

Abstract
Thanks to their ability to recognize biomolecular targets with high affinity and specificity, nucleic acid aptamers are increasingly investigated as diagnostic and therapeutic tools, particularly when their targets are cell-surface receptors. Here, we investigate the relationship between the folding of an anti-mouse transferrin receptor DNA aptamer and its interaction with the transferrin receptor both in vitro and in living cells. We identified and purified two aptamer conformers by means of chromatographic techniques. Fluorescence-anisotropy measurements showed that only one fold is able to bind mouse transferrin receptor. Besides displaying enhanced endocytosis in living mouse fibroblasts, the purified active fold is internalized also in human pancreatic cancer cells. Starting from these observations, we rationally designed variations of the parent sequence aimed at stabilizing the active fold, and consequently increase aptamer activity. A truncated version and full-length mutants with higher affinity than the parent sequence are shown.
Keywords: aptamer folding, aptamer-mediated endocytosis, aptamer rational engineering, transferrin receptor
Introduction
Nucleic acid aptamers are DNA or RNA oligonucleotides (typically single-stranded) that fold into well-defined three-dimensional structures and are able to recognize and bind a specific target molecule with high affinity and specificity.1,2 Aptamers binding to cell-surface receptors are increasingly investigated in nanomedicine mostly as potential vectors for intracellular delivery.3 DNA or RNA molecules have the ability to assume multiple folds via reversible pathways and the presence of these different conformations can sometimes be essential for the biological activity of the aptamer.4 The presence of misfolded material can, however, significantly lower the net activity of the aptamer. Other nucleic-acid molecules show conformation-dependent activity. This is the case of hairpin ribozymes, whose catalytic activity is modulated by the presence of inactive conformers.5,6
Investigation on the presence and activity of such different folds can help unveil the actual action mechanism and provide cues for the design of aptamers with optimized therapeutic efficiency.
Transferrin receptor (TfR) is a transmembrane protein essential for the delivery of ferric iron to all cells.7 TfR is ubiquitously expressed at low levels in normal cells and overexpressed (about 100-fold upregulation) in cells with a high proliferation rate, such as cancer cells, or in those requiring large amounts of iron.8 TfR is also highly expressed on the endothelium of the blood–brain barrier, where it plays a crucial role in transcytosis of iron-loaded transferrin across the blood–brain barrier.9 As a consequence, TfR is an ideal marker in cancer diagnosis10 and one of the most widely investigated tools for drug delivery across the blood–brain barrier.11
Transferrin (Tf) is the natural ligand of TfR and has been widely used as vector with promising results.12 Unfortunately transferrin receptors are nearly saturated by endogenous Tf in physiological conditions.13 Thus, exceedingly high concentrations of exogenous Tf derivatives are necessary to ensure adequate delivery of payloads to target tissues. A promising alternative is represented by aptamers since they can target different TfR sites, thus avoiding the saturation effect caused by endogenous Tf or even show higher affinity towards TfR than the natural ligand.14
Recently, Chen et al.15 developed natural DNA and RNA aptamers that selectively recognize and activate the extracellular domain of mouse transferrin receptor (mTfR), apparently with no competition with transferrin, and explored the suitability of a DNA aptamer (GS24) as vector for enzyme-replacement therapy.
Here, we show our results on the existence and activity of different GS24 conformers. We shall clarify aptamer structural properties and internalization process, and discuss the impact of GS24 folding on its interaction with the target both in vitro and in living cells. Furthermore, we report optimized GS24 sequences specifically designed to enhance the structural stability and consequently improve biological activity.
Results
Folding conformation analysis
In all our studies, anti-mTfR DNA aptamer GS24 was covalently conjugated to a fluorescent organic dye (ATTO 633). This small tag was chosen in order to minimize steric indrance and unwanted side-effects. Thanks to this tag we were able to accurately track the aptamer during endocytosis and perform reliable in vitro binding experiments.
It is known that both ends of GS24 can be modified without loss of binding affinity for the receptor.15,16 We employed a 5′-aminohexyl modified analog of GS24, suitable for fluorescent labeling or further derivatization.
Preliminary HPLC weak anion-exchange analysis, performed in native conditions at 25 °C, identified the presence of two main structures (black line in Figure 1a) at 20:80 molar ratio, as estimated from the chromatographic area of two peaks. Further chromatographic analyses were performed after thermal denaturation (unfolding) followed by a slow cooling to room temperature (refolding), in order to assess if the two peaks were distinct folding conformations.4,17 Indeed, if the native structures are different conformers, the same initial pattern must be observed after a denaturation/refolding process. Elution in denaturing conditions showed a total disappearance of native elution profile with a single structure detected at longer retention time (red line in Figure 1a) in keeping with the increased number of charges exposed by unfolded oligonucleotide.18 On the other hand, denatured aptamer showed a complete recovery of the native pattern after refolding in less than 12 hours at room temperature (blue line in Figure 1a). Overall, these analyses indicate the presence of two interconvertible folds, hereafter referred to as A-1F and A-2F, with a relative free energy difference ΔG2.1 of ~0.88 kcal/mol, as estimated from their 20:80 molar ratio.
Figure 1.

Folding conformation analysis. (a) HPLC analysis of 5′-aminohexyl GS24 performed in native conditions (black line) revealed the presence of two main structures in 20:80 molar ratio. Chromatographic run performed in denaturing conditions at 80 °C (red line) showed a single component corresponding to a completely unfolded structure. Analysis in native conditions performed after a slow refolding process at room temperature (blue line) showed a complete recovery of the initial pattern with a 20:80 molar ratio (named A-1F and A-2F on the basis of the different retention time). (b) HPLC analysis showed a remarkable temperature dependence of the folds molar ratio. The aptamer was heated and eluted at 25, 37, 40, 42, and 50 °C. Inset shows temperature dependence of A-1F percentage in the mixture as a function of temperature. Notably, A-1F percentage changed from the initial value of 0.20 at 25 °C to 0.31, 0.52, and 0.68 at 37, 40, and 42 °C respectively (c) Kinetics of the equilibrium recovery of the purified A-2F (left panel) showed a complete return to the initial molar ratio after 30 hours from folds purification. The energy landscape of two confomers (right panel) shows a free energy difference ΔG2.1 of ~0.88 kcal/mol.
A further chromatographic investigation was performed to evaluate the temperature dependence of the molar ratio between the two folds. Thus, five aptamer samples were heated and eluted using IE-HPLC at 25, 37, 40, 42, and 50 °C. Molar ratio between the conformers was strongly dependent on temperature, the most critical interval being 40–42 °C (gray and red lines of Figure 1b), where A-2F extensively interconverted into A-1F. Notably, at 37, 40, and 42 °C the A-1F:A-2F molar ratio values were 31:69, 52:48, and 68:32, respectively (Figure 1b, inset). The initial pattern virtually disappeared at 50 °C, due to oligonucleotide unfolding (blue line in Figure 1b).
Finally, we examined the interconversion kinetics of each fold. The two conformers were purified by IE-HPLC, dissolved in a suitable buffer, and maintained at 25 °C under stirring. We monitored equilibration of the two species and quantified interconversion kinetics and relative stability by analysis of samples collected at different times. The kinetics of recovery of the 20:80 molar ratio was described by a single exponential (Figure 1c, left panel). Conversion of A-2F into A-1F occurred with τ(2→1) = 5.33 hours, a sufficiently extended period to allow most biological studies to be performed without any appreciable conformational change. On the contrary, A-1F showed a significantly higher conversion rate (τ(1→2) = 1.33 hours) resulting markedly less stable than A-2F. The two different interconversion rates are consistent with the calculated value for ΔG2.1, i.e., τ(2→1)/τ(1→2) =exp(-ΔG2.1/RT) (Figure 1c, right panel).
Molecular dynamic simulations
Two possible folds (α and β) were obtained using the secondary-structure prediction server Mfold.19 On the basis of the predicted free energies (ΔGαβ = −1.53 kcal/mol at 25 °C, to be compared with our experimental estimate of 0.88 kcal/mol), we assigned the more stable α-fold to the more populated A-2F. Following the procedure described in the Methods section, the secondary structures were converted into two 3D conformations. Molecular dynamics simulations were then employed to assess the structural properties of the two folds. For each fold, we analyzed a combined trajectory formed by ten 10-ns explicit-solvent molecular dynamics simulations performed using different initial velocities. A representative structure was obtained from each combined trajectory using cluster analysis (Figure 2).
