xa |
| pGEX(+) | GGGCTGGCAAGCCACGTTTGGTG | Recloning of cspA A14S in pQE-30 Xab |
| pGEX-CSPA Pst(−) | CCGAAACGCGCGCTGCAGATCGTCAGTCAGTCACG | Recloning of cspA A14S in pQE-30 Xab |
| aadA+NdeI | CATATGAGGGAAGCGGTGATC | Amplification of aadA |
| aadR+AatII | GACGTCATTATTTGCCGACTACC | Amplification of aadA |
| Fla6 | AACACACCAGCATCGCTTTCAGGGTCT | Amplification of flaB |
| Fla7 | TATAGATTCAAGTCTATTTTGGAAAGCACCTA | Amplification of flaB |
Sequences of specific restriction endonuclease recognition sites are underlined.
Plasmid pGEXcspA14 (56) was used as the template for recloning of the cspA gene of B. spielmanii A14S into pQE-30 Xa.
Construction of shuttle vectors for ectopic production of CspA orthologs.
Vector pKFSS1 was used for ectopic production of CspA orthologs in B. garinii G1 (46). The respective cspA genes, including their native promoter regions, were amplified from B. burgdorferi LW2, B. afzelii PKo, and B. spielmanii A14S by PCR (Table 1). Amplicons generated were digested with the appropriate restriction endonucleases and cloned into pKFSS1, yielding shuttle vectors pCspA LW2, pCspA PKo, and pCspA A14S. The inserted sequences were subjected to nucleotide sequencing to ensure that no mutations were introduced during PCR and subsequent cloning procedures. In addition, sequence alignments have been performed to verify the identity of the cloned sequences with their genomic counterparts using DNASTAR software (Lasergene, Madison, WI).
Generation of transformed B. garinii G1 strains.
B. garinii strain G1 was grown in BSK medium and harvested at mid-exponential phase (1 × 108 cells/ml). Preparation and transformation of electrocompetent cells as well as the selection of positive clones by microdilution have been described previously (14). Briefly, 50-μl aliquots of competent B. garinii strain G1 cells were electroporated at 12.5 kV/cm in 2-mm cuvettes with 10 μg of pCspA LW2, pCspA PKo, and pCspA A14S, respectively. For control purposes, B. garinii strain G1 cells also were transformed with the empty shuttle vector pKFSS1 (46). Cells were immediately diluted into 10 ml BSK medium and incubated without antibiotic selection at 33°C for 48 to 72 h. Bacteria were then diluted into 100 ml BSK medium containing streptomycin (25 μg/ml), and 200-μl aliquots were plated into 96-well cell culture plates (Corning, Amsterdam, The Netherlands) for selection of transformants. Several clones selected were expanded in 1 ml fresh BSK medium without antibiotic selection for 7 days and then transferred into 10 ml fresh BSK medium containing streptomycin (50 μg/ml).
Characterization of transformed spirochetes.
Transformed spirochetes were characterized by amplification of the cspA, aadA, and flaB genes using primers listed in Table 1. Spirochetes (100 μl) were sedimented by centrifugation, washed with phosphate-buffered saline (PBS), and suspended in 50 μl of water. Five microliters of suspension was amplified by PCR using oligonucleotide primers at final concentrations of 100 nM each plus 200 μM deoxynucleoside triphosphates (dNTPs). PCR was carried out for 25 cycles using the following parameters: denaturation at 94°C for 1 min, annealing at 50°C for 1 min, and extension at 72°C for 1 min. Plasmid DNA was prepared from the presumptive E. coli transformants with a Wizard plus SV miniprep DNA purification system (Promega, Mannheim, Germany), and DNA inserts were sequenced by a commercial provider (GATC, Constance, Germany).
Ectopic expression of CspA orthologs in transformed spirochetes was verified by ligand affinity blotting. To confirm localization of native CspA orthologs on the spirochetal surface, protease accessibility assays using proteinase K (Sigma-Aldrich, Taufkirchen, Germany) were performed as previously described (11, 13, 47).
Enzyme-linked immunosorbent assay.
Microtiter plates were coated with His-tagged CspA orthologs (5 μg/ml each) overnight at 4°C. After blocking, 10 μg/ml of CFH or plasminogen (Hematologic Technologies, Essex Junction, VT) was added and incubated for 1 h at room temperature. Bound proteins were detected with an anti-CFH or anti-plasminogen antiserum, and protein complexes were identified using secondary horseradish peroxidase (HRP)-coupled antisera. The reaction was developed with 1,2-phenylenediamine dihydrochloride (Sigma-Aldrich, Taufkirchen, Germany), and absorbance was measured at 490 nm.
For analyzing interaction of CspA orthologs with complement components, microtiter plates were coated with His-tagged borrelial proteins as described above. After blocking, complement component C5b-6, C7, C8, C9, or TCC (5 μg/ml each) in 100 μl GVB++ was added and incubated for 1 h at room temperature. Bound protein complexes were detected using anti-C5, anti-C7, anti-C8, or anti-C5b-9 antibody (diluted 1:1,000). Following incubation with appropriate secondary antibodies, the reaction was developed as described above.
Serum susceptibility testing of borrelial strains.
Serum susceptibility of wild-type strains, as well as transformed spirochetes, to active human complement was assessed by a serum bactericidal assay (48, 49). Briefly, borrelial cells grown to mid-logarithmic phase were harvested, washed, and resuspended in fresh BSK medium. Spirochetes (1.25 × 107) diluted in a final volume of 100 μl in BSK medium containing 240 μg ml−1 phenol red were incubated with 50% NHS or 50% heat-inactivated NHS (hiNHS) in microtiter plates for 8 days at 33°C (Corning, Amsterdam, The Netherlands). BSK medium instead of NHS was included in all assays as an additional control. Multiplication of spirochetes was monitored by measuring the pH indicator color shift of the medium at 562 and 630 nm using an enzyme-linked immunosorbent assay (ELISA) reader (PowerWave HT; BioTek Instruments, Winooski, VT). For data analysis, Gen5 software, version 1.11.4 (BioTek Instruments), was used. Each experiment was conducted at least three times, and means ± standard deviations (SD) were calculated.
Binding of native CFH to the borrelial surface.
A serum adsorption assay was employed to assess binding of CFH to viable spirochetes (5 × 108 cells). After incubation with EDTA-treated NHS, bound proteins were eluted and the last wash and the eluate fractions were separated by SDS-PAGE under nonreducing conditions. Following Western blotting, CFH was detected with the monoclonal anti-CFH antibody IXF9 as described previously (12, 13).
C3b degradation assay.
The C3b inactivation capacity of spirochetes (4 × 107 cells) was assayed after preincubation with PBS supplemented with 1.25 μg/ml CFH for 1 h at room temperature. After washing, cells were resuspended in 50 μl PBS containing 10 μg/ml C3b and 20 μg/ml factor I and incubated for 1 h at 37°C. Cells were then sedimented and the supernatants were mixed with sample buffer.
To assess C3b degradation by activated plasmin(ogen), 1 μg His-tagged protein was immobilized on microtiter plates at 4°C overnight. Following washing and blocking, 2 μg of plasminogen in 50 mM Tris-HCl (pH 7.5) was added, and plates were incubated at room temperature for 1 h. Where indicated, 50 mM tranexamic acid was added as a negative control. After washing, 5 μg/ml of C3b as well as 0.16 μg/ml uPA in 50 mM Tris-HCl (pH 7.5) were added, and plates were incubated at 37°C for 24 h. Aliquots were taken at the indicated time points, and reactions were stopped by addition of SDS-PAGE sample buffer. The samples were then subjected to SDS-PAGE and transferred to nitrocellulose. C3b degradation products were detected by Western blotting.
Detection of deposited complement components on the borrelial surface.
Deposition of activated complement components C3, C6, and C5b-9 (TCC) on the borrelial surface was detected by immunofluorescence microscopy after incubation of spirochetes with 25% active NHS as previously described (13, 47).
Plasminogen activation assay.
