

Abstract
Multiple infections are intensively studied because of their consequences for the health of the host but also because they can radically alter the selective pressures acting on parasites. I discuss how multiple infections have been modelled in evolutionary epidemiology. First, I briefly mention within-host models, which are at the root of these epidemiological models. Then, I present the super-infection framework, with an original focus on how the definition of the super-infection function can lead to evolutionary branching. There are several co-infection models and, for each of them, I briefly go through the underlying mathematics (especially the invasion fitness of a mutant strain) and I discuss the biological assumptions they make and the questions they consequently may ask. In particular, I show that a widely used co-infection model should not be invoked for invasion analyses because it confers a frequency-dependent advantage to rare neutral mutants. Finally, I present more recent frameworks, such as the Price equation framework in epidemiology, that can account for increased parasite diversity. To conclude, I discuss some perspectives for the study of multiple infections in evolutionary epidemiology.
Keywords: epidemiology, evolutionary biology, multiple infections, virulence, trade-offs, adaptive dynamics
1. Introduction
Host infections by more than one parasite strain or species are ubiquitous in the wild [1–4]. Furthermore, the association between different parasite species in a host is not random as infection by some parasites can facilitate infections by other parasites [5]. Such ‘multiple infections’ (also referred to as mixed infections, diverse infections or polymicrobial infections) are of particular interest to medical doctors [6,7], because they tend to worsen human health compared with single infections [8]. They are also well studied by evolutionary biologists because they alter the selective pressures acting on parasite evolution by adding a level of selection [9–11]. Furthermore, understanding how multiple infections shape virulence may have direct applications for health [12].
In 2010, Rigaud et al. [13] concluded that diverse within-host assemblages make predictions extremely difficult. Indeed, it is necessary to take into account not only the nature of the co-infecting parasites, but also that these may interact in ways that are often unknown. However, these difficulties also emphasize the importance of developing mathematical approaches to improve our understanding of parasite dynamics in settings with multiple infections. This is all the more necessary because selective pressures are strongly affected by epidemiological feedbacks [11].
Epidemiological models that study disease evolution in response to multiple infections fall into two broad categories: super-infection models, which assume that parasites never coexist in a host [14,15], and co-infection models, which assume that strains always coexist in the host [16,17]. Arguably, there is a trade-off between complexity and biological realism, with super-infection models being easily tractable and co-infection models allowing for more details.
From a biological standpoint, most of the models described in this review have been used to investigate the evolution of parasite virulence, which is usually defined as the increase in host mortality owing to the infection [11]. Some studies consider other traits, such as the investment into sexual reproduction versus asexual reproduction [18] or the evolution of the sex ratio [19]. Another important trait is immune escape [20], but the associated models usually do not include co-infections per se (the interaction takes place between one strain and the host immunity elicited by another strain already cleared from the host). Furthermore, in addition to the variety of traits studied, models also differ in the type of questions asked; for example, short-term versus long-term evolutionary outcome or quantitative versus qualitative outcome.
There is a profusion of multiple infection models in epidemiology [16], some of which have uncovered striking results, such as the enhancement of the spread of one pathogen by another [21]. It would require at least a book to review all these models, and I will focus here on evolutionary frameworks, which typically follow the ability of a rare parasite strain (the ‘mutant’ strain) to invade and replace another parasite strain that is already established in the population (the ‘resident’ strain). Note that some frameworks, such as the ‘Price equation’ framework, can capture very diverse parasite populations [22].
This review was largely motivated by the fact that several models currently coexist without there being any clear discussion on their mathematical and biological differences. In fact, as shown in §4.1, one of these models should not be used to address evolutionary biology questions because it is inherently biased (a rare mutant always has a frequency-dependent advantage). Before presenting the super-infection framework, I first give an overview of within-host models, which are at the root of multiple infection models. Then, I discuss co-infection models, which are much more diverse. For instance, they can allow the study of co-infections by closely related parasites [17] or by different species [23]. Finally, I present some more recent models that allow the study of short-term evolutionary dynamics [24] or the study of n-infections, that is, co-infections by up to an arbitrary number of strains [25]. In §7, I compare the relative merits of each of these models and discuss their links with the biology as well as some perspectives for future research.
2. Kin selection and ecological models
Multiple infections were first studied in evolutionary biology by focusing on within-host interactions and using these to make inferences at the between-host level. This was largely motivated by work on the evolution of cooperation. Indeed, increasing the number of strains per host decreases the average level of relatedness between two co-infecting parasites. As originally formulated by Hamilton [26, p. 224]:
Drawing an analogy between a clone of parasitic bacteria and a long-lived organism, the argument suggests, that bacteria should most readily evolve benign relations with their hosts where it is members of the same clone that suffer most if the host (or local host population) is killed by the infection (or epidemic).