Figure 2.
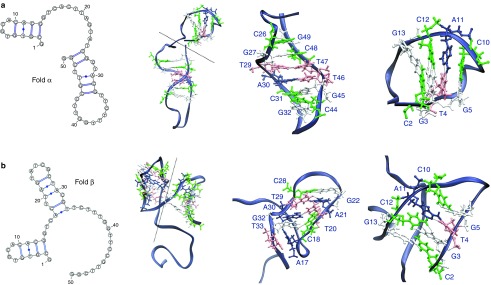
Mfold predictions of the two lowest-energy folds of GS24 sequence and corresponding structures from MD simulations (top, fold α; bottom, fold β). From left to right, secondary-structure diagrams, structure of representative conformation (centroid of the most populated cluster), magnification of the domains. A dashed line in the complete 3D structure separates the two domains.
The stated correspondence of conformations α and β to A-2F and A-1F respectively is further confirmed by relating HPLC retention times to the electrostatic properties of the two conformations. In order to establish this relationship, we computed an “effective total charge” for each conformation (see Materials and Methods section) and related it to HPLC retention times. The calculated “effective total charge” for conformer α is −13.9 ± 0.4 e and is greater than that calculated for conformer β (−11.7 ± 1.3 e).
Fluorescence anisotropy assay
The presence of two interconvertible folds at or near physiological temperature can represent a limit to the use of this aptamer in vivo. Indeed, changes in the three-dimensional conformation of an aptamer frequently impact its actual biological activity. Thus, we performed a fluorescence anisotropy (FA) assay20 with the two fluorescently labeled aptamer conformers, in order to determine the receptor-binding affinity of each fold. We performed all measurements at 25 °C consistently with the in vitro binding assay initially performed during the SELEX process leading to GS24 identification.15
The FA of the aptamer was measured in the absence or presence of increasing amounts of recombinant murine TfR (mTfR). We initially performed measurements on the mixture of the two folds (20/80 molar ratio as indicated by HPLC analyses). The amount of added protein ranged from 0 to 2,500 nmol/l. As expected, FA increased in the presence of mTfR, due to protein-aptamer complex formation (Figure 3a). The observed behavior of FA as a function of protein concentration is compatible with a monophasic ligand-receptor binding.21 We also measured the FA of the purified folds separately, at mTfR concentration of 600 nmol/l (Figure 3b). During the course of FA assays, a partial re-equilibration of the two populations took place. Indeed the A-1F and A-2F samples contained 55:45 and 3:97 molar fractions, respectively, as measured by HPLC after anisotropy measurements (Supplementary Figure S1). As shown in Figure 3b, A-2F measured anisotropy exceeded that found for GS24, which in turn was higher than that relative to the A-1F sample, corresponding to a fraction of bound aptamer (fB) of 0.85, 0.69, and 0.46, respectively (see supplemental data for further details). These fB values are proportional to the measured population of A-2F in the three samples (i.e., 97%, 80%, and 45%). These results led us to conclude that A-2F is the only fold able to bind mTfR. In fact, binding of A-1F with a different Kd would result in a biphasic curve of the anisotropy.21
Figure 3.
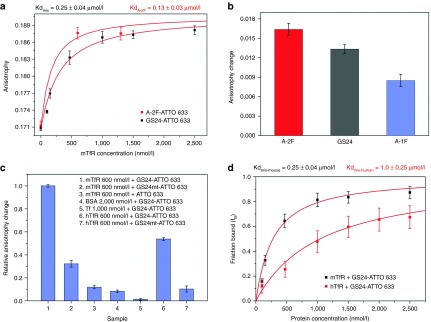
Fluorescence anisotropy assay. (a) Titration of GS24-ATTO 633 (50 nmol/l) with mTfR (0–2,500 nmol/l) at 25 °C. A-2F binding was examined at 600 and 1,300 nmol/l, the latter considered as concentration close to the saturation. The curve of the mixture (black squares) was obtained by fitting the anisotropy values as a function of mTfR concentration. The fit afforded both Kd and AB (anisotropy in saturating condition). The curve of A-2F (red squares) was obtained as above, using the value of AB obtained from the fitting of GS24 anisotropy curve. (b) Binding efficiency of the two folds and GS24 was measured at [mTfR] = 600 nmol/l. In this case the anisotropy change is defined as the difference between aptamer anisotropy in the presence and in absence of protein. (c) Aptamer selectivity control experiments. Anisotropy change was measured for GS24 mutant (GS24mt) and unconjugated ATTO 633 (50 nmol/l) in presence of 600 nmol/l mTfR respectively. Next, GS24 aptamer selectivity was tested incubating the sample (50 nmol/l labeled GS24) with saturating amounts of BSA, Tf and hTfR. The anisotropy change of each sample was compared with the anisotropy measured in the presence of 600 nmol/l mTfR. GS24mt was also tested in presence of 600 nmol/l of hTfR (d) Fraction bound of aptamer (fB) for mouse (black squares) and human (red squares) TfR was calculated from the corresponding anisotropy titration curves. The mixture of two folds (GS24) showed a different affinity for each protein.
The anisotropy of the A-2F sample in saturating conditions ([mTfR] = 1,300 nmol/l) is compatible with the measured anisotropy of GS24 (Figure 3a) and thus implies that the unbound fraction eventually equilibrates between the two folds during the course of FA measurements. Within this assumption, the Kd of the mixture and that of the purified A-2F should be related by Kdmix = (1 + L)KdA-2F, L being the molar ratio between A-1F and A-2F (L = 0.25 from the HPLC assay).
Fitting the FA data yields a dissociation constant for the mixture Kdmix = 0.25 ± 0.04 μmol/l (Figure 3a), and a theoretical estimate for KdA-2F ≈ 0.2 μmol/l. The FA results obtained from the A-2F sample yield an estimate of a lower KdA-2F = 0.13 ± 0.03 µmol/l. This discrepancy probably stems from an incompletely equilibrated molar fraction during the titration of the GS24 mixture.
Next, we examined aptamer specificity. A control sequence (GS24mt)16 which is known to be unable to recognize the transferrin receptor was incubated with 600 nmol/l of mTfR, and revealed a remarkably lower anisotropy change as compared with the GS24 value (Figure 3c). Additionally, we further examined the aptamer selectivity towards different proteins. The addition of saturating amounts of bovine serum albumin (BSA) and transferrin (Tf) to GS24 sample did not affect aptamer anisotropy, and we observed no significant anisotropy change. Overall these results, together with the anisotropy variations recorded for unconjugated fluorophores with 600 nmol/l of mTfR, suggest that the anisotropy enhancement observed during FA is mainly dependent on aptamer structure (Figure 3c).
Notably, a substantial anisotropy change was observed also with human TfR (600 nmol/l) (Figure 3c) suggesting that GS24 could be active also against the human receptor, possibly via its active form A-2F. This is supported by the greater change in FA shown by a sample enriched in A-2F, compared with GS24 (Supplementary Figure S2). Moreover, addition of hTfR to GS24mt under identical conditions caused only a minor anisotropy change, in keeping with the specific binding of GS24 to the human protein (Figure 3c). Thus, a second titration experiment was performed using increasing concentrations of hTfR against labeled GS24. By applying the same model described above the calculated Kd for human receptor was 1.0 ± 0.25 µmol/l, i.e., about fourfold higher than the measured Kdmix for mTfR (Figure 3d).
Internalization assay in living cells
Internalization assays were performed in order to assess whether the different binding activities of the two folds observed in vitro lead to different aptamer activity in living cells. To this end, we first performed internalization assays on mouse fibroblasts (NIH 3T3) using the purified folds. Each sample was incubated in cultured cells at a concentration of 1 µmol/l, and aptamer endocytosis was monitored by confocal fluorescence microscopy. BSA and calf thymus DNA were added to the medium during incubation in order to minimize nonspecific binding.22,23
Figure 4 shows that A-1F internalization (panel b) is not significantly different from that of a control constituted by the incubation with the labeled scrambled sequence, the GS24mt-ATTO 633 probe (panel a). This is not the case for A-2F (panel c). The incubation of this fold promotes intense vesicular fluorescence in the perinuclear region that partially colocalizes with lysosomes after 120 minutes (Supplementary Figure S3a) and is still detectable in the cells at longer times (180 minutes, Supplementary Figure S3b). Notably, A-2F promotes endocytosis even at lower concentration (200 nmol/l) (Supplementary Figure S3c).
