

Abstract
Context:
Aberrant DNA methylation is known to be a major factor in oncogenesis and cancer progression, but effects of methylation in papillary thyroid cancer (PTC) are not well defined.
Objective:
The objective of the study was to identify altered methylation patterns, which may be associated with PTC disease behavior.
Design:
This study was a genome-wide methylation analysis of PTC.
Setting:
The study was conducted at the National Institutes of Health Clinical Center.
Patients:
PTC tissue from 51 patients were analyzed and compared with normal thyroid tissue from seven patients.
Interventions:
CpG methylation status was assessed using advanced genome-wide methylation bead chips.
Outcome Measures:
Altered methylation patterns in PTC were analyzed by stage, recurrence, histological subtype of tumor, and tumor genotype.
Results:
PTC is globally hypomethylated compared with normal thyroid with 2837 differentially methylated CpG sites. The follicular variant of PTC demonstrated less differential methylation with only 569 differentially methylated CpG sites. Tumors with mutations in BRAF, RET/PTC, and RAS demonstrated a 3.6-fold increase in the number of differentially methylated sites compared with wild-type tumors. The differentially methylated genes were associated with oncological pathways including cellular movement, growth, and proliferation.
Conclusion:
PTC is epigenetically distinct from the follicular variant of PTC and by gene mutation status (BRAF, RET/PTC, and RAS).
Thyroid cancer is the most common endocrine malignancy with an increasing incidence over the last 3 decades (1, 2). Papillary thyroid cancer (PTC) accounts for approximately 80% of all thyroid cancer cases (3). There have been consistent and largely mutually exclusive genetic alterations associated with PTC (4). Activating somatic mutations involving the MAPK pathway, most commonly involving BRAF, RAS, or RET/PTC rearrangements, are present in three fourths of PTC (5). These driver mutations have been associated with aberrant methylation in several genes involved in thyroid cancer differentiation and progression, including NIS, TSHR, RARβ2, and RASSF1A (6, 7). Gene specific methylation studies have also identified several other genes that are differentially methylated in PTC but are not associated with the common PTC-related somatic mutations, including ECAD, ATM, and DAPK (8).
The mechanism by which aberrant methylation can alter cancer phenotype is well described (9–11). Methylation patterns are heritable, and CpG island hypermethylation in the promoter region of a gene is a well-described mechanism of gene silencing in cancer (12, 13). Hypermethylation in other locations, such as the first exon of a gene, may also lead to silencing of gene expression (14, 15). Additionally, global hypomethylation is well described in human cancers and is thought to predispose to chromosomal double-strand breaks and rearrangements (16, 17).
An altered methylome can have overarching effects on disease phenotype and progression, as originally demonstrated in colorectal cancer by Toyota et al in 1999 (18). The identification of altered genome-wide methylation patterns have been shown to have potential diagnostic and prognostic applications in several cancers, including adrenocortical cancer, prostate cancer, and myeloproliferative neoplasms (19–22). Although there have been gene- or pathway-specific methylation analysis among different subtypes of thyroid cancer, to date there has not been a comprehensive genome-wide methylome study in PTC (23). An unbiased, genome-wide methylation analysis of PTC could provide important insights into the pathobiology of PTC with possible applications for improving diagnosis and prognostication of PTC and in identifying new therapeutic targets. Thus, the purpose of this study was to identify altered methylation patterns, which may be associated with PTC genetics and behavior.
Materials and Methods
Patients and tissue procurement
Thyroid samples were obtained at the time of surgical resection, snap frozen, and immediately stored at −80°C. All patient data collection was performed prospectively after written informed consent on an institutional review board-approved protocol. Patient information was prospectively collected with annual follow-up. Tissue sections adjacent to those used for nucleic acid isolation were stained using hematoxylin-eosin to confirm the diagnosis and tumor cell content greater than 50%. All tissues included in the study underwent secondary histological review by a single pathologist (M.J.M.). Tissues were classified as PTC when there was classic papillary architecture and histological features of PTC in the sample and follicular variant of papillary thyroid cancer (FVPTC) when prominent follicular structure was present within the tumor architecture. Normal thyroid tissues were obtained at thyroidectomy for noncancer indications or from the contralateral lobe of a patient with thyroid cancer. Normal samples were also rereviewed to ensure that no microcarcinoma or lymphocytic infiltrate was present in the tissue. Extent of disease at diagnosis was assessed using the tumor node metastasis system and staged according to the American Joint Committee on Cancer system (24). Forty-three cancer samples were tested for mutations in exon 15 of BRAF, RET/PTC1, and RET/PTC3 rearrangements, and hot spot mutations in N-RAS, H-RAS, and K-RAS by direct Sanger sequencing and nested PCR, as previously described (25).