A plasminogen activation assay was performed by coating microtiter plates with 1 μg of CspA orthologs or bovine serum albumin (BSA). Following blocking, 2 μg of plasminogen was added. Where indicated, wells were supplemented with 50 mM tranexamic acid as a negative control. Plates were incubated for 3 h at room temperature, and 96 μl reaction buffer containing 50 mM Tris-HCl (pH 7.5), 300 mM NaCl, 0.003% Triton X-100, and 0.3 mg/ml S-2251 (D-Val-Leu-Lys-p-nitroanilide dihydrochloride) (Sigma-Aldrich) was added. Finally, 4 μl of 2.5 μg/ml uPA (Chemicon) was used to activate plasminogen. Microtiter plates were then incubated at room temperature, and the absorbance at 405 nm was measured every 30 min over a time period of 24 h using an ELISA reader (PowerWave HT; BioTek Instruments, Winooski, VT).
Cell-based hemolytic assays.
Hemolytic assays were performed to examine the inhibitory potential of CspA orthologs on the AP, CP, and TP. Briefly, NHS was preincubated with increasing concentrations of CspA orthologs for 30 min at 37°C. RE (AP) or amboceptor-coated SE (CP) (1 × 107 each) were incubated with NHS or CspA preincubated with NHS for 30 min at 37°C in either Mg-EGTA buffer (AP) or GVB++ buffer (CP/TP). Inhibition of the TP was analyzed by using SE (1.5 × 107 cells) preincubated with C5b-6 (1.5 μg/ml) for 10 min at room temperature. C7 (2 μg/ml), C8 (0.4 μg/ml), and C9 (2 μg/ml) were preincubated with or without increasing concentrations (8 to 42 μg/ml) of CspA orthologs for 5 min at room temperature. After 30 min at 37°C, erythrocytes were sedimented and hemolysis was determined by measuring the absorbance of the supernatant at 414 nm.
Statistical analyses.
Both the unpaired Student's t test and one-way analysis of variance (ANOVA) test with Bonferroni's multiple-comparison posttest were employed for statistical analysis using GraphPad Prism, version 4. Results were deemed statistically significant for the following P values: P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***).
RESULTS
Generation and characterization of transformed spirochetes.
To investigate the role of CspA orthologs of B. burgdorferi, B. afzelii, and B. spielmanii in facilitating complement resistance independently of other borrelial CFH-binding proteins, serum-sensitive B. garinii strain G1 was transformed with the respective shuttle vectors. Characterization of the selected clones revealed the presence of the introduced cspA as well as the streptomycin-encoding aadA resistance genes (Fig. 1A). Additionally, production of CspA orthologs in B. garinii G1 was assessed indirectly by detecting binding of CFH from NHS or purified CFH using ligand affinity blotting. Compared to wild-type B. burgdorferi LW2, B. afzelii PKo, and B. spielmanii A14S, all transformants produced the CspA proteins while the B. garinii G1 wild-type strain (Fig. 1B) or G1/pKFFS1, containing the empty shuttle vector, did not (data not shown). Moreover, protease susceptibility assays confirmed surface localization of CspA orthologs, as all molecules were completely degraded after incubation of viable spirochetes with 100 μg/ml proteinase K (Fig. 1C).
FIG 1.
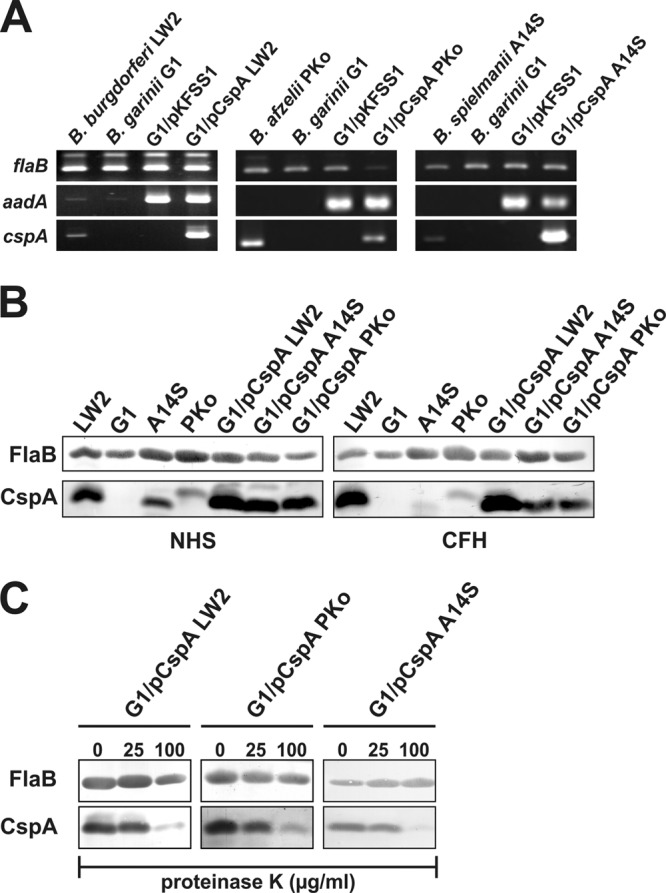
Characterization of B. garinii G1 transformants producing CspA orthologs. (A) B. burgdorferi LW2, B. afzelii PKo, B. spielmanii A14S, B. garinii G1, and transformed strains G1/pKFSS1, G1/pCspA LW2, G1/pCspA PKo, and G1/pCspA A14S were characterized by PCR amplification using flaB-, aadA-, and cspA-specific primers, as listed in Table 1. (B) Production of CspA orthologs by wild-type as well as transformed strains was assessed using ligand affinity blotting. Whole-cell lysates (15 μg each) of B. burgdorferi LW2, B. afzelii PKo, B. spielmanii A14S, and B. garinii G1 as well as transformants G1/pCspA LW2, G1/pCspA PKo, and G1/pCspA A14S were separated by SDS-PAGE and transferred to nitrocellulose. After incubation with NHS (left middle) or purified CFH (right middle), binding of CFH to CspA orthologs was identified using a polyclonal anti-CFH antiserum (dilution, 1/1,000). A monoclonal antibody, L41 1C11, specific for the flagellin protein FlaB, was applied to show equal loading of borrelial lysates. (C) Surface localization of CspA orthologs in transformed borrelial cells. Native spirochetes were incubated with or without proteinase K (25 and 100 μg/ml) and lysed by sonication, and total proteins were separated by SDS-PAGE. CspA orthologs were identified by ligand affinity analysis as described above. Flagellin (FlaB) was detected with MAb L41 1C11 (dilution, 1/1,000) by Western blotting.
CspA orthologs impart resistance to complement-mediated bacteriolysis.
To investigate CspA-mediated serum resistance, the wild-type as well as the transformed strains were incubated in the presence of 50% NHS or 50% hiNHS, and color changes in the medium as a consequence of the multiplication of viable spirochetes were monitored over an incubation period of 8 days. Different levels of serum susceptibility were observed among the various strains (Fig. 2). All CspA-producing transformants as well as wild-type B. burgdorferi LW2, B. afzelii PKo, and B. spielmanii A14S resist complement-mediated bacteriolysis, as demonstrated by a continuous decrease in absorbance due to acidification of the medium. In contrast, growth of B. garinii G1 and the transformant G1/pKFSS1 carrying the empty vector was completely inhibited. When using hiNHS instead of NHS, growth of all borrelial strains remained unaffected (Fig. 2).
FIG 2.

Serum susceptibility testing of transformed B. garinii G1. A growth inhibition assay was used to investigate susceptibility to human serum of B. garinii G1, B. burgdorferi LW2, B. afzelii PKo, B. spielmanii A14S, G1/pCspA PKo, and G1/pCspA A14S. Spirochetes were incubated in either 50% NHS (filled squares) or 50% hiNHS (filled triangles) for a cultivation period of 8 days at 33°C. Color changes were monitored by measurement of the absorbance at 562 and 630 nm. All experiments were performed three times, with each test conducted in triplicate with very similar results. For clarity, only data from one representative experiment are shown. Error bars represent ±SD.
CspA-producing spirochetes exhibit limited deposition of complement components.