Figure 4.

Internalization assay of the labelled aptamer folds in mouse fibroblasts (NIH 3T3). Each sample was incubated in cultured cells at a concentration of 1 µmol/l and endocytosis was monitored by confocal fluorescence microscopy. (a) GS24mt-ATTO 633 probe was not able to recognize and bind mTfR, consequently only weak vesicular signal was detected into the cells. (b) Vesicular signal of A-1F-ATTO 633 was weakly detected into the cell cytoplasm, indicating a negligible activity, likely due to partial conversion in the active fold A-2F during incubation. (c) A-2F-ATTO 633 showed strong perinuclear vesicular signals. Scale bars: 10 μm.
In order to verify that the observed internalization is an active, receptor-mediated process, we performed time-lapse imaging at low temperature24 after treatment with both aptamer and Tf-AF488; the latter was used as control to verify that endocytosis is inhibited by temperature. As expected from an energy-driven process,25 no internalization was observed while cells were kept on ice; on the contrary, both A-2F and Tf were rapidly internalized following warming to 37 °C (Supplementary Figure S4). Tf and aptamer showed remarkably different internalization kinetics. The natural ligand displayed greater efficiency in keeping with the higher affinity of transferrin (monoferric Tf Kd~200 nmol/l and diferric forms Kd~10 nmol/l)14,26 compared with that of the aptamer. This resulted in a substantial lack of colocalization between A-2F and Tf detected during the initial phase of endocytosis process, in agreement with previous GS24 observations.15 Colocalization was observed at longer times (Supplementary Figure S4), however, and indicates that aptamer and the nonrecycled fraction of Tf share the same internalization pathway and final fate (i.e., lysosomes).
Furthermore, we performed another assay to establish if binding of the aptamer is followed by activation of the internalization process both in mTfR and in hTfR. We examined A-2F internalization efficiency in a human pancreatic carcinoma cell line (MIA PaCa-2), which is known to overexpress hTfR.27,28 In this case, a higher concentration of aptamer (3 µmol/l) was used, with the aim of balancing the lower binding efficiency of the oligonucleotide towards hTfR with respect to its murine counterpart. Upon incubation, fluorescence was observed mainly from the plasma membrane (Figure 5a), while spotty vesicular signal from the cytoplasm was detected only at longer times (60 minutes), with a significant amount of aptamer still present on cell surface (Figure 5b). Fluorescence completely disappeared from the plasma membrane only after 140 minutes; by this time, most of the signal was localized in the perinuclear region (Figure 5c). Contrarily, virtually no intracellular fluorescence was observed when the scrambled control GS24mt was used (Supplementary Figure S5), in keeping with endocytosis in mouse cells (see above) and previous observations.16 Finally, we performed colocalization experiments with the monoclonal antibody anti-hTfR (mAb anti-CD71) in order to assess whether the internalization of A-2F in human cells is a receptor-mediated process. Cells were incubated with both A-2F and mAb anti-CD71; after 60 minutes, we observed extensive, although not complete, colocalization between the aptamer and the antibody in many vesicular compartments, as assessed by a Pearson's correlation coefficient of 0.63 (Supplementary Figure S6). Overall, these results suggest that activation of endocytosis pathway by the active fold of GS24 aptamer occurs also in human cells, although with slower kinetics.
Figure 5.

Endocytosis of the A-2F in human pancreatic tumor cells (MIA PaCa-2). 3 µmol/l A-2F-ATTO 633 was incubated in cultured cells. (a) After incubation, fluorescence signal was mainly present on plasma membrane. (b) Significant vesicular signals could be observed into cell cytoplasm after 60 minutes since incubation, but A-2F fluorescence was still present on the cell surface. (c) At longer times (140 minutes) all A-2F was internalized into the cells and perinuclear signals were observed. Scale bars: 10 μm.
Rational engineering of GS24 aptamer
We first searched for the substructure responsible for the binding affinity of GS24, in order to select a minimized sequence with a well-defined secondary structure.
As stated above, a close inspection of the conformations of GS24 folds resulting from the secondary-structures predictions and our molecular dynamics simulations revealed a significant difference in the region including nucleotides 16–50. Notably, A-2F displays a folded domain comprising residues 26 to 50, which is completely absent in the A-1F conformation.
Importantly, Chen et al. selected both GS24 and an RNA aptamer (FB4) against the extracellular domain of mouse transferrin receptor. The latter showed the same properties of GS24. Thus, we examined and compared the secondary structures prediction of DNA and RNA aptamers. Notably, DNA and RNA aptamer GS24 and FB4, and their extended versions share a highly conserved stem-loop domain, approximately located in a central region, which contains a conserved nonamer (CGTTSTYTG; where S = G or C, Y = T or C. Note that in the RNA structure U replaces T; Supplementary Figure S7a).
A comparison of these structures, together with MD analysis on the conformation of the two folds, suggested that the first portion at 5′-end of the aptamer sequences could play a relatively minor role, and highlighted the importance of the conserved stem-loop region formed by the nonamer sequence in order to preserve aptamer activity. Thus, using the conserved stem-loop region within the 26–50 sequence of A-2F as a guide, we designed a truncated version of GS24 comprised of 35 nucleotides, hereafter referred to as minimal GS24 (GS24min). A comparison between GS24 (A-2F) and GS24min secondary structure showed a remarkable structural homology and GS24min secondary structure still displayed the conserved stem-loop regions (Supplementary Figure S7b).
In order to evaluate if GS24min internalization is as effective as for the original sequence, we generated it and tested its activity in living cells. The internalization assay performed on mouse fibroblasts (NIH 3T3) showed that the labeled GS24min is still active. It displays an endocytosis pattern similar to that of A-2F (Figure 6a) and shows partial colocalization with lysosomal compartments (Supplementary Figure S8). On the contrary, GS24min showed very weak vesicular signals when tested in human MIA PaCa-2 cells (Figure 6a) possibly owing to specific internalization similar to that shown by the scrambled sequence (Supplementary Figure S5).
Figure 6.
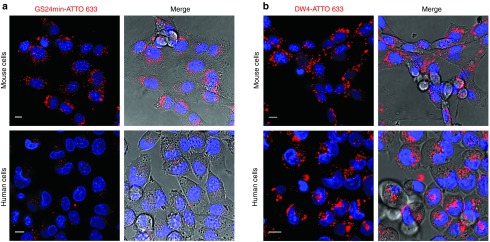
Endocytosis of GS24min and DW4 in mouse fibroblasts (NIH 3T3, upper panels) and in human pancreatic tumor cells (MIA PaCa-2, lower panels). Aptamer concentration during incubation with NIH 3T3 and MIA PaCa-2 cells was 1 and 3 µmol/l respectively. (a) Significant vesicular signals could be observed into cell cytoplasm after incubation in mouse cells with labeled GS24min. On the contrary weak spotty cytoplasmic signals were detected into human cells after incubation of GS24min (b) DW4 shows efficient internalization both in mouse and human cells. Scale bars: 10 μm.
Importantly, the GS24min activity observed with mouse cells was related to a unique conformer. Indeed, the chromatographic investigation of the labeled GS24min in native conditions is compatible with a single fold, contrarily to what shown by the aptamer extended sequence (Supplementary Figure S9a).
Next, we performed a FA assay in order to calculate the in vitro affinity of GS24min towards the recombinant mouse receptor. Although GS24min was still able to bind TfR, the calculated Kd was 1.2 ± 0.39 µmol/l, nearly five times higher than the measured Kd of GS24 towards mTfR (Supplementary Figure S9b).
These results clearly evidence that correct folding of sequence 26–50 is essential to maintain recognition capability towards TfR. We thus moved to a more conservative approach based on the stabilization of the desired fold (A-2F) by means of punctual mutations. We focused our attention to two base pairs: T29-T47 (which, in our simulations, showed a noncanonical base-pairing) (see Figure 2), and A30-T46. These two base pairs were selected as the most critically involved in the stabilization of the folded domain comprising bases 26–50, simultaneously destabilizing the inactive conformer. Most importantly, mutations at these sites did not change the conserved nonamer sequence in the stem-loop region. Thus, we designed four mutants: T29A; T29G T47C; A30G T46C; T29G A30G T46C T47C; hereafter called DW1, DW2, DW3 and DW4, respectively. These mutants were chosen as candidates capable of stabilizing the active fold by either transforming a noncanonical pairing into a more stable Watson-Crick coupling, or strengthening an A-T base pair by mutation into a stronger G-C.