DNA extraction and methylation analysis
Frozen thyroid tissue was sectioned and DNA was extracted using the DNeasy blood and tissue kit (QIAGEN) according to the manufacturer's protocol. DNA quality was determined using a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific Inc) and quantified using the Quant-iT PicoGreen double-stranded DNA assay (Life Technologies). One microgram of DNA was bisulfate converted using the EZ DNA methylation-gold kit (Zymo Research Corp) with the manufacturer's thermocycle protocol modified (16 cycles of 95°C for 30 sec, 50°C for 60 min) as recommended by Illumina.
Six hundred nanograms of converted DNA were assayed on Infinium Human Methylation 450 BeadChips using the Illumina Infinium HD methylation kit (Illumina). Each sample underwent overnight isothermal whole-genome amplification and was then fragmented, precipitated, and resuspended. Hybridization occurred overnight at 48°C in an Illumina hybridization oven. Using a Tecan Evo robot protocol (Tecan Group Ltd), hybridized arrays were processed through a single-base extension reaction on the probe sequence using DNP-or biotin-labeled nucleotides and immunostained. BeadChips were then coated, dried, and imaged with an Illumina HiScanSQ. Images were extracted using the Genome Studio version 2010.3 methylation module (Illumina). β-Values were calculated at each locus (β = (intensity of methylated allele/intensity of unmethylated allele + intensity of methylated allele) × 100), and the final report file was exported from Genome Studio for further data analysis. Methylation data from the X and Y chromosomes were excluded from the results. Quality control inclusion depended on hybridization detection values of P < .05. All samples met quality control (QC) standards and were included in the analysis.
Data interpretation and statistical analysis
Methylation analysis and data QC were performed using R package (lumi and methylumi), perl scripts, and Partek software (Partek Inc). Comparison of different tissue groups and driver mutations were analyzed using ANOVA based on M values converted from corresponding β-values at each locus. The Benjamini-Hochberg method was used to adjust P values for multiple comparisons and minimum absolute β-value difference cutoff of β = .2 was used for generating the final target list.
Pathway and functional analysis was performed using Ingenuity Pathway Analysis software (Ingenuity Systems Inc). CpG sites were considered to be significantly differentially expressed when the adjusted value was P < .05.
Results
Study cohort characteristics
Tissue analysis included eight normal and 51 PTC tissue samples. Among PTC samples, 29 were classical PTC primary tumors, 15 were FVPTC primary tumors, and seven were recurrent tumors of classical PTC. Recurrent tumors were not from the same patients as primary PTC tumor samples. Genetic testing revealed BRAF mutations in 21 samples, RET/PTC3 rearrangements in six samples, and RAS mutation in three samples. Three samples had multiple mutations (two BRAF and RET/PTC3 and one BRAF, RET/PTC3, and RAS). Detailed clinical characteristics of the study cohort are shown in Table 1.
Table 1.