Since all CspA-producing transformants resist complement-mediated killing, we next assessed complement activation at different levels of the complement cascade, namely, C3, C6, and C5b-9 (TCC). Spirochetes were incubated in 25% NHS or 25% hiNHS, and deposition of activated complement components on the bacterial surface was analyzed by immunofluorescence microscopy. In agreement with previous studies (13, 16), only a few cells of wild-type B. burgdorferi LW2, B. afzelii PKo, and B. spielmanii A14S stained positive for C3, C6, and C5b-9, while the majority of cells were negative for deposited complement components (see Fig. S1 in the supplemental material). Furthermore, a remarkable reduction of cells positive for C6 and C5b-9 was observed when transformants G1/pCspA LW2, G1/pCspA PKo, and G1/pCspA A14S were assayed under identical conditions, suggesting that CspA orthologs protect spirochetes from complement activation (Fig. 3). In contrast, almost all cells of serum-sensitive G1/pKFSS1 and B. garinii G1 (see Fig. S1 in the supplemental material) stained positive and were covered with complement. As expected, when applying hiNHS instead of NHS, no complement deposition could be detected on the surface of serum-resistant as well as serum-sensitive strains (data not shown).
FIG 3.
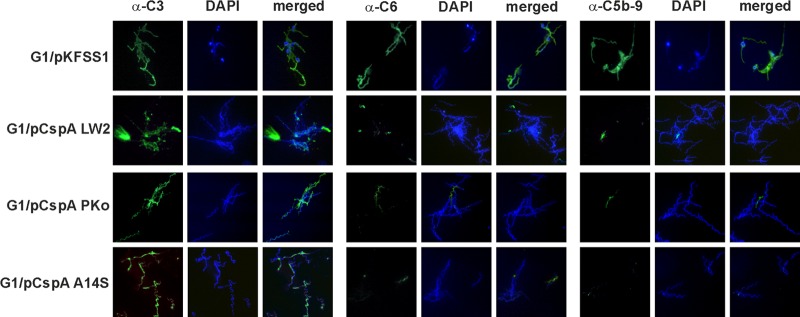
Deposition of complement components C3, C6, and TCC on the surface of spirochetes. Complement components deposited on the surface of transformants G1/pKFSS1, G1/pCspA LW2, G1/pCspA PKo, and G1/pCspA A14S were visualized by indirect immunofluorescence microscopy. Spirochetes (6 × 106) were incubated with 25% NHS for 30 min at 37°C with gentle agitation, and bound C3, C6, and C5b-9 (TCC) were analyzed with specific antibodies against each component and appropriate Alexa Fluor 488-conjugated secondary antibodies. For visualization of the spirochetes in a given microscopic field, the DNA-binding dye 4′,6-diamidino-2-phenylindole (DAPI) was used. The spirochetes were observed at a magnification of ×100. The data were recorded with an Axio Imager M2 fluorescence microscope (Zeiss) equipped with a Spot RT3 camera (Visitron Systems). Each panel is representative of at least 20 microscope fields.
Inactivation of C3b by binding of CFH to the borrelial surface.
To gain further insight into the molecular mechanism of serum resistance mediated by CspA orthologs, we first investigated binding of native CFH to viable spirochetes. As depicted in Fig. 4A, all serum-resistant spirochetes producing CspA orthologs on their surface recruited CFH from human serum, while serum-sensitive G1 and G1/pKFSS1 did not. More importantly, CFH bound to the surface of serum-resistant spirochetes retained its full complement regulatory activity in the presence of factor I, as shown by the appearance of characteristic C3b inactivation products α′ 68 kDa, α′ 43 kDa, and α′ 41 kDa (Fig. 4B). In the absence of CFH, no degradation of C3b could be observed, indicating that spirochetes lack intrinsic proteolytic activity.
FIG 4.

Binding of CFH by B. garinii transformants and functional analysis of cell-bound CFH. (A) Binding of CFH to spirochetes was determined by serum adsorption assays. Wild-type B. burgdorferi LW2, B. garinii G1, B. afzelii PKo, and B. spielmanii A14S as well as transformants G1/pKFSS1, G1/pCspA LW2, G1/pCspA PKo, and G1/pCspA A14S were incubated in NHS-EDTA to prevent complement activation and washed, and then bound proteins were eluted using 0.1 M glycine (pH 2.0). Both the last wash (w) and the eluate (e) fractions obtained from each strain were separated by 12.5% SDS-PAGE and transferred to nitrocellulose. Membranes were probed with monoclonal anti-CFH antibody IXF9 and an HRP-conjugated secondary antiserum. Mobility of the molecular mass standard is shown to the left. (B) Schematic representation of the α- and β-chains of C3b and the cleavage fragments of the α-chain generated by CFH and factor I. (C) Factor I-mediated conversion of C3b to iC3b was analyzed by detection of C3b cleavage fragments after incubation of spirochetes (4 × 107) with purified CFH. Spirochetes were incubated with CFH for 60 min at room temperature. After extensive washing with PBS, C3b (10 ng/ml) and factor I (20 ng/ml) were added, and the mixture was incubated for 30 min at 37°C. For control purposes, all reactions were also performed in the absence of CFH. Subsequently, samples were boiled for 5 min, subjected to 12.5% SDS-PAGE, and transferred onto a nitrocellulose membrane. The various C3b degradation products (α′ 68-, α′ 43-, and α′ 41-kDa bands) were visualized by Western blotting using a polyclonal goat anti-human C3 antiserum. As additional controls, reaction mixtures containing C3b and factor I were incubated with (+) or without (−) purified CFH.
CspA orthologs inhibit activation of the alternative pathway.
Assuming that CspA orthologs also directly influence complement activation, NHS was preincubated with recombinant proteins, and complement regulatory activity was analyzed in a cell-based hemolytic assay. In this system, reduction of erythrocyte lysis directly mirrors the impact of CspA orthologs on AP- and CP-induced complement activation. Investigating AP activation, rabbit erythrocytes were assayed with NHS preincubated with increasing concentrations of CspA orthologs, and the amount of erythrocyte lysis was measured. All three proteins act as potent inhibitors of the AP, as hemolysis was significantly inhibited in a dose-dependent manner (Fig. 5A). When SE were used for analyzing inhibition of CP activation by the CspA orthologs, no effect could be observed at all (Fig. 5B). These findings suggest that all three CspA orthologs mainly affect AP-induced hemolysis.
FIG 5.
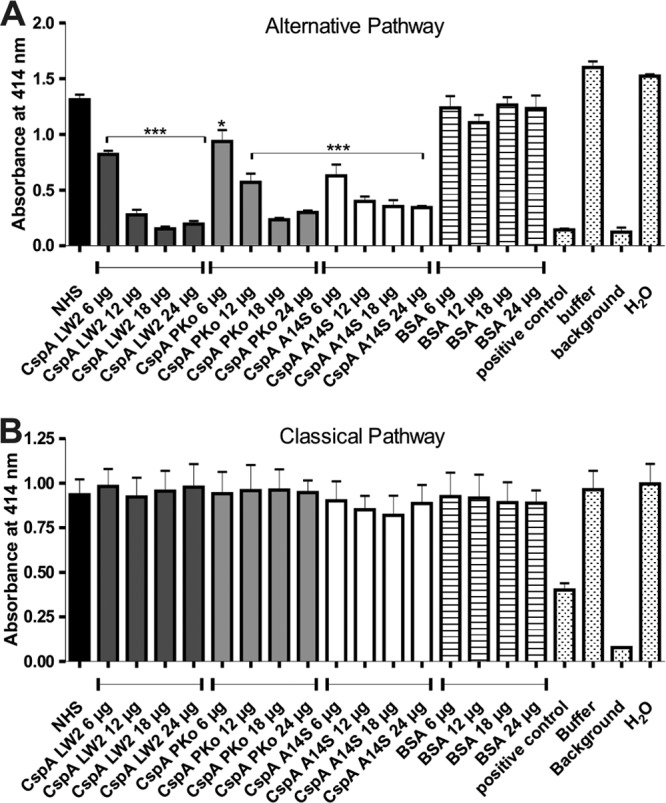
CspA orthologs inhibit TCC deposition induced by the AP and CP. In order to analyze the inhibitory effect of CspA orthologs on the AP (A) and CP (B), a hemolytic assay was employed. RE (AP) or amboceptor-coated SE (CP) was incubated with NHS or with NHS preincubated with CspA orthologs or BSA in either Mg-EGTA buffer (AP) or GVB++ buffer (CP). Following incubation, erythrocyte lysis was detected at 414 nm. Means from three separate experiments are shown, and error bars correspond to SD. Raw data were analyzed using one-way ANOVA (A and B). **, P < 0.01; *, P < 0.05.