Interestingly, all mutants displayed an increased difference in the ΔΔG of the two conformers as predicted by MFold (Table 1). We then analyzed by HPLC the percentage of inactive fold for each structure, both at 25 and at 37 °C (Table 1). Interestingly, the active fold percentage is higher in all mutants, compared with the parent structure GS24. In two cases (DW2 and 4), the percentage of the inactive fold was nearly identical to that attained immediately after HPLC purification of GS24 (3%). Insertion of a fluorophore at 5′ end of the sequences did not alter significantly the ratio between the two folds (data not shown).
Table 1. Fold molar ratio and anisotropy change comparison of GS24 mutants.
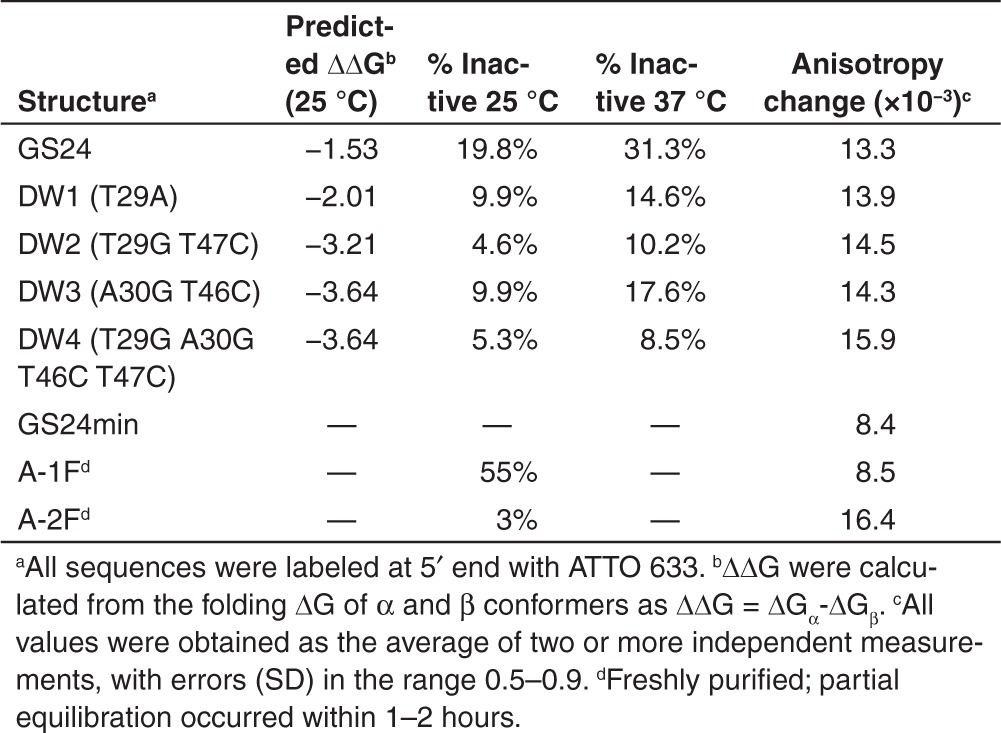
Next, we administered the labeled mutants to NIH-3T3 cells, in order to assess whether the mutations affected the recognition capability towards the mouse receptor in its native form. All sequences were able to promote internalization in mouse fibroblasts. Interestingly, we observed no significant difference in internalization kinetics or efficiency, compared with the active fold of the parent aptamer. Then, we assessed the in vitro binding affinity of all mutants towards mTfR by FA measurements. Firstly, we evaluated the anisotropy change at a single protein concentration (600 nmol/l) by using the same protocol adopted in the case of the purified folds (Table 1). All mutations showed increased binding efficiency compared with GS24. We selected the most promising DW4 and determined its Kd towards mTfR (Supplementary Figure S10). Our results yielded Kd = 0.19 ± 0.01 µmol/l, 25% lower than the value found with GS24 (0.25 ± 0.4 µmol/l). Notably, although this value is somewhat higher than the calculated value for pure A-2F (0.13 ± 0.03 µmol/l), one must consider that the present mutant outperforms the purified fold in terms of thermal stability and ease of manipulation.
Finally, we tested DW4 for further evaluation in human pancreatic tumor cells. Contrarily to what observed for the truncated form GS24min, our mutant sequence could efficiently promote internalization also in MIA PaCa-2 cells at the same concentration of A-2F. Interestingly, intense vesicular signal was already present immediately after incubation, suggesting faster endocytosis kinetics for DW4, compared with A-2F (Figure 6b). This result is supported by the higher anisotropy change (11.8 ± 0.6 and 9.2 ± 0.4 respectively, Supplementary Figure S2) measured upon interaction with the hTfR under the same experimental conditions previously used (600 nmol/l).
Discussion
Transferrin receptor is a transmembrane protein highly overexpressed in a number of cancer-cell types and crucially present in the endothelium of the blood–brain barrier; hence, targeting molecular payloads to TfR may lead to useful strategies of interest in nanomedicine.8 In this context, anti-TfR aptamers are promising candidates for diagnostic and therapeutic purposes, given their known high affinity and selectivity in target recognition. Unfortunately, aptamer activity is largely influenced by changes in its three dimensional structure.4,29 Thus, the precise knowledge and control of aptamer conformation is needed to optimize activity and selectivity (e.g., capability to discriminate between cancerous and healthy cells) both in living cells and in vivo.
In this work, we explored the relationship between the folding and the activity of GS24, a DNA aptamer recently selected to target mouse TfR.15 In order to evaluate its physico-chemical properties, we covalently labeled the oligonucleotide with ATTO 633, a small fluorescent dye. Compared with the previously used biotin-streptavidin labeling approach,15 the use of organic dyes has the advantage of a reduced interference on the three-dimensional structure, and hence on the biological activity of the aptamer. This strategy was chosen to allow an unbiased evaluation of the activity of GS24 conformers.
A chromatographic investigation was performed both in native and denaturing conditions and revealed an important structural feature, namely the presence of two distinct structures: A-1F and A-2F (Figure 1a). Different interconversion times between A-1F and A-2F were measured at 25 °C (τ1→2 = 1.33 hours and τ2→1 = 5.33 hours for A-1F and A-2F, respectively). These interconversion rates indicate that A-2F is thermodynamically more stable than A-1F. Importantly, conversion of A-2F into A-1F occurs over a period of time sufficiently extended to allow most biological studies to be performed without any appreciable conformation change.
Fold molar ratio in the mixture is dependent on temperature, most critically around 40–42 °C, where the two folds become equally populated (Figure 1b). Such temperature dependence is particularly relevant for biological purposes, since in vivo and living-cell studies are performed near 37 °C. Importantly, interconversion between two folds with different structure can lead to significant changes in aptamer biological activity.
Secondary-structure predictions yield two folds with relative stability comparable to the experimental ΔG (Figure 2). Comparison between the net exposed charge calculated from MD simulations and HPLC retention times provides further evidence of our structural assignment (folds α = A-2F and fold β = A-1F).
The measured interconversion rate is sufficiently slow to allow accurate fluorescence anisotropy measurements in solutions where isolated A-1F and A-2F are incubated with purified mTfR. We were thus able to precisely quantify the binding efficiency of each fold for TfR (Figure 3b). Figure 3a,b present our FA assays and indicate that only A-2F does recognize mTfR, while A-1F is inactive. Not surprisingly, A-2F displays higher affinity for mouse TfR than GS24 since the latter is actually a mixture of the two folds (Figure 3a). Coherently with the different binding affinities observed in vitro, A-1F and A-2F also show remarkably different internalization efficiency in living mouse fibroblast cells. Upon cell incubation with the two folds, A-2F signal is efficiently detected in endocytic vesicles, while internalization of A-1F is barely detectable (Figure 4). We believe that the weak vesicular signal displayed upon treatment with A-1F (Figure 4b) stems from the small fraction of A-1F converting to A-2F during incubation.