Clinical and Genetic Characteristics of Study Cohort
| Variable | n (%) |
|---|---|
| Gender (Female:Male) | |
| PTC | 40:11 (21.6% M) |
| Normal thyroid | 7:1 (12.5% M) |
| Age at surgery, y (mean ± SD) | |
| PTC | 42.7 ± 18.2 |
| Range | 14–86 |
| Normal thyroid | 39.9 ± 12.9 |
| Range | 20–62 |
| Disease characteristics | |
| Stage 1 | 28 (54.9%) |
| Stage 2 | 2 (3.9%) |
| Stage 3 | 5 (9.8%) |
| Stage 4 | 6 (11.8%) |
| Recurrent classical PTC | 7 (13.7%) |
| Classical papillary histology | 29 (56.9%) |
| Follicular variant histology | 15 (29.4%) |
| BRAF mutation | 21 (41.2%) |
| RAS mutation | 3 (5.9%) |
| RET/PTC rearrangement | 6 (11.8%) |
| Multiple mutations | 3 (5.9%) |
| No mutation identified | 25 (49.0%) |
| Follow-up and clinical outcomesa | |
| Median follow-up, mo | 71 |
| Follow-up range, mo | 5–120 |
| Patients developing recurrent disease | 10 (19.8%) |
| Patients with disease-related morality | 7 (13.7%) |
After the primary operation.
Methylome of PTC is distinct from normal thyroid tissue
Methylation array data met QC standards for all samples. Using an adjusted P ≤ .05 and β-value differences (δβ) of at least 20% (δβ < −0.20 or δβ > +0.20), PTC was globally hypomethylated compared with normal thyroid (Figure 1). There were 2837 differentially methylated CpG sites between PTC and normal tissue, of which 2585 were hypomethylated and 252 were hypermethylated relative to normal thyroid.
Figure 1.
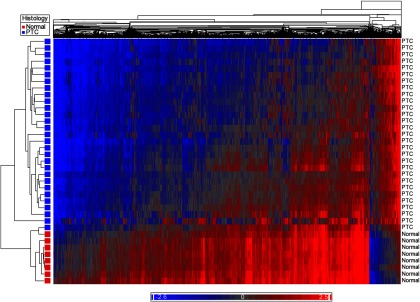
Heat map demonstrating global hypomethylation of PTC compared with normal thyroid tissue. Blue represents hypomethylated loci, and red represents hypermethylation.
Genome-wide methylation pattern is distinct based on histological subtype
When comparing the methylome based on histology, FVPTC had the fewest methylation differences when compared with normal tissue (569 differentially methylated sites; 164 hypermethylated and 405 hypomethylated). Primary classic PTC tumors demonstrated more aberrant methylation compared with normal tissue (2837 differentially methylated sites), 91% of which were hypomethylated. The methylome of recurrent classic PTC tumors was most different from normal tissue, with 3819 differentially methylated sites (73% hypomethylated, 27% hypermethylated). There were fewer significant differentially methylated sites when cancer subtypes were compared directly. The comparison of FVPTC and primary PTC showed 330 differentially methylated sites (315 hypermethylated, 15 hypomethylated). There were no significant differentially methylated CpG sites between recurrent and primary PTC when compared directly. A summary of aberrant methylation by histological subtype can be found in Supplemental Table 1, published on The Endocrine Society's Journals Online web site at http://jcem.endojournals.org. Unsupervised principal component analysis demonstrated that primary and recurrent PTC samples clustered closely with modest variation. Normal samples clustered separately from the PTC samples. FVPTC had greater variability (Figure 2A).
Figure 2.

A, Principal component analysis of normal, FVPTC, primary PTC, and recurrent PTC, using ANOVA and adjusted P ≤ .01. Primary and recurrent PTC samples cluster separately from normal samples. B, Distribution of differentially methylated CpG sites between promoter, gene body, 3′UTR, and intergenic regions by tumor histology. C, Distribution of differentially methylated sites relative to CpG islands by histology. RPTC, recurrent PTC.
Methylation changes can be found throughout the genome, including in gene promoter regions, gene bodies, and untranscribed intergenic regions. The distribution of differentially methylated sites between promoters, gene bodies, 3′-untranslated regions (UTRs), and untranscribed regions was similar between histological subtypes (Figure 2B). Although there were fewer differentially methylated sites in FVPTC overall, FVPTC demonstrated a higher percentage of differentially methylated sites in CpG islands (9.3%) compared with classic PTC (3.1%) and recurrent PTC (6.7%) (Figure 2C).