CspA orthologs inhibit TCC-mediated lysis by binding of C7, C8, and C9.
To gain further insight into the inhibitory capacity of CspA orthologs on TCC formation, SE were preincubated with the C5b-6 complex, and increasing concentrations of recombinant proteins preincubated with C7, C8, and C9 were added. Following activation of the TP, all three CspA orthologs protect erythrocytes from complement-mediated lysis in a dose-dependent fashion (Fig. 6A). Apparently, among the three borrelial proteins investigated, CspA PKo displayed the strongest inhibitory activity. These findings indicate that CspA orthologs differ in their potential to inhibit TCC-mediated lysis.
FIG 6.
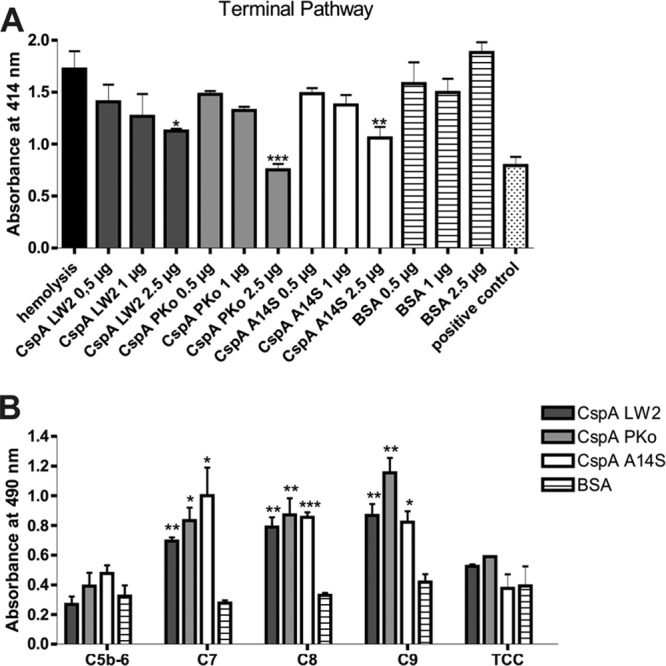
CspA orthologs inhibit TCC formation by interaction with complement components of the terminal pathway. (A) Inhibition of the TP was investigated using SE preincubated with C5b-6 employing hemolytic assays. A reaction mixture containing C7, C8, and C9 was preincubated with increasing concentrations of CspA orthologs or BSA. Following incubation, erythrocyte lysis was detected at 414 nm. Means from three separate experiments are shown, and error bars correspond to SD. Raw data were analyzed using one-way ANOVA with a post hoc Bonferroni correction. **, P < 0.01, *, P < 0.05. (B) Binding of complement components was verified for CspA orthologs using ELISA. Hexahistidine-tagged recombinant proteins or BSA (5 μg/ml each) was immobilized onto microtiter plates and incubated with purified C5b-6, C7, C8, C9, or TCC. Bound complement components were detected using specific antibodies and HRP-conjugated anti-goat immunoglobulins. Absorbance was measured at 490/560 nm. Experiments were performed in triplicate, and graphs represent means from at least three independent experiments. Error bars indicate standard errors of the means. ***, P < 0.001; **, P < 0.01; *, P < 0.05 (Student t test).
We next sought to identify the complement component(s) of the terminal pathway interacting with CspA orthologs using ELISA. Recombinant proteins were immobilized, and C5b-6, C7, C8, and C9 were added. As shown in Fig. 6B, all CspA orthologs bound to C7, C8, and C9 but not to the C5b-6 complex or the fully formed TCC. Slight differences occur for binding of CspA orthologs to C7 and to C9, as CspA A14S tends to bind C7 more strongly than CspA LW2 and CspA Pko, whereas CspA PKo has a tendency to bind C9 more strongly than CspA LW2 and CspA A14S.
CspA orthologs simultaneously bind CFH and plasminogen.
Recently, we showed that CspA of B. burgdorferi bound plasminogen (30). Since plasmin(ogen) acts as a complement inhibitor (40), we sought to examine the capability of CspA PKo and CspA A14S to bind plasminogen. Both CspA PKo and A14S orthologs exhibited a stronger binding capacity than CspA LW2 (P < 0.001) (Fig. 7A). To rule out possible inhibition of the CspA-CFH interaction by competition with plasminogen at the same binding site, we initially assessed binding of CFH to all three CspA orthologs. Using both purified and serum-derived CFH, no differences were observed in the binding capacity of this complement regulator for the CspA proteins (Fig. 7B and C). CspA orthologs were immobilized next, and binding of CFH in the presence of increasing concentrations of plasminogen was assayed. CFH binding remained unaffected by increasing concentrations of plasminogen (Fig. 7D to F), indicating that both host proteins simultaneously bind to the borrelial proteins.
FIG 7.

CspA orthologs simultaneously bound to CFH and plasminogen. Hexahistidine-tagged recombinant proteins or BSA (5 μg/ml each) was immobilized onto microtiter plates and incubated with 10 μg/ml purified plasminogen (PLG) (A), 10 μg/ml purified CFH (B), or 100 μl 3% NHS (C). Simultaneous binding of plasminogen and CFH to CspA LW2 (D), CspA PKo (E), or CspA A14S (F) was assayed by ELISA. CFH and increasing amounts of plasminogen were added to the immobilized CspA orthologs (molar ratios are indicated). Protein-protein complexes formed in the assays were detected using either a polyclonal goat anti-plasminogen antibody or a polyclonal goat anti-CFH antibody. Absorbance of each test was measured at 490 nm. Experiments were performed in triplicate. Data represent means and SEM from three separate experiments, each performed at least in triplicate. Raw data were analyzed by one-way ANOVA with a post hoc Bonferroni correction. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
Interaction of plasminogen with CspA orthologs is not influenced by electrostatic forces.
To further assess the nature of the CspA-plasminogen interaction, binding of plasminogen to CspA orthologs was examined in the presence of increasing concentrations of salt. CspA orthologs were immobilized, and increasing salt concentrations were applied. Even at six times the physiological concentration of NaCl, binding of CspA orthologs to plasminogen remained unaffected, suggesting that electrostatic forces are not involved in the CspA-plasminogen interaction (data not shown).
CspA-bound plasminogen is accessible to urokinase-type plasminogen activator.
Accessibility of cell-bound plasminogen to uPA is a prerequisite for the conversion of plasminogen to proteolytically active plasmin (Fig. 8A). Employing the chromogenic substrate S-2251, we demonstrated that plasminogen bound to CspA orthologs is accessible to uPA and converted to active plasmin (Fig. 8C to E), while no cleavage of the chromogenic substrate was observed when using BSA as a negative control (Fig. 8B). Additional control reactions with the plasminogen inhibitor tranexamic acid, and omitting either plasminogen or uPA, did not result in degradation of the chromogenic substrate.
FIG 8.

CspA-bound plasminogen is converted to plasmin by uPA. Microtiter plate wells were coated with 10 μg/ml purified plasminogen (PLG) (A), BSA (B), CspA LW2 (C), CspA PKo (D), or CspA A14S (E). BSA and CspA orthologs were subsequently incubated with 20 μg/ml plasminogen. Upon washing, the plasminogen activator uPA was added (2.5 μg/ml) together with the chromogenic substrate S-2251 (D-Val-Leu-Lys-p-nitroanilide dihydrochloride) (■). As negative controls, either uPA (▲) or plasminogen (●) was omitted or the plasminogen inhibitor tranexamic acid (T) (50 mM) was added to plasminogen (◆). Microtiter plates were incubated for approximately 15 h, and absorbance at 405 nm was measured at 30-min intervals. Three separate experiments were performed; data shown are from a representative experiment.