The significant anisotropy change observed in vitro when using hTfR (Figure 3c) prompted us to further investigate binding affinity of GS24 towards hTfR. Accordingly, we performed FA measurements in which GS24 and its folds were incubated with human TfR (Supplementary Figure S2). Our results indicate that GS24 via A-2F is indeed able to bind both hTfR and mTfR. The comparable, albeit lower, affinity of GS24 towards hTfR (Figure 3d), whose homology with mTfR is about 77%, may indicate that the recognition site is located near the active site of the receptor, usually highly conserved portion. In previous works when a mixture of conformers (i.e., GS24) was employed, no binding to human TfR was observed.15,16 On the basis of these results, and given the lower affinity of the aptamer towards hTfR, we administered pure A-2F to human pancreatic tumor cells. We found that A-2F can indeed promote internalization through aptamer-mediated endocytosis also in human cells (Figure 5). The significantly slower kinetics of internalization observed in this case suggests that the Kd in vivo be higher than the affinity measured in vitro by FA assay. Indeed, the high A-2F concentration incubated in the MIA PaCa-2 cells is not sufficient to balance the lower affinity towards hTfR, allowing a fast internalization also in human cells.
Notably, no recycling of the aptamer was observed either in mouse or human cell lines, contrary to what shown by transferrin in mouse fibroblasts. This suggests either aptamer separation from TfR in the endosomial compartment or enzymatic degradation at the later lysosomial stage.
Importantly, secondary and tertiary structure predictions suggest that the folded domain (sequence 26–50) of the active conformer A-2F have a major influence on the biological activity of GS24. This hypothesis is further supported by the fact that A-1F and A-2F share the same structure only at the 5′-end. The comparison of the sequences and secondary structures of the previously selected aptamers (full length and minimal DNA aptamer GS24, and RNA aptamer FB4) shows a conserved stem-loop region, within the 26–50 region, containing a conserved nonamer (Supplementary Figure S7a). Based on these results, we generated engineered sequences according to two different approaches: truncation—leading to a minimal sequence—and punctual mutations performed on the basis of secondary-structures predictions and molecular modeling approaches.
The truncated version of GS24 reduced to 35 residues (GS24min) showed a high structural homology with the parent sequence. In particular, the conserved stem-loop was still present in its secondary structure (Supplementary Figure S7b). Thus, we tested this sequence in living cells and in vitro by FA assay. Interestingly, GS24min is still able to promote internalization in mouse fibroblasts with an endocytosis pattern similar to what shown by the extended sequence. No significant internalization was observed in human pancreatic cancer cells (Figure 6a), however, suggesting that GS24min is not able to recognize and bind hTfR, contrary to what observed for the active fold of GS24 (A-2F). This behavior is consistent with the low in vitro affinity calculated by FA assay towards mTfR, i.e., the natural target of SELEX process (Supplementary Figure S9b). Indeed, GS24min showed a Kd approximately five times higher than the measured Kd for GS24. This decrease in aptamer affinity following the minimization process is not particularly surprising and the same reduction was reported for a recently discovered anti-hTfR RNA aptamer.14
Rational engineering afforded sequences with interesting properties. Indeed, molecular modeling led to the identification of two critical base pairs involved in the stability of the two folds. This observation coupled with the result obtained with the truncated sequence GS24min prompted us to target the stabilization of the folded domain comprising bases 26–50 in order to increase A-2F stability and minimize the occurrence of misfolded structures. Modification of bases in these critical sites yielded aptamers with very low (usually <10%) percentages of inactive fold (Table 1). The engineered structures fully retained their capability to recognize mTfR in physiological conditions. Additionally, FA measurements highlighted the most promising mutant (DW4), which displayed a Kd towards mTfR (~190 nmol/l) superior to that of GS24 and comparable with freshly purified A-2F (Supplementary Figure S10). Most importantly, DW4 displayed a higher anisotropy change upon interaction with hTfR (Supplementary Figure S2) and was able to promote efficient internalization in human cell lines (Figure 6b).
The main conclusion of this work is that the biological activity of aptamer GS24 is affected by the presence of two different aptamer folds only one of which (A-2F) is able to recognize its target. This observation suggests that the main conformational difference between the two folds occurs in a region critically involved in binding to TfR. The results obtained with the engineering of GS24, and in particular those related to GS24min and DW4, confirm this hypothesis.
Most importantly, the two folds become nearly equimolar in the 40–42 °C interval. These data imply that the actual active form present when aptamer GS24 is administered in living cells or in vivo is markedly lower than expected from the nominal aptamer concentration. The presence of the two folds is easily rationalized by examining the SELEX strategy that led to GS24 selection.15 Only in vitro binding assays at 25 °C were used to evaluate aptamer affinity for the receptor. This strategy only provides information regarding the average activity of a given mixture of conformers, but is intrinsically unable to report on the presence, and hence the activity, of different conformers sharing the same primary structure. Our identification of two folds with markedly different activity for aptamer GS24 highlights the need to perform during SELEX processes additional functional assays capable of assessing oligonucleotide suitability for the desired biological function.
Finally, our engineering of GS24 led to a novel sequence (DW4), which shows an enhanced activity compared to the parent molecule. Thermal dependence of molar ratio showed that with this sequence the inactive fold is less than 10% both at 25 °C and 37 °C. In perspective, this feature promotes DW4 as a valid tool for targeted drug delivery and in vivo applications.
Additionally, the analysis of the secondary and tertiary structures of DW4 and other mutants presented in this work, together with our experimental data on binding affinity provide useful hints on which aptamer domains are most involved in binding to mouse and human TfR. Thus, DW4 may represent a useful starting point for further optimization of the primary sequence for applications both in mouse and in humans.
Materials and methods
Materials. DNA aptamer GS24, its minimal version (GS24min) and mutants (DW1, DW2, DW3, DW4) and a scrambled aptamer sequence (GS24 mutant or GS24mt) were purchased from Integrated DNA Technologies (IDT, Coralville, IA) and HPLC purified.
All oligonucleotide sequences contained a 5′amino group attached by a C-6 alkyl chain. The sequence of full-length DNA aptamer (GS24) was the following: 5′-GCGTGTGCACACGGTCACTTAGTATCGCTACGTTCTTTGGTTCCGTTCGG-3′
The minimal sequence of GS24 aptamer (GS24min) was the following: 5′-CACTTAGTATCGCTACGTTCTTTGGTTCCGTTCGG-3′
The mutant sequences of GS24 were the following:
DW1 (T29A): 5′-GCGTGTGCACACGGTCACTTAGTATCGCAACGTTCTTTGGTTCCGTTCGG-3′
DW2 (T29G T47C): 5′-GCGTGTGCACACGGTCACTTAGTATCGCGACGTTCTTTGGTTCCGTCCGG-3′
DW3 (A30G T46C): 5′-GCGTGTGCACACGGTCACTTAGTATCGCTGCGTTCTTTGGTTCCGCTCGG-3′
DW4 (T29G A30G T46C T47C): 5′-GCGTGTGCACACGGTCACTTAGTATCGCGGCGTTCTTTGGTTCCGCCCGG-3′
The scrambled sequence of DNA aptamer (GS24mt) was the following: 5′-GCCATTGCCATTGCCATTGCCATTGCCATTGCCATTGCCATTGCCATTGCCATTG-3′.
Synthetic aptamer samples were dissolved in DNase free water and stored at −20 °C. Recombinant mouse and human transferrin receptor (TFRC/CD71) were purchased from Sino Biological (Beijing, China). Proteins were dissolved in 20 mmol/l Hepes-EDTA (pH 7.4). BSA and calf thymus DNA were purchased from Sigma Aldrich (St Louis, CA), Transferrin-Alexa Fluor 488 conjugate, monoclonal antibody anti-human CD71-FITC conjugate (clone T56/14), Lysotracker green and Hoechst dye were purchased from Invitrogen. All other reagents were purchased from Sigma Aldrich and used as received.
HPLC analyses. All HPLC analyses were performed on a Dionex Ultimate 3000 HPLC, equipped with automated analytical Autosampler with Fraction Collector WPS-3000FC, UltiMate 3000 Diode Array and Multiple-Wavelength Detectors, UltiMate 3000 Thermostated Column Compartment Series and controlled by Chromeleon Management Software.
Absorption and fluorescence measurements. Absorption data were recorded at 25 °C in a JASCO V550 spectrophotometer (JASCO Europe, Cremello, Italy) using 1 nm band-pass, 0.5 nm step-size and 0.25 second integration time. Appropriate quartz cuvettes (Hellma, Milan, Italy) displaying an absorption/excitation optical path of 1 cm were used in all experiments. Fluorescence intensity and anisotropy measurements were carried out with a Cary Eclipse fluorometer (Varian, Palo Alto, CA), using 1 nm step-size and 1.0 second integration time. Slits for both excitation and emission were set at 10 nm. A 100 µl quartz cuvette (Hellma, Milan, Italy) with an optical path of 1 cm was used in all experiments.