Tumors with common somatic mutations (BRAF, RET/PTC, and RAS) in PTC have an altered methylome
Methylation analysis based on driver somatic mutation in PTC also revealed differential methylation based on tumor genetics. Cancers with RAS and BRAF mutations demonstrated the largest number of differentially methylated sites. Tumors with RAS mutations were differentially methylated at 7892 sites (4008 hypermethylated, 3884 hypomethylated), whereas tumors with BRAF mutations were differentially methylated at 4068 sites (464 hypermethylated, 3604 hypomethylated). Wild-type (WT) tumors and those with RET/PTC rearrangements had fewer differentially methylated sites when compared with normal thyroid (WT: 826 sites, 154 hypermethylated, 672 hypomethylated; RET/PTC: 973 total, 263 hypermethylated, 710 hypomethylated). Similarly when all mutations were grouped together and compared with normal thyroid, there were 3003 differentially methylated sites observed in tumors (368 hypermethylated, 2635 hypomethylated). A summary of differential methylation by genotypes can be found in Table 2. The methylation difference between the WT tumors and those with mutations in the genes involved in the MAPK pathway is demonstrated in Figure 3, A and B.
Table 2.
Differentially Methylated CpG Loci by Histological Subtype and Driver Mutation
| Comparisona | Hypermethylated Loci | Hypomethylated Loci | Total Significant Loci |
|---|---|---|---|
| Follicular variant PTC | 164 | 405 | 569 |
| Classic PTC | 255 | 2582 | 2837 |
| Recurrent classic PTC | 1023 | 2796 | 3819 |
| BRAF mutant | 464 | 3604 | 4068 |
| RAS mutant | 4008 | 3884 | 7892 |
| RET/PTC rearrangement | 263 | 710 | 973 |
| All mutants (BRAF, RAS, and RET/PTC) | 368 | 2635 | 3003 |
| WT classical PTC | 154 | 672 | 826 |
Comparison with normal thyroid tissue.
Figure 3.


Heat maps comparing methylome of normal thyroid with MAPK mutant tumors (A) and MAPK WT tumors (B). Blue represents hypomethylated loci, and red represents hypermethylation. C, Distribution of differentially methylated CpG sites between promoter, gene body, 3′UTR, and intergenic regions based on driver mutation status. D, Distribution of differentially methylated sites relative to CpG islands based on driver mutation status. E, Heat map demonstrating differential methylation between MAPK mutant and MAPK WT tumor samples.
When analyzed by driver mutation status, RAS mutants had the highest percentage of differentially methylated CpG sites in promoter regions (30.8%), whereas WT tumors had the lowest percentage within promoters (22.7%) (Figure 3C). Tumors with RAS mutations also had the highest percentage of differentially methylated sites located in CpG islands (14.9%) when compared with BRAF (4.3%), RET/PTC (7.5%), and WT tumors (3.0%) (Figure 3D).
When comparing all mutant vs WT PTC samples, there were 2260 differentially methylated CpG sites. Of these CpG sites, 1993 (88.2%) were hypomethylated in mutant tumors, whereas 267 (11.8%) were hypermethylated. Analysis of differential methylation status by genomic site revealed that 25.3% of the CpG sites were within promoters, 39.1% within gene bodies, 3.4% in 3′UTR regions, and 32.3% in intergenic regions. When analyzed by distance from CpG islands, 4.3% of CpG sites were in islands, 18.2% in shores, 11.4% in shelves, and 66.1% were away from CpG islands. The methylation distribution is demonstrated in Figure 3E.
No difference in methylation patterns by cancer stage and recurrence
Primary tumor samples were also stratified based on cancer stage and recurrence status. Using adjusted P ≤ .05 and δβ of at least 20%, there were no significant differentially methylated sites by cancer stage (stages 1 and 2 vs stages 3 and 4). There were also no sites demonstrating significantly different methylation in the primary tumor by recurrence.
Methylation status of genes known to be aberrantly methylated
The methylation status of five genes (ECAD, DAPK, NIS, TIMP3, and TSHR) that had been previously identified to be hypermethylated in PTC was also analyzed (6, 8, 26). Hypermethylation was observed in 31.4% of PTC in ECAD, 56.9% of PTC in DAPK, 31.4% of PTC in NIS, and 35.3% of PTC in TIMP3. These rates of hypermethylation are similar to those described previously in the literature. No hypermethylation was observed in TSHR.