Activated plasmin(ogen) bound to CspA orthologs mediates C3b inactivation.
To further assess the contribution of CspA-bound plasminogen in mediating C3b degradation, CspA orthologs or BSA were immobilized and incubated with plasminogen, which was activated to plasmin using uPA. CspA-bound plasmin cleaved C3b, as indicated by the appearance of degradation products in the range of 30 to 40 kDa after 24 h of incubation (Fig. 9, arrowheads). These cleavage products were absent from experiments which omitted plasminogen or upon inclusion of tranexamic acid. No prominent degradation of C3b was observed with BSA used as a negative control, indicating that CspA orthologs also inhibit complement to some extent by binding plasmin(ogen).
FIG 9.
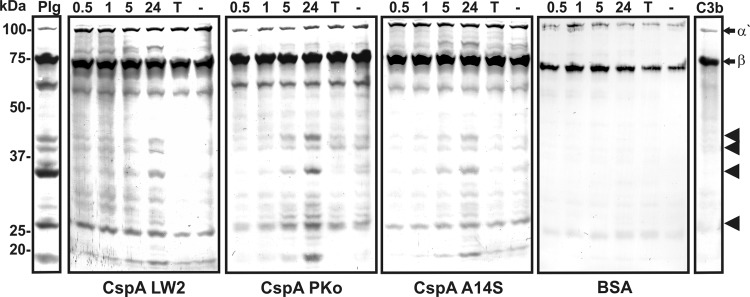
Plasmin bound to CspA orthologs degrades complement component C3b. Microtiter plates were coated with recombinant CspA orthologs and BSA (10 μg/ml). Following incubation with 20 μg/ml plasminogen (PLG), uPA (2.5 μg/ml) and purified C3b (5 μg/ml) were added. Microtiter plates were incubated at 37°C for 24 h, and aliquots were taken at different time intervals (0.5, 1, 5, and 24 h). Following separation by 12.5% SDS-PAGE, proteins were blotted onto nitrocellulose membranes, and C3b and its degradation products were detected by employing a polyclonal goat antiserum raised against C3b and a corresponding HRP-conjugated secondary antibody. Representative results from two independent experiments are shown. Arrows mark C3b α′- and β-chains, and arrowheads mark C3b degradation products with a molecular mass in the range of 25 to 40 kDa. Negative controls include addition of 50 mM tranexamic acid (T) as well as omission of plasminogen (−).
DISCUSSION
Many important human pathogens overcome the destructive effects of complement by recruitment of fluid-phase complement regulators (50–53). In particular, B. burgdorferi, B. afzelii, and B. spielmanii resist complement-mediated killing by binding CFH and FHL-1 (11, 12, 15, 16, 54–56). Recently, the CFH- and FHL-1-binding CspA protein has been identified as the key molecule of B. burgdorferi conferring resistance to complement-mediated killing (41, 42). Owing to their ability to bind complement regulators, it was hypothesized that CspA proteins of B. afzelii and B. spielmanii also was responsible for serum resistance (12, 15, 56), but unambiguous proof-of-principle studies with viable spirochetes producing individual CspA orthologs are still lacking. Regrettably, available molecular tools that were successfully engineered for gene inactivation in B. burgdorferi completely failed in B. afzelii and B. spielmanii. However, previous studies conclusively demonstrated that lipoproteins from B. burgdorferi retained their biological function and still interacted with various host proteins when ectopically produced in B. garinii G1 (13, 14, 47). Thus, we decided to employ this serum-sensitive B. garinii strain as a surrogate to overcome the aforementioned technical limitations and investigate CspA-mediated serum resistance of viable cells. Following transformation of serum-sensitive B. garinii G1, all CspA-producing spirochetes and their corresponding wild-type isolates multiplied equally well in the presence of 50% NHS (Fig. 2) and showed only limited deposition of harmful complement components on the bacterial surface (Fig. 3). These findings indicate that CspA of B. burgdorferi, B. afzelii, and B. spielmanii, when membrane exposed, impart spirochetes with comprehensive protection from complement-mediated killing. Moreover, transformed cells bound functionally active CFH, thereby successfully inactivating C3b in the presence of factor I, as demonstrated by the appearance of characteristic degradation products (Fig. 4).
The role of CspA in facilitating complement resistance has recently been demonstrated using an avirulent and a virulent cspA-deficient mutant of B. burgdorferi B31 (41, 42). Lack of CspA results in a strong susceptibility to complement-mediated killing accompanied with a simultaneous increase of activated complement components on the surface of CspA-deficient cells. Comparative analysis also revealed that the CspA mutant displayed a serum-sensitive phenotype that is similar to that of B. garinii G1, which was used as a surrogate in this study (data not shown). In addition, no difference was observed in the deposition of C3, C6, and C5b-9 when analyzed under identical conditions.
Binding of complement regulators seems to be the most prominent mechanism of serum-resistant Borreliae for evading complement attack in host compartments where CFH and FHL-1 are present in adequate concentrations. In the absence of, or with only limited access to, complement regulators, CspA orthologs are able to inactivate complement by an additional mode of action, namely, by direct interaction with components of the terminal pathway, C7, C8, and C9 (Fig. 6B). Interactions with various complement proteins of the terminal pathway target TCC assembly at different levels, e.g., formation and insertion of the membrane-bound tetrameric C5b-8 complex into the spirochetal membrane, recruitment of C9 molecules by the C5b-8 complex, and autopolymerization of C9 (43). Binding to the TCC family of homologous proteins (57) has been used as an efficient strategy for protecting invading microorganisms against complement attack, including the parasites Trypanosoma cruzi, Entamoeba histolytica, Schistosoma mansoni, and Trichinella spiralis, but only fragmentary information is available regarding the underlying molecular mechanisms (58–62). Previous studies illustrate how paramyosin of Schistosoma mansoni and Trichinella spiralis interacts specifically with C8 and C9, leading to an efficient inhibition of TCC assembly (62, 63). Concerning bacteria, TraT of Escherichia coli binds C6 and inhibits subsequent accumulation of C7, C8, and C9, while Rck of Salmonella enterica serovar Typhimurium counteracts C9 polymerization by direct binding of the pore-forming complement component (64, 65). Furthermore, Pausa et al. described a yet-unidentified CD59-mimiking protein of 80 kDa in serum-resistant B. burgdorferi and B. afzelii strains which preferentially binds to C8 and C9 and thereby efficiently inhibits assembly of the TCC (66). Due to the noticeable difference between the 80-kDa molecular mass of the CD59-like protein and the ∼28-kDa mass of the CspA orthologs, it is highly unlikely that these molecules are identical, although they apparently exhibit similar binding capabilities.
Concerning the capacity of CspA orthologs for complement inactivation, CspA of B. afzelii PKo appeared to be a more potent inhibitor of TCC formation than CspA orthologs of B. burgdorferi LW2 (43) and B. spielmanii A14S. Nevertheless, all three proteins significantly blocked activation of the alternative pathway in a dose-dependent fashion. Despite a sequence identity of only ∼47%, protein modeling revealed a structural organization and fold of CspA PKo and CspA A14S similar to that of the refined structure of CspA of B. burgdorferi ZS7 (27, 67, and P. Kraiczy, unpublished data), suggesting a comparable mode of action on human complement. Moreover, all three proteins possess almost identical binding properties for CFH and FHL-1 (15, 19, 56).