Cell culture. Mouse embryonic fibroblast cells (NIH-3T3) and human pancreatic carcinoma cells (MIA PaCA-2) were purchased from the American Type Culture Collection (ATCC, Manassas, VA). NIH-3T3 cells were grown in Dulbecco's modified Eagle Medium (DMEM) purchased from Invitrogen (Carlsbad,CA) with 10% calf serum (CS) and MIA PaCa-2 were grown in DMEM with 10% fetal bovine serum (FBS). Culture media were supplemented with 4 mmol/l L-Glutamine, 100 U/ml penicillin and 100 mg/ml streptomycin (Invitrogen). Cells were maintained at 37 °C in a humidified 5% CO2 atmosphere. For live cell microscopy cells were plated onto glass-bottom petri dishes (WillCo-dish GWSt-3522) and imaged at 37 °C, 5% CO2.
Confocal imaging of cells. Cells were imaged using a Leica TCS SP5 SMD inverted confocal microscope (Leica Microsystems AG) interfaced with a diode laser (Picoquant) for excitation at 403 nm, with an Ar laser for excitation at 488 and with a HeNe laser for excitation at 633 nm. Glass bottom Petri dishes containing cells were mounted in a thermostated chamber at 37 °C (Leica Microsystems) and viewed with a 63 × 1.2 NA water immersion objective (Leica Microsystems). The pinhole aperture was set to 1.0 Airy. All data collected were analyzed by ImageJ software version 1.44o.
Chromatographic analyses. Analytical analysis of oligonucleotide samples and purification of labeled GS24 aptamer folds (labeled A-1F and A-2F) GS24min and DW1, DW2, DW3, DW4 aptamer mutants were performed with an IE-HPLC on a Clarity 10 µm Oligo-WAX, LC Column 100 × 4.6 mm (Phenomenex) at 2.2 ml/minute, using Tris HCl 20 mmol/l pH 8.0, ACN 10% (Eluent A), and NaCl 1.2 mol/l, Tris HCl 20 mmol/l pH 8.0, ACN 10% (Eluent B) as mobile phase. Chloride gradient was 0.36–1.2 mol/l NaCl during 15 minutes. Native conditions analysis was performed at 25 °C. Analyses in denaturing condition were performed at 80 °C. In this experiment aptamer samples were heated in a thermoblock at 95 °C for 5 minutes, and then analyzed by IE-HPLC. Absorbance was measured between 200 and 650 nm.
Aptamer labeling. The amino residue at 5′-end of oligonucleotide sequences (GS24, GS24mt, truncated and mutants GS24) was conjugated to the ATTO 633 NHS fluorophore by means of standard NHS coupling procedures between the primary amine of aptamer and NHS-derivative of the fluorophore. The labeling reaction was performed as follows: 10 nmoles of aptamer sample were dissolved in sodium bicarbonate buffer 0.15 mol/l at pH 9 and mixed with 20-fold molar excess (100 nmol, 75 µl in DMSO) of ATTO 633 NHS (ATTO-TEC GmbH, Germany), in a final volume of 200 µl. The reaction mixture was stirred overnight at 4 °C. Analytical evaluation of labeling reaction and purity of the labeled aptamer samples was performed at 25 °C using a RP-HPLC on a Clarity 5 µm Oligo-RP, LC Column 250 × 4.6 mm (Phenomenex) using Triethylammonium bicarbonate (TEAB) 50 mmol/l pH 9 (Eluent A) and acetonitrile (Eluent B) at 1.0 ml/minute. Purified labeled aptamer was freeze-dried, resuspended in DNase-free water and quantified by UV-VIS. Dye-to-aptamer ratio was evaluated from absorbance measured at 260 and 633 nm and was 1 within experimental error.
Purification of labeled folds. Purification of the labeled folds was performed as follows: the folds were separated by means of IE-HPLC, and collected. Each fold was then rapidly desalted on illustra NAP-25 Columns containing Sephadex G-25 DNA grade (GE Healthcare, Piscataway, NJ) equilibrated with DNase free water. The collected fractions were freeze-dried, resuspended in appropriate volume of DNase free water and stored at −20 °C. The whole process, from separation to freeze-drying, usually lasted less than 30 minutes. Finally, an aliquot of each sample was quantified by UV-VIS analysis and analyzed in IE-HPLC to check the purity of each fold.
Dye-to-aptamer fold ratio was evaluated from absorbance measured at 260 and 633 nm and was 1 within experimental error.
Fluorescence anisotropy assay. Excitation was set at 630 nm and emission at 650 nm. Slits for both excitation and emission were set at 10 nm. A 100 µl quartz cuvette (Hellma, Milan, Italy) was used in all experiments. The G factor, i.e., the ratio of IHV to IHH, where IHV and IHH are the observed intensities at horizontal or vertical orientations of the polarizers, was determined before the first experiment and this value was used throughout the experiments in which instrumental factors (emission wavelength and spectral bandpass) were kept constant.
During fluorescence-anisotropy measurements, a highly concentrated (~15 µmol/l) solution of mouse and human TfR was used for each titration to minimize dilution effects (final volume after titration was 1.15 times the initial volume). Equilibrium binding isotherms were constructed by titrating 50 nmol/l of aptamer-ATTO 633 probe (labeled GS24, GS24min, and DW4) with increasing concentration of TfR in PBSM buffer (PBS containing 1 mmol/l MgCl2) at 25 °C. The anisotropy value was automatically calculated by the instrument. The integration time was set to 1.0 second for each anisotropy measurement, while the acquisition time for each protein concentration was 30 minutes for mouse TfR and 60 minutes for human protein. The average anisotropy-value used for data processing was calculated on at least 40 data points for each protein concentration. Equilibrium binding isotherms were evaluated also for the labeled GS24, A-1F, DW1, DW2, DW3, DW4 at [mTfR] = 600 nmol/l and for A-2F at 600 nmol/l and 1,300 nmol/l.
Data represent the average of two or more independent experiments. Error bars represent the SD from two or more independent experiments.
Fluorescence anisotropy data analysis. The aptamer fraction bound (fB) is related to the measured anisotropy by:
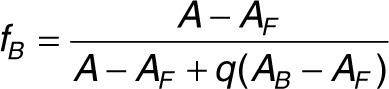 |
where A, AF, and AB are the anisotropies of the sample, free aptamer, and aptamer saturated with protein, respectively. The factor q takes into account fluorescence quantum yield changes upon binding30 and is given by:
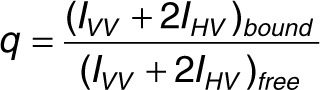 |
where IVV and IHV are the observed intensities at different orientations of the polarizers as described earlier. The value of q was 0.89 for GS24 and 0.82 both for minimal GS24 and DW4. Binding was assumed to be described by a 1:1 binding model: L + P → LP where L, P, and LP are free aptamer, TfR, and aptamer/TfR complex, respectively. The aptamer fraction bound (fB) is given by:
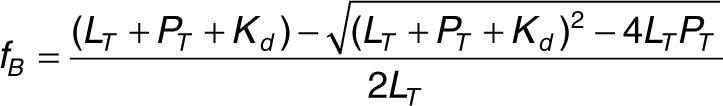 |
where LT and PT are the total aptamer and TfR concentrations, respectively.
Frequently, the aptamer anisotropy in saturating condition (AB) cannot be measured directly. In order to avoid the self-consistent evaluation of AB,31 we preferred to fit directly the anisotropy A as a function of TfR concentration. This fit directly gave both Kd and AB, and was performed using the following expression:
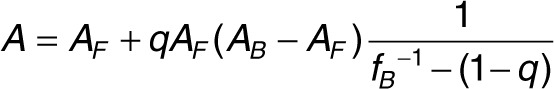 |
where fB is taken from Eq. 3. This procedure yielded both a better estimate of AB and its error. Using the experimentally determined values of A, AF, and q, the anisotropy data were fitted by nonlinear least squares regression to Eq. 4 with both Kd and AB as parameters using Origin Pro 8S0 (One Roundhouse Plaza, Northampton, MA).