Biological functions associated with aberrant methylation
Ingenuity pathway analysis was used to assess the biologic function of the differentially methylated genes based on histology and driver mutation. This analysis focused on aberrantly methylated genes in all PTC samples as well as sites unique to BRAF, RET/PTC, and RAS mutant samples. Cancer was the most significant disease category associated with aberrant PTC methylation, with significant molecular functions including cellular movement, growth, and proliferation. A summary of the affected cellular functions, predicted regulators, and disease relationships is summarized in Tables 3 and 4.
Table 3.
Genes Differentially Methylated in PTC and Pathway Analysis Results: Diseases and Disorders Associated With Differentially Methylated Genes
| All PTCsa |
Mutant (BRAF, RAS, and RET/PTC) Onlyb |
||
|---|---|---|---|
| Disease | P Value | Disease | P Value |
| Cancer | 1.43 × 10−18 | Cancer | 1.19 × 10−12 |
| Cardiovascular | 1.27 × 10−14 | Inflammation | 1.49 × 10−9 |
| Respiratory | 1.55 × 10−12 | Cardiovascular | 4.78 × 10−9 |
All classic PTC samples were compared with normal thyroid.
All samples with BRAF and RAS mutations (and RET/PTC rearrangements) were compared with normal thyroid.
Table 4.
Genes Differentially Methylated in PTC and Pathway Analysis Results: Molecular and Cellular Functions Associated With Differentially Methylated Genes
| All PTCs |
Mutant (BRAF, RAS, and RET/PTC) Only |
||
|---|---|---|---|
| Function | P Value | Function | P Value |
| Cellular movement | 5.37 × 10−25 | Cellular movement | 8.79 × 10−18 |
| Cellular growth and proliferation | 7.85 × 10−15 | Cellular growth and proliferation | 1.28 × 10−11 |
| Cellular assembly and organization | 4.37 × 10−13 | Cellular morphology | 1.24 × 10−10 |
Discussion
In this study, we analyzed the genome-wide methylation pattern of PTC based on histological subtype, genetic mutation, and clinical course. PTC was found to be globally hypomethylated compared with normal thyroid tissue, whereas FVPTC demonstrated a variable methylome not dramatically different from normal thyroid tissue samples. Tumor samples with a somatic mutation in BRAF, RAS, and RET/PTC rearrangements demonstrated more aberrant methylation than WT tumors. There was no significant difference in global methylation pattern based on cancer stage or PTC recurrence. Differentially methylated genes clustered into molecular pathways known to affect cancer development, with predicted effects on cellular movement, growth, proliferation, and morphology. To our knowledge, this study represents the most comprehensive analysis of the PTC methylome to date.
In our findings, the differential methylation in the setting of somatic mutation in BRAF, RAS, and RET/PTC rearrangements is most striking. Aberrant methylation, either hypomethylation or hypermethylation, was identified at 3003 sites in mutants compared with only 826 sites in WT tumors, a 3.64-fold increase. This finding is particularly interesting when considering that several of the differentially methylated genes identified previously were associated with BRAF mutation (6, 26). The down-regulation of these genes, specifically NIS, RARβ2, and TIMP3, is involved in tumor dedifferentiation and metastasis, and dysregulation of these genes is thought to be a major event in PTC progression. The role of differential methylation in these events, and its association with BRAF mutation status, is especially important in light of growing evidence that tumors with BRAF mutation are associated with more aggressive PTC (7, 27, 28).
These genome-wide data indicate that differential methylation in BRAF mutants may not be the result of targeted effects but rather are secondary to generalized dysregulation of the epigenetic monitoring systems in the setting of activating mutations known to affect the MAPK pathway. Although MAPK activation was not specifically tested for in these tumor samples, tumors with mutations known to activate MAPK were found to be more globally hypomethylated than WT tumors (2635 vs 672 sites, 3.92-fold increase), which is consistent with previous in vitro data (29). This generally dysregulated methylation phenotype could account for a substantial portion of the more aggressive characteristics of BRAF mutant tumors.