This idea was formalized by Frank [27,28]. Later on, kin selection models were developed that included other types of within-host interactions, such as production of public goods [10,29,30] or interference competition [31]. In short, these kin selection models proceed by expressing the fitness of a focal pathogen W as a function of its phenotypic trait (x) and of the average trait value in the population, that is, within the host ( ). They then adopt an optimization approach to find the value of x* that satisfies the two conditions
). They then adopt an optimization approach to find the value of x* that satisfies the two conditions
| 2.1a |
and
| 2.1b |
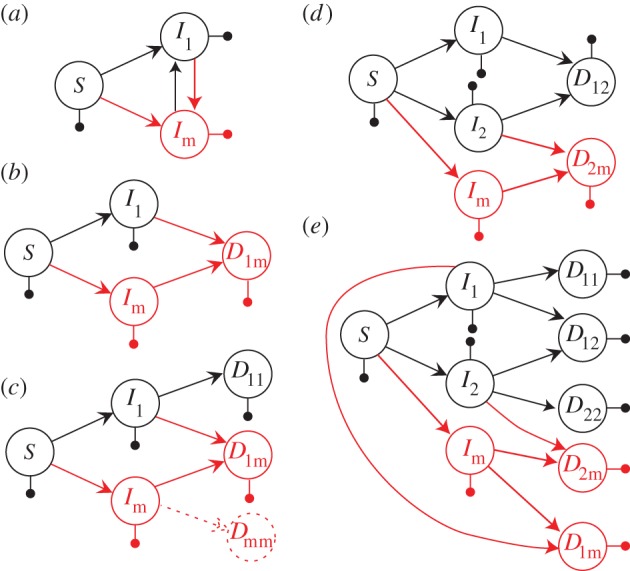
The first condition states that x* is associated with a fitness extremum, and the second condition that this extremum is a maximum. These two conditions define an evolutionary stable strategy (ESS), which is a strategy that is robust to invasion by any mutant with a different trait value. Here, this W can be seen as R0 (table 1), i.e. as the number of secondary infections produced by an infected individual over the course of an infection [28,30]. The relatedness between co-infecting parasites (r) typically emerges in these calculations through the assumption that 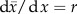 [32]. Practically, r is often assumed to be the inverse of the number of co-infecting strains (i.e. r = 1/n).
[32]. Practically, r is often assumed to be the inverse of the number of co-infecting strains (i.e. r = 1/n).
Table 1.
Notations used in the article. v indicates variables and equilibrium values are indicated with a tilde, e.g.  .
.
| symbol | description |
|---|---|
| R0 | basic reproduction number (parasite between-host fitness in a naive population) |
| Rm | between-host invasion fitness of a rare mutant strain |
| μ | baseline mortality rate |
| S | density of susceptible hosts |
| Ii | density of hosts singly infected by i |
| Dij | density of hosts co-infected by i and j |
| ρ | host input function |
| αi | virulence of a host infected by i |
| αij | overall virulence of a host co-infected by i and j |
| βi | transmission rate of i in single infection |
| βij | transmission rate of strain i in a host co-infected by i and j |
| λi | force of infection of parasite strain i |
| ϕi →j | super-infection function: the rate at which strain i replaces strain j |
| σS | vulnerability of susceptible hosts to infection |
| σI | vulnerability of infected hosts to a new infection |
Kin selection models were instrumental in understanding the expression of traits in co-infected hosts. However, when it comes to trait evolution, they are limited by the fact that they tend to lack an epidemiological dimension. They do offer a between-host perspective, but their structure is equivalent to assuming that all the hosts in the population are co-infected by exactly the same number of strains (n). For some within-host interactions such as the production of public goods, adding epidemiological dynamics can qualitatively affect model predictions [33].
It is worth mentioning that several evolutionary ecology models do include such feedbacks. For instance, Eshel [34] modelled the evolution of altruist and selfish genotypes in a metapopulation setting and allowed genotype frequencies to vary among patches. His model shares many features with the co-infection models described below as it shows that there are two conflicting selective pressures. Within a patch, altruists are counter-selected because they are exploited by cheaters at the between-patch level, patches with many altruists are favoured because they produce more offspring. More recent ecological models, such as the ‘milker–killer’ predator–prey model, also share similarities with the super-infection models that will be described later. In these models, there is competition between predators (i.e. the pathogen in epidemiological models) for access to preys and this competition takes place locally within patches (i.e. within a host in epidemiology) or between patches [35]. In addition to ecology, other insights came from game theory. Bremermann & Pickering [36] developed a within-host competition model, which they then included in an epidemiological model in a way that resembles more recent so-called nested models, which are discussed in §6 [37]. As I show below, when discussing recent work by Lion [25], it is possible to reconcile epidemiology-based and kin selection approaches.