Secondary and tertiary structure generation. Secondary structure prediction was obtained using the webserver Mfold19 with the default settings. Using these parameters, the two lowest free-energy structures were retained.
The Mfold generated secondary structures were used to obtain two coarse three-dimensional structures using the software RNA2D3D.32
Simulated annealing. In order to refine the two folds obtained with RNA2D3D, we performed a simulated annealing scheme imposing Watson and Crick hydrogen bond constraints.
Simulated annealing were performed using AMBER software version 12.33 The AMBER forcefield ff12SB was used. Each fold was solvated in a dodecahedral box of minimum 0.8 nm distance from the aptamer using TIP3P water model. Sodium and Chloride ions were added to reach an ionic strength of 150 mmol/l reproducing the experimental environment. The solvent was minimized using 1,000 steepest-descent steps keeping the nucleic acid structure restrained. Then the full system was minimized by 500 conjugate-gradient steps. The minimized structure was subjected to a simulated annealing procedure in the NVT ensemble. The temperature was first raised to 600 K and slowly decreased to 300 K in 50 ns. Temperature was kept constant using Langevin dynamics with the collision frequency parameter set to 2 ps−1, The SHAKE algorithm was used to constraint bonds length, allowing to use a 2 fs integration time step. Van der Waals and Coulomb interactions were truncated at 0.8 nm. Long range electrostatic interactions where calculated using the Particle-mesh Ewald (PME) summations scheme. Distance restraints were applied on the Watson and Crick hydrogen bonds (using AMBER parameters nmropt = 1 and ipnlty = 1).
Molecular dynamics simulations. The stability of two folds given as output from simulated annealing procedure was assessed by performing NPT molecular dynamics (MD) simulations without hydrogen bond distance restraints. The same parameters described for the simulated annealing were used here with the addition of the pressure coupling. Pressure was kept constant at 1.013 bar using a relaxation time of 1.0 ps. To improve conformational sampling, ten 10 ns simulations were carried out starting from different initial velocities. The ten trajectories were then combined into a single trajectory, which was used for all subsequent analysis.
Hierarchical cluster analysis was performed using the average linkage criteria.34 The root mean squared deviation between the backbone atoms was used as distance metric.
Electrostatic properties. Effective net charge was obtained by computing the electrostatic potential of 100 conformations within each cluster solving the nonlinear Poisson-Boltzmann equation using the UHBD software.35 Each electrostatic potential was radially averaged and then averaged over the ensemble of 100 conformations. The Debye-Hückel potential was fitted on the average monodimensional electrostatic potential leaving the total charge (z) as free parameter.
According to the Debye-Hückel theory of dilute electrolyte solutions, all ions in the solvent are treated as point charges while each macromolecule is treated as a sphere with diameter a (the radius of gyration was used) and net charge z. The electrostatic potential is given by:
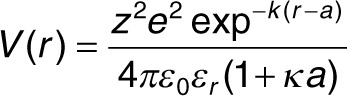 |
where e is the elementary charge, r is the distance form the center, ε0 is the vacuum permittivity, εr is the relative permittivity of the solvent and κ is the inverse of the Debye length and is proportional to the ionic strength.
Endocytosis assays in living cells. In a typical endocytosis assay in mouse fibroblasts, NIH-3T3 cells were seeded 24 hours before the experiment in WillCo dishes to reach 80–90% confluence. Standard conditions for incubation consisted in 25-minutes incubation at 37 °C, 5% CO2 in DMEM containing 1% BSA, 0.2 mg/ml calf thymus DNA, 5 µg/ml of Hoechst dye and 1 µmol/l of aptamer-ATTO 633 probe (labeled A-1F, A-2F, GS24 mt, GS24min, DW1, DW2, DW3 and DW4) in a total volume of 500 µl. After incubation, cells were washed three times with PBS, fresh serum-containing medium was added and the sample was imaged by confocal microscopy. All experiments were performed in triplicate.
Colocalization experiments in mouse fibroblasts were performed as follows:
NIH-3T3 cells were coincubated for 25 minutes at 37 °C with aptamer-ATTO 633 probe (A-2F and GS24min) and Lysotracker green to a final concentration of 1 µmol/l and 70 nmol/l, respectively. After a 20 minutes long incubation, the sample was washed three times with PBS, fresh serum-containing medium was added and the sample was imaged by confocal microscopy.
In order to verify that aptamer endocytosis was an active, receptor-mediated process and to compare the internalization kinetics of A-2F-ATTO 633 probe and transferrin, NIH-3T3 cells were left 12 hours before the experiment in DMEM with 2% serum. Cells were then kept on ice for 15 minutes, and coincubated for 20 minutes on ice with 1 µmol/l of A-2F-ATTO 633 and 0.16 µmol/l of human transferrin-Alexa Fluor-488. After incubation, cells were washed with cold PBS and fresh serum-containing medium (10% serum) preheated at 37 °C was added. Real-time monitoring of endocytosis was performed by confocal microscopy.
In internalization assays in human pancreatic carcinoma cells, MIA-PaCa-2 cells were seeded 24 hours before experiment in WillCo dishes to reach 80–90% confluence. Six hours before each experiment the complete growth medium was replaced with a 2% serum-containing medium. At time of assay, cells were incubated for 20 minutes with a medium composed of DMEM containing 1% BSA, 5 µg/ml of Hoechst dye and 3 µmol/l of aptamer-ATTO 633 probe (A-2F, GS24mt, GS24min, and DW4) in a total volume of 500 µl. After incubation, cells were washed three times with PBS then fresh 10% serum-containing medium was added and the sample was imaged by confocal microscopy.
Colocalization experiments in human tumor cells were performed as follows:
MIA PaCa-2 cells, growth in WillCo dishes, were coincubated for 60 minutes at 37 °C with A-2F-ATTO 633 (3 µmol/l) and monoclonal antibody anti-CD71-FITC conjugate following manufacturer's protocol with minor modification (1:250 diluition, Invitrogen). At the end of incubation, the cells were washed with PBS, and then fixed with 4% paraformaldehyde and 4% sucrose in PBS for 20 minutes. Fixed cells were washed with PBS and the sample was imaged by confocal microscopy. All experiments were performed in triplicate. Pearson's coefficient was calculated by using the JaCoP ImageJ plugin.
SUPPLEMENTARY MATERIAL Figure S1. HPLC analysis of aptamer folds after FA measurements. Figure S2. Binding efficiency of aptamer samples towards human transferrin receptor. Figure S3. A-2F internalization in mouse fibroblasts. Figure S4. Time-lapse imaging of transferrin and A-2F endocytosis in mouse fibroblasts. Figure S5. Endocytosis of GS24mt in human pancreatic cancer cells. Figure S6. Colocalization assay between A-2F and mAb anti-CD71 in human pancreatic cancer cells. Figure S7. Analysis of the secondary-structure predictions of DNA aptamer GS24, GS24min and RNA aptamer FB4. Figure S8. Intracellular fate of GS24min in mouse fibroblasts. Figure S9. HPLC analysis and fluorescence anisotropy assay of GS24min. Figure S10. Fluorescence anisotropy assay of DW4.
Acknowledgments
The authors thank Francesco Cardarelli for useful discussions. The authors declare no conflict of interest.
Supplementary Material
HPLC analysis of aptamer folds after FA measurements.
Binding efficiency of aptamer samples towards human transferrin receptor.
Binding efficiency of aptamer samples towards human transferrin receptor.
Binding efficiency of aptamer samples towards human transferrin receptor.
Binding efficiency of aptamer samples towards human transferrin receptor.
Binding efficiency of aptamer samples towards human transferrin receptor.
Binding efficiency of aptamer samples towards human transferrin receptor.
Binding efficiency of aptamer samples towards human transferrin receptor.
Binding efficiency of aptamer samples towards human transferrin receptor.
Binding efficiency of aptamer samples towards human transferrin receptor.