The correlation between somatic mutation in BRAF, RAS, and RET/PTC rearrangements and aberrant methylation could have clinical implications as modulation of abnormal hypermethylation is possible pharmacologically (30, 31). This has been explored previously in PTC using the demethylating agent 5-aza-2′deoxycytidine as well as histone deacetylase inhibitors. These studies have demonstrated efficacy in correcting epigenetic dysregulation in PTC, with applications focused on reversal of dedifferentiation to improve response to radioiodine therapy, although toxicity has been a limiting factor (32, 33). Furthermore, use of drugs that target pathways activated by mutations in BRAF, RAS, and RET/PTC rearrangements have been shown to induce redifferentiation in preclinical models and in isolated patient cases (34, 35). These data provide a possible mechanism by which these drugs may reverse thyroid cancer dedifferentiation.
These data demonstrate that the genome-wide methylation pattern is not different by extent of disease (stages 1 and 2 vs stage 3 and 4) or between primary vs recurrent PTC. However, these findings could miss subtle differences that would require a much larger sample set to achieve statistical significance. Only 11 patients had advanced disease (stages 3 or 4), and only 10 experienced recurrence during follow-up. It is also intuitive that if PTC is monoclonal that the epigenetic differences between primary and recurrent tumors would be less stark than differences observed based on histology or the driver mutation. Nonetheless, the potential link between epigenetic abnormalities and PTC aggressiveness should still be further explored in a larger study cohort.
The finding of differential methylation in PTC compared with FVPTC is also surprising. FVPTC has traditionally been regarded as an isolated histological finding and little to no treatment modifications are made compared with classic PTC. Recently, however, there is emerging evidence that it is a distinct disease with clinical outcomes between follicular thyroid cancer and PTC (36). Several molecular and genetic associations have been described in FVPTC, including the presence of higher rates of RAS mutations and lower rates of BRAF mutations and a distinct microRNA profile compared with PTC (37–39). This study contributes to the growing molecular and clinical evidence that FVPTC is a biologically distinct entity based on methylation pattern.
The data from this study must also be contrasted with those reported by Rodriguez-Rodero et al (23), which demonstrated much lower overall differences in methylation between histological types of thyroid cancer. There are two main reasons for this difference. First, in the study by Rodriquez-Roder et al, only eight thyroid cancer samples (two PTC) were compared with two normal thyroid samples and four thyroid cancer cell lines. Our study has a much larger sample sizes and is thus powered to find more variations in the methylome. Second, the platform used in our study assessed nearly 500 000 independent CpG sites in the methylome, compared with 27 578 in the platform used by Rodriquez-Rodero and associates. Thus, the combination of a larger sample size and a more comprehensive interrogation of the methylome most likely explain the differences in the reported data.
There are some limitations to this study. First, no studies were performed to validate activation of the MAPK pathway in tumors with mutations in genes that regulate the MAPK pathway, and thus, our results show an association only between the presence of gene mutations and methylation status. Furthermore, these data do not demonstrate whether mutation in BRAF, RAS, and RET/PTC rearrangements cause a deranged methylome or vice versa. Second, integration of the methylation data presented with gene expression data was not performed. Although this study is descriptive only of the PTC methylome, we believe these data facilitate future integrated methylation expression analyses to further define loci whose expression is regulated by the described aberrant methylome. Such an analysis should also integrate data on driver mutation status in the same tumor sample to further elucidate the relationship between mutations and the PTC methylome. Finally, although the number of tumor samples analyzed is the largest to be studied for genome-wide methylation changes in PTC, the sample size may still be too limited to detect small methylation changes.
In conclusion, our data represent the most robust genome-wide characterization of epigenetic alterations in PTC. Like other cancers, PTC is characterized by global hypomethylation compared with normal thyroid tissue, although approximately 8.9% of differentially methylated CpG sites were hypermethylated compared with normal tissue. Mutations in BRAF and RAS and RET/PTC rearrangements were associated with a markedly different methylome, with 3.64-fold more differentially methylated sites observed as compared with WT tumors, whereas FVPTC was more similar to normal thyroid. Pathway analysis revealed that differentially methylated sites were involved in known oncologic pathways and were associated with dysregulated cellular movement, growth, and proliferation. These findings greatly enhance our understanding of PTC biology and may provide insights into mechanisms by which BRAF mutant tumors become more aggressive than other WT PTC.