Recently, CspA has been identified as a novel plasminogen-binding protein of B. burgdorferi which, upon activation to plasmin, degrades the natural substrate fibrinogen, suggesting that interactions with plasminogen play a role in pathogen adhesion and tissue dissemination (30). Here, we showed for the first time that CspA orthologs of B. afzelii and B. spielmanii bind plasminogen more efficiently (P < 0.001) than CspA of B. burgdorferi. Binding of plasminogen is independent of CFH binding, so that under physiological conditions (plasminogen-to-CFH molar ratio of 1:1.4), both serum proteins simultaneously bind to a single CspA molecule. In addition, binding of plasminogen remained unaffected in the presence of increasing concentrations of NaCl, indicating that changes in ionic strength do not affect plasminogen binding, and that higher-order structural determinants also contribute to the CspA-plasminogen interaction. Similarly, high salt concentrations do not affect binding of plasminogen to ErpP, ErpC, or ErpA or to enolase of B. burgdorferi (68, 69). Beyond its role in degradation of extracellular matrix components, plasminogen bound to CspA orthologs, upon activation by uPA, cleaved and inactivated the key complement component C3b to some extent (Fig. 8 and 9). This cleavage of C3b by CspA-bound plasmin represents yet another mode of action by which CspA orthologs inhibit the complement system and reiterates the manifold functions CspA orthologs have in serum-resistant spirochetes. As this degradation of C3b by CspA-bound plasmin appears somewhat limited, it is tempting to speculate that the primary mechanism of complement inactivation by CspA orthologs is the binding of CFH as well as the inhibitory capacity on the assembly of the terminal complement complex. However, a small amount of C3b cleavage by CspA-bound plasmin could well serve as an ancillary mechanism, supporting the other means of complement inactivation.
Spirochetes, like other important pathogens, have developed a variety of strategies to overcome the immune system and to survive in the human host. In particular, outer surface proteins interacting with multiple host molecules possessing regulatory or proteolytic function are of significant importance for those microorganisms that lack certain virulence factors. Here, we showed that CspA orthologs were able to bind CFH and plasmin(ogen) simultaneously and also interact with complement components of the terminal complement complex. Of note, concentrations used to analyze protein-protein interaction by biochemical approaches did not entirely reflect physiological conditions prevailing in vivo but can provide important information concerning the underlying molecular mechanisms. As the concentration of CFH as well as plasminogen in human blood is quite high (500 and 200 μg/ml, respectively), limited access to these molecules should play a subordinate role. In addition, CspA of B. burgdorferi forms biologically functional homodimers (27), theoretically enabling the bacteria to bind more host molecules on their surface. Computer modeling revealed that CspA orthologs of B. afzelii and B. spielmanii most likely are also assembled as homodimeric complexes (P. Kraiczy, unpublished). Binding of CFH is thought to occur at the interdimeric cleft, while plasminogen-binding sites are located at two distinct regions, at the N terminus and C terminus of CspA (30). Thus, despite the relatively large dimensions of both host proteins, 92 kDa for plasminogen and 150 kDa for CFH, concurrent binding of these proteins to CspA appears quite feasible.
In conclusion, the present study demonstrates that CspA orthologs act as multifunctional immune evasion molecules, inhibiting complement by at least three different modes of action: (i) recruitment of CFH, followed by inactivation of C3b in the presence of factor I; (ii) direct interaction with complement components C7, C8, and C9, resulting in the inhibition of TCC formation; and (iii) degradation of C3b by binding of plasmin(ogen). Thus, CspA orthologs represent the spirochete's multipurpose tools of complement evasion.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the Deutsche Forschungsgemeinschaft (Kr3383/1-2).
We thank Christa Hansen-Hübner, Jessica Günnewig, and Axel Teegler for skillful and expert technical assistance.
This work forms part of the doctoral thesis of C.H.
Footnotes
Published ahead of print 4 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01094-13.
REFERENCES
- 1.Stanek G, Reiter M. 2011. The expanding Lyme Borrelia complex-clinical significance of genomic species? Clin. Microbiol. Infect. 17:487–493. 10.1111/j.1469-0691.2011.03492.x [DOI] [PubMed] [Google Scholar]
- 2.Steere AC, Coburn J, Glickstein L. 2004. The emergence of Lyme disease. J. Clin. Investig. 113:1093–1101. 10.1172/JCI21681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franke J, Hildebrandt A, Dorn W. 2013. Exploring gaps in our knowledge on Lyme borreliosis spirochaetes–updates on complex heterogeneity, ecology, and pathogenicity. Ticks Tick-Borne Dis. 4:11–25. 10.1016/j.ttbdis.2012.06.007 [DOI] [PubMed] [Google Scholar]
- 4.Estrada-Pena A, Ortega C, Sanchez N, Desimone L, Sudre B, Suk JE, Semenza JC. 2011. Correlation of Borrelia burgdorferi sensu lato prevalence in questing Ixodes ricinus ticks with specific abiotic traits in the western palearctic. Appl. Environ. Microbiol. 77:3838–3845. 10.1128/AEM.00067-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richter D, Schlee DB, Allgower R, Matuschka F-R. 2004. Relationships of a novel Lyme disease spirochete, Borrelia spielmani sp. nov., with its hosts in Central Europe. Appl. Environ. Microbiol. 70:6414–6419. 10.1128/AEM.70.11.6414-6419.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Dam AP, Kuiper H, Vos K, Widjojokusumo A, Jongh BM, Spanjaard L, Ramselaar AC, Kramer MD, Dankert J. 1993. Different genospecies of Borrelia burgdorferi are associated with distinct clinical manifestations of Lyme borreliosis. Clin. Infect. Dis. 17:708–717. 10.1093/clinids/17.4.708 [DOI] [PubMed] [Google Scholar]
- 7.Ohlenbusch A, Matuschka FR, Richter D, Christen HJ, Thomssen R, Spielman A, Eiffert H. 1996. Etiology of the acrodermatitis chronica atrophicans lesion in Lyme disease. J. Infect. Dis. 174:421–423. 10.1093/infdis/174.2.421 [DOI] [PubMed] [Google Scholar]
- 8.Fingerle V, Schulte-Spechtel UC, Ruzic-Sabljic E, Leonhard S, Hofmann H, Weber K, Pfister K, Strle F, Wilske B. 2008. Epidemiological aspects and molecular characterization of Borrelia burgdorferi s.l. from southern Germany with special respect to the new species Borrelia spielmanii sp. nov. Intern. J. Med. Microbiol. 298:279–290. 10.1016/j.ijmm.2007.05.002 [DOI] [PubMed] [Google Scholar]
- 9.Földvari G, Farkas R, Lakos A. 2005. Borrelia spielmanii erythema migrans, Hungary. Emerg. Infect. Dis. 11:1794–1795. 10.3201/eid1111.050542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maraspin V, Ruzic-Sabljic E, Strle F. 2006. Lyme borreliosis and Borrelia spielmanii. Emerg. Infect. Dis. 12:1177. 10.3201/eid1207.060077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kraiczy P, Skerka C, Brade V, Zipfel PF. 2001. Further characterization of complement regulator-acquiring surface proteins of Borrelia burgdorferi. Infect. Immun. 69:7800–7809. 10.1128/IAI.69.12.7800-7809.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kraiczy P, Skerka C, Kirschfink M, Brade V, Zipfel PF. 2001. Immune evasion of Borrelia burgdorferi by acquisition of human complement regulators FHL-1/reconectin and factor H. Eur. J. Immunol. 31:1674–1684. [DOI] [PubMed] [Google Scholar]