References
- Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nat Rev Drug Discov. 2010;9:537–550. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Pei W, Han Y, Jayaseelan S, Shekhtman A, Shi H, et al. One RNA aptamer sequence, two structures: a collaborating pair that inhibits AMPA receptors. Nucleic Acids Res. 2009;37:4022–4032. doi: 10.1093/nar/gkp284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang X, Kim H, Pereira MJ, Babcock HP, Walter NG, Chu S. Correlating structural dynamics and function in single ribozyme molecules. Science. 2002;296:1473–1476. doi: 10.1126/science.1069013. [DOI] [PubMed] [Google Scholar]
- Esteban JA, Walter NG, Kotzorek G, Heckman JE, Burke JM. Structural basis for heterogeneous kinetics: reengineering the hairpin ribozyme. Proc Natl Acad Sci USA. 1998;95:6091–6096. doi: 10.1073/pnas.95.11.6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin Immunol. 2006;121:144–158. doi: 10.1016/j.clim.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Daniels TR, Delgado T, Helguera G, Penichet ML. The transferrin receptor part II: targeted delivery of therapeutic agents into cancer cells. Clin Immunol. 2006;121:159–176. doi: 10.1016/j.clim.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Roberts RL, Fine RE, Sandra A. Receptor-mediated endocytosis of transferrin at the blood-brain barrier. J Cell Sci. 1993;104 (Pt 2):521–532. doi: 10.1242/jcs.104.2.521. [DOI] [PubMed] [Google Scholar]
- Högemann-Savellano D, Bos E, Blondet C, Sato F, Abe T, Josephson L, et al. The transferrin receptor: a potential molecular imaging marker for human cancer. Neoplasia. 2003;5:495–506. doi: 10.1016/s1476-5586(03)80034-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian ZM, Li H, Sun H, Ho K. Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol Rev. 2002;54:561–587. doi: 10.1124/pr.54.4.561. [DOI] [PubMed] [Google Scholar]
- Li H, Qian ZM. Transferrin/transferrin receptor-mediated drug delivery. Med Res Rev. 2002;22:225–250. doi: 10.1002/med.10008. [DOI] [PubMed] [Google Scholar]
- Widera A, Norouziyan F, Shen WC. Mechanisms of TfR-mediated transcytosis and sorting in epithelial cells and applications toward drug delivery. Adv Drug Deliv Rev. 2003;55:1439–1466. doi: 10.1016/j.addr.2003.07.004. [DOI] [PubMed] [Google Scholar]
- Wilner SE, Wengerter B, Maier K, de Lourdes Borba Magalhães M, Del Amo DS, Pai S, et al. An RNA alternative to human transferrin: a new tool for targeting human cells. Mol Ther Nucleic Acids. 2012;1:e21. doi: 10.1038/mtna.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, Dellamaggiore KR, Ouellette CP, Sedano CD, Lizadjohry M, Chernis GA, et al. Aptamer-based endocytosis of a lysosomal enzyme. Proc Natl Acad Sci USA. 2008;105:15908–15913. doi: 10.1073/pnas.0808360105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MZ, Yu RN, Chen J, Ma ZY, Zhao YD. Targeted quantum dots fluorescence probes functionalized with aptamer and peptide for transferrin receptor on tumor cells. Nanotechnology. 2012;23:485104. doi: 10.1088/0957-4484/23/48/485104. [DOI] [PubMed] [Google Scholar]
- Thayer JR, Wu Y, Hansen E, Angelino MD, Rao S. Separation of oligonucleotide phosphorothioate diastereoisomers by pellicular anion-exchange chromatography. J Chromatogr A. 2011;1218:802–808. doi: 10.1016/j.chroma.2010.12.051. [DOI] [PubMed] [Google Scholar]
- Buncek M, Backovská V, Holasová S, Radilová H, Safárová M, Kunc F, et al. Unusual chromatographic behavior of oligonucleotide sequence isomers on two different anion exchange HPLC columns. Anal Biochem. 2006;348:300–306. doi: 10.1016/j.ab.2005.10.047. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potty AS, Kourentzi K, Fang H, Schuck P, Willson RC. Biophysical characterization of DNA and RNA aptamer interactions with hen egg lysozyme. Int J Biol Macromol. 2011;48:392–397. doi: 10.1016/j.ijbiomac.2010.12.007. [DOI] [PubMed] [Google Scholar]
- Green LS, Jellinek D, Jenison R, Ostman A, Heldin CH, Janjic N. Inhibitory DNA ligands to platelet-derived growth factor B-chain. Biochemistry. 1996;35:14413–14424. doi: 10.1021/bi961544+. [DOI] [PubMed] [Google Scholar]
- Kusano T, Steinmetz D, Hendrickson WG, Murchie J, King M, Benson A, et al. Direct evidence for specific binding of the replicative origin of the Escherichia coli chromosome to the membrane. J Bacteriol. 1984;158:313–316. doi: 10.1128/jb.158.1.313-316.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le NC, Gubala V, Gandhiraman RP, Daniels S, Williams DE. Evaluation of different nonspecific binding blocking agents deposited inside poly(methyl methacrylate) microfluidic flow-cells. Langmuir. 2011;27:9043–9051. doi: 10.1021/la2011502. [DOI] [PubMed] [Google Scholar]
- Ng PP, Dela Cruz JS, Sorour DN, Stinebaugh JM, Shin SU, Shin DS, et al. An anti-transferrin receptor-avidin fusion protein exhibits both strong proapoptotic activity and the ability to deliver various molecules into cancer cells. Proc Natl Acad Sci USA. 2002;99:10706–10711. doi: 10.1073/pnas.162362999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid SL, Carter LL. ATP is required for receptor-mediated endocytosis in intact cells. J Cell Biol. 1990;111 6 Pt 1:2307–2318. doi: 10.1083/jcb.111.6.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JH, Kushner JP, Kaplan J. Transferrin receptors of human fibroblasts. Analysis of receptor properties and regulation. Biochem J. 1982;208:19–26. doi: 10.1042/bj2080019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vittorio O, Cirillo G, Iemma F, Di Turi G, Jacchetti E, Curcio M, et al. Dextran-catechin conjugate: a potential treatment against the pancreatic ductal adenocarcinoma. Pharm Res. 2012;29:2601–2614. doi: 10.1007/s11095-012-0790-9. [DOI] [PubMed] [Google Scholar]
- Ryschich E, Huszty G, Knaebel HP, Hartel M, Büchler MW, Schmidt J. Transferrin receptor is a marker of malignant phenotype in human pancreatic cancer and in neuroendocrine carcinoma of the pancreas. Eur J Cancer. 2004;40:1418–1422. doi: 10.1016/j.ejca.2004.01.036. [DOI] [PubMed] [Google Scholar]
- Vento MT, Iuorio M, Netti PA, Ducongè F, Tavitian B, Franciscis V, et al. Distribution and bioactivity of the Ret-specific D4 aptamer in three-dimensional collagen gel cultures. Mol Cancer Ther. 2008;7:3381–3388. doi: 10.1158/1535-7163.MCT-08-0580. [DOI] [PubMed] [Google Scholar]
- Lakowicz JR. Principles of Fluorescence Spectroscopy. 2006. pp. 353–381.
- Tetin SY, Hazlett TL. Optical spectroscopy in studies of antibody-hapten interactions. Methods. 2000;20:341–361. doi: 10.1006/meth.1999.0927. [DOI] [PubMed] [Google Scholar]
- Martinez HM, Maizel JV, Jr, Shapiro BA. RNA2D3D: a program for generating, viewing, and comparing 3-dimensional models of RNA. J Biomol Struct Dyn. 2008;25:669–683. doi: 10.1080/07391102.2008.10531240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, Darden TA, Cheatham TE, 3rd, Simmerling CL, Wang J, Duke RE, et al. 2012AMBER 12 < http://ambermd.org/ >.
- Rencher AC.2002Methods of Multivariate Analysis. Wiley-Interscience: Hoboken; IIE Transactions 37. [Google Scholar]
- Madura JD, Briggs JM, Wade RC, Davis ME, Luty B, Ilin A, et al. Electrostatics and diffusion of molecules in solution: simulations with the University of Houston Brownian Dynamics program. Comp Phys Comm. 1995;91:57–95. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
HPLC analysis of aptamer folds after FA measurements.
Binding efficiency of aptamer samples towards human transferrin receptor.
Binding efficiency of aptamer samples towards human transferrin receptor.
Binding efficiency of aptamer samples towards human transferrin receptor.
Binding efficiency of aptamer samples towards human transferrin receptor.
Binding efficiency of aptamer samples towards human transferrin receptor.
Binding efficiency of aptamer samples towards human transferrin receptor.
Binding efficiency of aptamer samples towards human transferrin receptor.
Binding efficiency of aptamer samples towards human transferrin receptor.
Binding efficiency of aptamer samples towards human transferrin receptor.