Acknowledgments
This work was supported by the Intramural Research Program at the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- FVPTC
- follicular variant of PTC
- PTC
- papillary thyroid cancer
- QC
- quality control
- UTR
- untranslated region
- WT
- wild type.
References
- 1. Chen AY, Jemal A, Ward EM. Increasing incidence of differentiated thyroid cancer in the United States, 1988–2005. Cancer. 2009;115(16):3801–3807 [DOI] [PubMed] [Google Scholar]
- 2. Jung KW, Park S, Kong HJ, et al. Cancer statistics in Korea: incidence, mortality and survival in 2006–2007. J Korean Med Sci. 2010;25(8):1113–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Romei C, Elisei R. RET/PTC translocations and clinico-pathological features in human papillary thyroid carcinoma. Front Endocrinol (Lausanne). 2012;3:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pacini F, Castagna MG. Approach to and treatment of differentiated thyroid carcinoma. Med Clin North Am. 2012;96(2):369–383 [DOI] [PubMed] [Google Scholar]
- 5. Li X, Abdel-Mageed AB, Kandil E. BRAF mutation in papillary thyroid carcinoma. Int J Clin Exp Med. 2012;5(4):310–315 [PMC free article] [PubMed] [Google Scholar]
- 6. Hu S, Liu D, Tufano RP, et al. Association of aberrant methylation of tumor suppressor genes with tumor aggressiveness and BRAF mutation in papillary thyroid cancer. Int J Cancer. 2006;119(10):2322–2329 [DOI] [PubMed] [Google Scholar]
- 7. Xing M, Alzahrani AS, Carson KA, et al. Association between BRAF V600E mutation and mortality in patients with papillary thyroid cancer. JAMA. 2013;309(14):1493–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eze OP, Starker LF, Carling T. The role of epigenetic alterations in papillary thyroid carcinogenesis. J Thyroid Res. 2011;2011:895470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res. 1998;72:141–196 [PubMed] [Google Scholar]
- 10. Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 2000;16(4):168–174 [DOI] [PubMed] [Google Scholar]
- 11. Clark SJ, Melki J. DNA methylation and gene silencing in cancer: which is the guilty party? Oncogene. 2002;21(35):5380–5387 [DOI] [PubMed] [Google Scholar]
- 12. Bird AP, Wolffe AP. Methylation-induced repression—belts, braces, and chromatin. Cell. 1999;99(5):451–454 [DOI] [PubMed] [Google Scholar]
- 13. Burri N, Shaw P, Bouzourene H, et al. Methylation silencing and mutations of the p14ARF and p16INK4a genes in colon cancer. Lab Invest. 2001;81(2):217–229 [DOI] [PubMed] [Google Scholar]
- 14. Wong L, Zhou J, Anderson D, Kratzke RA. Inactivation of p16INK4a expression in malignant mesothelioma by methylation. Lung Cancer. 2002;38(2):131–136 [DOI] [PubMed] [Google Scholar]
- 15. Brenet F, Moh M, Funk P, et al. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS One. 2011;6(1):e14524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene. 2002;21(35):5400–5413 [DOI] [PubMed] [Google Scholar]
- 17. Lin CH, Hsieh SY, Sheen IS, et al. Genome-wide hypomethylation in hepatocellular carcinogenesis. Cancer Res. 2001;61(10):4238–4243 [PubMed] [Google Scholar]
- 18. Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA. 1999;96(15):8681–8686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rechache NS, Wang Y, Stevenson HS, et al. DNA methylation profiling identifies global methylation differences and markers of adrenocortical tumors. J Clin Endocrinol Metab. 2012;97(6):E1004–E1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luo JH, Ding Y, Chen R, et al. Genome-wide methylation analysis of prostate tissues reveals global methylation patterns of prostate cancer. Am J Pathol. 2013;182(6):2028–2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nischal S, Bhattacharyya S, Christopeit M, et al. Methylome profiling reveals distinct alterations in phenotypic and mutational subgroups of myeloproliferative neoplasms. Cancer Res. 2013;73(3):1076–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kaiser MF, Johnson DC, Wu P, et al. Global methylation analysis identifies prognostically important epigenetically inactivated tumour suppressor genes in multiple myeloma. Blood. 2013;122(2):219–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rodriguez-Rodero S, Fernandez AF, Fernandez-Morera JL, et al. DNA methylation signatures identify biologically distinct thyroid cancer subtypes. J Clin Endocrinol Metab. 2013;98(7):2811–2821 [DOI] [PubMed] [Google Scholar]