- 13.Siegel C, Hallström T, Skerka C, Eberhardt H, Uzonyi B, Beckhaus T, Karas M, Wallich R, Stevenson B, Zipfel PF, Kraiczy P. 2010. Complement factor H-related proteins CFHR2 and CFHR5 represent novel ligands for the infection-associated CRASP proteins of Borrelia burgdorferi. PLoS One 5:e13519. 10.1371/journal.pone.0013519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siegel C, Schreiber J, Haupt K, Skerka C, Brade V, Simon MM, Stevenson B, Wallich R, Zipfel PF, Kraiczy P. 2008. Deciphering the ligand-binding sites in the Borrelia burgdorferi complement regulator-acquiring surface protein 2 required for interactions with the human immune regulators factor H and factor H-like protein 1. J. Biol. Chem. 283:34855–34863. 10.1074/jbc.M805844200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wallich R, Pattathu J, Kitiratschky V, Brenner C, Zipfel PF, Brade V, Simon MM, Kraiczy P. 2005. Identification and functional characterization of complement regulator-acquiring surface protein 1 of the Lyme disease spirochetes Borrelia afzelii and Borrelia garinii. Infect. Immun. 73:2351–2359. 10.1128/IAI.73.4.2351-2359.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herzberger P, Siegel C, Skerka C, Fingerle V, Schulte-Spechtel U, van Dam A, Wilske B, Brade V, Zipfel PF, Wallich R, Kraiczy P. 2007. Human pathogenic Borrelia spielmanii sp. nov. resists complement-mediated killing by direct binding of immune regulators factor H and factor H-like protein 1. Infect. Immun. 75:4817–4825. 10.1128/IAI.00532-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bykowski T, Woodman ME, Cooley AE, Brissette CA, Brade V, Wallich R, Kraiczy P, Stevenson B. 2007. Coordinated expression of Borrelia burgdorferi complement regulator-acquiring surface proteins during the Lyme disease spirochete's mammal-tick infection cycle. Infect. Immun. 75:4227–4236. 10.1128/IAI.00604-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hartmann K, Corvey C, Skerka C, Kirschfink M, Karas M, Brade V, Miller JC, Stevenson B, Wallich R, Zipfel PF, Kraiczy P. 2006. Functional characterization of BbCRASP-2, a distinct outer membrane protein of Borrelia burgdorferi that binds host complement regulators factor H and FHL-1. Mol. Microbiol. 61:1220–1236. 10.1111/j.1365-2958.2006.05318.x [DOI] [PubMed] [Google Scholar]
- 19.Kraiczy P, Hellwage J, Skerka C, Becker H, Kirschfink M, Simon MM, Brade V, Zipfel PF, Wallich R. 2004. Complement resistance of Borrelia burgdorferi correlates with the expression of BbCRASP-1, a novel linear plasmid-encoded surface protein that interacts with human factor H and FHL-1 and is unrelated to Erp proteins. J. Biol. Chem. 279:2421–2429. 10.1074/jbc.M308343200 [DOI] [PubMed] [Google Scholar]
- 20.Kraiczy P, Hellwage J, Skerka C, Kirschfink M, Brade V, Zipfel PF, Wallich R. 2003. Immune evasion of Borrelia burgdorferi: mapping of a complement-inhibitor factor H-binding site of BbCRASP-3, a novel member of the Erp protein family. Eur. J. Immunol. 33:697–707. 10.1002/eji.200323571 [DOI] [PubMed] [Google Scholar]
- 21.McDowell JV, Wolfgang J, Tran E, Metts MS, Hamilton D, Marconi RT. 2003. Comprehensive analysis of the factor H binding capabilities of Borrelia species associated with Lyme disease: delineation of two distinct classes of factor H binding proteins. Infect. Immun. 71:3597–3602. 10.1128/IAI.71.6.3597-3602.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Metts MS, McDowell JV, Theisen M, Hansen PR, Marconi RT. 2003. Analysis of the OspE determinants involved in binding of factor H and OspE-targeting antibodies elicited during Borrelia burgdorferi infection in mice. Infect. Immun. 71:3587–3596. 10.1128/IAI.71.6.3587-3596.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.von Lackum K, Miller JC, Bykowski T, Riley SP, Woodman ME, Brade V, Kraiczy P, Stevenson B, Wallich R. 2005. Borrelia burgdorferi regulates expression of complement regulator-acquiring surface protein 1 during the mammal-tick infection cycle. Infect. Immun. 73:7398–7405. 10.1128/IAI.73.11.7398-7405.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller JC, Stevenson B. 2004. Increased expression of Borrelia burgdorferi factor H-binding surface proteins during transmission from ticks to mice. Int. J. Med. Microbiol. 293(Suppl 37):120–125 [DOI] [PubMed] [Google Scholar]
- 25.Miller JC, von Lackum K, Babb K, McAlister JD, Stevenson B. 2003. Temporal analysis of Borrelia burgdorferi Erp protein expression throughout the mammal-tick infectious cycle. Infect. Immun. 71:6943–6952. 10.1128/IAI.71.12.6943-6952.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caesar JJ, Johnson S, Kraiczy P, Lea SM. 2013. ErpC, a member of the complement regulator-acquiring family of surface proteins from Borrelia burgdorferi, possesses an architecture previously unseen in this protein family. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 69:624–628. 10.1107/S1744309113013249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cordes FS, Roversi P, Kraiczy P, Simon MM, Brade V, Jahraus O, Wallis R, Skerka C, Zipfel PF, Wallich R, Lea SM. 2005. A novel fold for the factor H-binding protein BbCRASP-1 of Borrelia burgdorferi. Nat. Struct. Mol. Biol. 12:276–277. 10.1038/nsmb902 [DOI] [PubMed] [Google Scholar]
- 28.Bhattacharjee A, Oeemig JS, Kolodziejczyk R, Meri T, Kajander T, Lehtinen MJ, Iwai H, Jokiranta TS, Goldman A. 2013. Structural basis for complement evasion by Lyme disease pathogen Borrelia burgdorferi. J. Biol. Chem. 288:18685–18695. 10.1074/jbc.M113.459040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kraiczy P, Stevenson B. 2013. Complement regulator-acquiring surface proteins of Borrelia burgdorferi: structure, function and regulation of gene expression. Ticks Tick-Borne Dis. 4:26–34. 10.1016/j.ttbdis.2012.10.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hallström T, Haupt K, Kraiczy P, Hortschansky P, Wallich R, Skerka C, Zipfel PF. 2010. Complement regulator-acquiring surface protein 1 of Borrelia burgdorferi binds to human bone morphogenic protein 2, several extracellular matrix proteins, and plasminogen. J. Infect. Dis. 202:490–498. 10.1086/653825 [DOI] [PubMed] [Google Scholar]
- 31.Walport MJ. 2001. Complement–first of two parts. N. Engl. J. Med. 344:1058–1066. 10.1056/NEJM200104053441406 [DOI] [PubMed] [Google Scholar]
- 32.Harris CL, Rushmere NK, Morgan BP. 1999. Molecular and functional analysis of mouse decay accelerating factor (CD55). Biochem. J. 341(Part 3):821–829. 10.1042/0264-6021:3410821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zipfel PF. 2009. Complement and immune defense: from innate immunity to human diseases. Immunol. Lett. 126:1–7. 10.1016/j.imlet.2009.07.005 [DOI] [PubMed] [Google Scholar]
- 34.Zipfel PF, Skerka C. 2009. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 9:729–740. 10.1038/nri2620 [DOI] [PubMed] [Google Scholar]
- 35.Makou E, Herbert AP, Barlow PN. 2013. Functional anatomy of complement factor H. Biochemistry (Moscow) 52:3949–3962. 10.1021/bi4003452 [DOI] [PubMed] [Google Scholar]
- 36.Coleman JL, Gebbia JA, Piesman J, Degen JL, Bugge TH, Benach JL. 1997. Plasminogen is required for efficient dissemination of Borrelia burgdorferi in ticks and for enhancement of spirochetemia in mice. Cell 89:1111–1119. 10.1016/S0092-8674(00)80298-6 [DOI] [PubMed] [Google Scholar]
- 37.Coleman JL, Sellati TJ, Testa JE, Kew RR, Furie MB, Benach JL. 1995. Borrelia burgdorferi binds plasminogen, resulting in enhanced penetration of endothelial monolayers. Infect. Immun. 63:2478–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu LT, Perides G, Noring R, Klempner MS. 1995. Binding of human plasminogen to Borrelia burgdorferi. Infect. Immun. 63:3491–3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klempner MS, Noring R, Epstein MP, McCloud B, Hu R, Limentani SA, Rogers RA. 1995. Binding of human plasminogen and urokinase-type plasminogen activator to the Lyme disease spirochete, Borrelia burgdorferi. J. Infect. Dis. 171:1258–1265. 10.1093/infdis/171.5.1258 [DOI] [PubMed] [Google Scholar]