- 24. Edge SB, American Joint Committee on Cancer et al. AJCC Cancer Staging Manual. 7th ed New York: Springer; 2010:xiv. [DOI] [PubMed] [Google Scholar]
- 25. Mathur A, Moses W, Rahbari R, et al. Higher rate of BRAF mutation in papillary thyroid cancer over time: a single-institution study. Cancer. 2011;117(19):4390–4395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xing M. Gene methylation in thyroid tumorigenesis. Endocrinology. 2007;148(3):948–953 [DOI] [PubMed] [Google Scholar]
- 27. Wang W, Zhao W, Wang H, et al. Poorer prognosis and higher prevalence of BRAF (V600E) mutation in synchronous bilateral papillary thyroid carcinoma. Ann Surg Oncol. 2012;19(1):31–36 [DOI] [PubMed] [Google Scholar]
- 28. O'Neill CJ, Bullock M, Chou A, et al. BRAF(V600E) mutation is associated with an increased risk of nodal recurrence requiring reoperative surgery in patients with papillary thyroid cancer. Surgery. 2010;148(6):1139–1145; discussion 1145–1146 [DOI] [PubMed] [Google Scholar]
- 29. Hou P, Liu D, Xing M. Endocr Relat Cancer. 2011;18(6):687–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vivaldi A, Miasaki FY, Ciampi R, et al. Mol Cell Endocrinol. 2009,307(1–2):142–148 [DOI] [PubMed] [Google Scholar]
- 31. Dom G, Galdo VC, Tarabichi M, et al. 5-Aza-2′-deoxycytidine has minor effects on differentiation in human thyroid cancer cell lines, but modulates genes that are involved in adaptation in vitro. Thyroid. 2013;23(3):317–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dong X, Korch C, Meinkoth JL. Histone deacetylase inhibitors upregulate Rap1GAP and inhibit Rap activity in thyroid tumor cells. Endocr Relat Cancer. 2011;18(3):301–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sherman EJ, Su YB, Lyall A, et al. Evaluation of romidepsin for clinical activity and radioactive iodine reuptake in radioactive iodine-refractory thyroid carcinoma. Thyroid. 2013;23(5):593–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chakravarty D, Santos E, Ryder M, et al. Small-molecule MAPK inhibitors restore radioiodine incorporation in mouse thyroid cancers with conditional BRAF activation. J Clin Invest. 2011;121(12):4700–4711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ho AL, Grewal RK, Leboeuf R, et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N Engl J Med. 2013;368(7):623–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yu XM, Schneider DF, Leverson G, Chen H, Sippel RS. Follicular variant of papillary thyroid carcinoma is a unique clinical entity: a population-based study of 10,740 cases. Thyroid. 2013;23(10):1263–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dettmer MS, Perren A, Moch H, Komminoth P, Nikiforov YE, Nikiforova MN. Comprehensive microRNA expression profiling identifies novel markers in follicular variant of papillary thyroid carcinoma. Thyroid. 2013;23(11):1383–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gupta N, Dasyam AK, Carty SE, et al. RAS mutations in thyroid FNA specimens are highly predictive of predominantly low-risk follicular-pattern cancers. J Clin Endocrinol Metab. 2013;98(5):E914–E922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Howitt BE, Jia Y, Sholl LM, Barletta J. Molecular alterations in partially-encapsulated/well-circumscribed follicular variant of papillary thyroid carcinoma. Thyroid. 2013;23(10):1256–1262 [DOI] [PubMed] [Google Scholar]