- 40.Barthel D, Schindler S, Zipfel PF. 2012. Plasminogen is a complement inhibitor. J. Biol. Chem. 287:18831–18842. 10.1074/jbc.M111.323287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brooks CS, Vuppala SR, Jett AM, Alitalo A, Meri S, Akins DR. 2005. Complement regulator-acquiring surface protein 1 imparts resistance to human serum in Borrelia burgdorferi. J. Immunol. 175:3299–3308 [DOI] [PubMed] [Google Scholar]
- 42.Kenedy MR, Vuppala SR, Siegel C, Kraiczy P, Akins DR. 2009. CspA-mediated binding of human factor H inhibits complement deposition and confers serum resistance in Borrelia burgdorferi. Infect. Immun. 77:2773–2782. 10.1128/IAI.00318-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hallström T, Siegel C, Morgelin M, Kraiczy P, Skerka C, Zipfel PF. 2013. CspA from Borrelia burgdorferi inhibits the terminal complement pathway. mBio 13:e00481–13. 10.1128/mBio.00481-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prodinger WM, Hellwage J, Spruth M, Dierich MP, Zipfel PF. 1998. The C-terminus of factor H: monoclonal antibodies inhibit heparin binding and identify epitopes common to factor H and factor H-related proteins. Biochem. J. 331:41–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hauser U, Lehnert G, Wilske B. 1999. Validity of interpretation criteria for standardized Western blots (immunoblots) for serodiagnosis of Lyme Borreliosis based on sera collected throughout Europe. J. Clin. Microbiol. 37:2241–2247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frank KL, Bundle SF, Kresge ME, Eggers CH, Samuels DS. 2003. aadA confers streptomycin resistance in Borrelia burgdorferi. J. Bacteriol. 185:6723–6727. 10.1128/JB.185.22.6723-6727.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hammerschmidt C, Hallstrom T, Skerka C, Wallich R, Stevenson B, Zipfel PF, Kraiczy P. 2012. Contribution of the infection-associated complement regulator-acquiring surface protein 4 (ErpC) to complement resistance of Borrelia burgdorferi. Clin. Dev. Immunol. 2012:349657. 10.1155/2012/349657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Breitner-Ruddock S, Würzner R, Schulze J, Brade V. 1997. Heterogeneity in the complement-dependent bacteriolysis within the species of Borrelia burgdorferi. Med. Microbiol. Immunol. 185:253–260 [DOI] [PubMed] [Google Scholar]
- 49.Kraiczy P, Hunfeld KP, Breitner-Ruddock S, Wurzner R, Acker G, Brade V. 2000. Comparison of two laboratory methods for the determination of serum resistance in Borrelia burgdorferi isolates. Immunobiology 201:406–419. 10.1016/S0171-2985(00)80094-7 [DOI] [PubMed] [Google Scholar]
- 50.Blom AM, Hallström T, Riesbeck K. 2009. Complement evasion strategies of pathogens-acquisition of inhibitors and beyond. Mol. Immunol. 46:2808–2817. 10.1016/j.molimm.2009.04.025 [DOI] [PubMed] [Google Scholar]
- 51.Kraiczy P, Wurzner R. 2006. Complement escape of human pathogenic bacteria by acquisition of complement regulators. Mol. Immunol. 43:31–44. 10.1016/j.molimm.2005.06.016 [DOI] [PubMed] [Google Scholar]
- 52.Lambris JD, Ricklin D, Geisbrecht BV. 2008. Complement evasion by human pathogens. Nat. Rev. Microbiol. 6:132–142. 10.1038/nrmicro1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rooijakkers SH, van Strijp JA. 2007. Bacterial complement evasion. Mol. Immunol. 44:23–32. 10.1016/j.molimm.2006.06.011 [DOI] [PubMed] [Google Scholar]
- 54.Alitalo A, Meri T, Ramo L, Jokiranta TS, Heikkila T, Seppala IJT, Oksi J, Viljanen M, Meri S. 2001. Complement evasion by Borrelia burgdorferi: serum-resistant strains promote C3b inactivation. Infect. Immun. 69:3685–3691. 10.1128/IAI.69.6.3685-3691.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hellwage J, Meri T, Heikkila T, Alitalo A, Panelius J, Lahdenne P, Seppala IJT, Meri S. 2001. The complement regulator factor H binds to the surface protein OspE of Borrelia burgdorferi. J. Biol. Chem. 276:8427–8435. 10.1074/jbc.M007994200 [DOI] [PubMed] [Google Scholar]
- 56.Herzberger P, Siegel C, Skerka C, Fingerle V, Schulte-Spechtel U, Wilske B, Brade V, Zipfel PF, Wallich R, Kraiczy P. 2009. Identification and characterization of the factor H and FHL-1 binding complement regulator-acquiring surface protein 1 of the Lyme disease spirochete Borrelia spielmanii sp. nov. Int. J. Med. Microbiol. 299:141–154. 10.1016/j.ijmm.2008.06.005 [DOI] [PubMed] [Google Scholar]
- 57.Lovelace LL, Cooper CL, Sodetz JM, Lebioda L. 2011. Structure of human C8 protein provides mechanistic insight into membrane pore formation by complement. J. Biol. Chem. 286:17585–17592. 10.1074/jbc.M111.219766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Albrecht JC, Fleckenstein B. 1992. New member of the multigene family of complement control proteins in herpesvirus saimiri. J. Virol. 66:3937–3940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Braga LL, Ninomiya H, McCoy JJ, Eacker S, Wiedmer T, Pham C, Wood S, Sims PJ, Petri WA., Jr 1992. Inhibition of the complement membrane attack complex by the galactose-specific adhesion of Entamoeba histolytica. J. Clin. Investig. 90:1131–1137. 10.1172/JCI115931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deng J, Gold D, LoVerde PT, Fishelson Z. 2003. Inhibition of the complement membrane attack complex by Schistosoma mansoni paramyosin. Infect. Immun. 71:6402–6410. 10.1128/IAI.71.11.6402-6410.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Iida K, Whitlow MB, Nussenzweig V. 1989. Amastigotes of Trypanosoma cruzi escape destruction by the terminal complement components. J. Exp. Med. 169:881–891. 10.1084/jem.169.3.881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Z, Yang J, Wei J, Yang Y, Chen X, Zhao X, Gu Y, Cui S, Zhu X. 2011. Trichinella spiralis paramyosin binds to C8 and C9 and protects the tissue-dwelling nematode from being attacked by host complement. PLoS Negl. Trop. Dis. 5:e1225. 10.1371/journal.pntd.0001225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deng J, Gold D, LoVerde PT, Fishelson Z. 2007. Mapping of the complement C9 binding domain in paramyosin of the blood fluke Schistosoma mansoni. Int. J. Parasitol. 37:67–75. 10.1016/j.ijpara.2006.09.011 [DOI] [PubMed] [Google Scholar]
- 64.Heffernan EJ, Reed S, Hackett J, Fierer J, Roudier C, Guiney D. 1992. Mechanism of resistance to complement-mediated killing of bacteria encoded by the Salmonella typhimurium virulence plasmid gene rck. J. Clin. Investig. 90:953–964. 10.1172/JCI115972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pramoonjago P, Kaneko M, Kinoshita T, Ohtsubo E, Takeda J, Hong KS, Inagi R, Inoue K. 1992. Role of TraT protein, an anticomplementary protein produced in Escherichia coli by R100 factor, in serum resistance. J. Immunol. 148:827–836 [PubMed] [Google Scholar]
- 66.Pausa M, Pellis V, Cinco M, Giulianini PG, Presani G, Perticarari S, Murgia R, Tedesco F. 2003. Serum-resistant strains of Borrelia burgdorferi evade complement-mediated killing by expressing a CD59-like complement inhibitory molecule. J. Immunol. 170:3214–3222 [DOI] [PubMed] [Google Scholar]
- 67.Caesar JJ, Wallich R, Kraiczy P, Zipfel PF, Lea SM. 2013. Further structural insights into the binding of complement factor H by complement regulator-acquiring surface protein 1 (CspA) of Borrelia burgdorferi. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 69:629–633. 10.1107/S1744309113012748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brissette CA, Haupt K, Barthel D, Cooley AE, Bowman A, Skerka C, Wallich R, Zipfel PF, Kraiczy P, Stevenson B. 2009. Borrelia burgdorferi infection-associated surface proteins ErpP, ErpA, and ErpC bind human plasminogen. Infect. Immun. 77:300–306. 10.1128/IAI.01133-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Floden AM, Watt JA, Brissette CA. 2011. Borrelia burgdorferi enolase is a surface-exposed plasminogen binding protein. PLoS One 6:e27502. 10.1371/journal.pone.0027502 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.