3. The super-infection framework
The first introduction of multiple infections in evolutionary epidemiology is usually attributed to Levin & Pimentel [14]. They formalized what is now known as the ‘super-infection’ hypothesis, which states that, when an infected host encounters a host already infected by another strain, it can still infect this host (figure 1a). In other words, a strain can ‘steal’ hosts from another strain. The key assumption here is that strain replacement is immediate, so that there is no coexistence inside a host.
Figure 1.
Epidemiological models with multiple infections. (a) A super-infection model, (b) a biased model that should not be used for invasion analyses, (c) a co-infection model with parasites from the same species, (d) a co-infection model with parasites from different species and (e) a co-infection model with parasites from the same species in a dimorphic population. The resident system is in black. The host classes related to the mutant (m) are in grey (red online). Arrows indicate infections and lines with a dot indicate virulence events (host death). In (c), a host co-infected twice by the mutant (dashed line) can be neglected because they are rare. In (d), I show only an invasion analysis for a mutant strain of species 1. (Online version in colour.)
The super-infection model is interesting because it already provides us with an example where the basic reproduction ratio (R0), which is often used as a measure of parasite fitness at the between-host level, is not maximized by evolution (see also [38]). One of the underlying assumptions of R0 is that the host population is completely susceptible, and this is not the case in an evolutionary epidemiology model because the mutant strain is competing against a resident strain. In many models, it does not affect the results [38] but it matters when super-infection is allowed.
To write the invasion fitness of a mutant strain, we need to find the equilibrium state of the resident population. There are two types of hosts before the emergence of the mutant strain: susceptible hosts, with density S, and hosts infected by the resident strain, with density I1 (for the sake of clarity, we leave aside the trivial equilibrium where there are no infections by the resident strain). Their dynamics are governed by the following ordinary differential equations (ODEs):
| 3.1a |
and
| 3.1b |
where ρ(S, I1) is a general function for the host input rate, μ is the host base-line mortality, β1 is the transmission rate of strain 1 and α1 is the disease-induced mortality the resident strain causes (i.e. its virulence). For simplicity, we assume here that there is no recovery (see §7). The equilibrium state of interest of the system is
 |
3.2 |
Note that this solution is not explicit if ρ is a function of S and I1.
The dynamics of a mutant strain (referred to by ‘m’) is given by
| 3.3 |
where ϕi →j is the rate at which strain i replaces strain j (assuming that there is contact and transmission between the two hosts). If constrained in [0,1], then it can also be interpreted as a probability of replacement. In a way, ϕi →j captures two factors: the ability of strain i to replace strain j and also the resistance of strain j to super-infection by strain i.
The growth of the mutant strain is simply given by
| 3.4 |
For consistency, we express this fitness in the way we would derive it using the next-generation theorem [39,40],
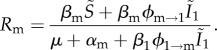 |
3.5 |
While the invasion condition for the fitness is Wm > 0, the invasion condition for the reproductive ratio is Rm > 1 (i.e. each infection should generate on average more than one infection). The rationale for preferring the latter formulation is that it bears many similarities to the R0 threshold commonly used in epidemiology. Furthermore, equation (3.5) nicely captures the effects of super-infection. On the one hand, it increases the ‘births’ of new infection (via ϕm → 1 on the numerator) and, on the other hand, it decreases the duration of the infection, because super-infection by other strains can occur (ϕ1 → m on the denominator).
If the mutant is identical to the resident, then the super-infection terms cancel out and Rm = 1 as 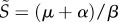 . This is known as the neutrality condition: a mutant with the same trait values as the resident cannot have a fitness advantage and is most likely to go extinct by drift because it is assumed to be very rare.
. This is known as the neutrality condition: a mutant with the same trait values as the resident cannot have a fitness advantage and is most likely to go extinct by drift because it is assumed to be very rare.
Super-infection is predicted to allow for the persistence of more virulent strains provided that they are more competitive at the within-host level [14], which has been shown experimentally for rodent malaria for instance [41]. As also stressed in [42], the super-infection hypothesis is independent from the transmission–virulence trade-off hypothesis, which states that virulence can be selected for if it correlates with increased transmission rate. This can be seen by assuming that ϕi → j = cαi (where c is a constant capturing the strength of super-infection) and that βm = β1 in a population with constant population size normalized to 1 (i.e. 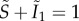 ). After some simple calculations involving the results from equation (3.2), the fitness of the mutant strain then becomes
). After some simple calculations involving the results from equation (3.2), the fitness of the mutant strain then becomes
| 3.6 |
The evolutionary stable level of virulence is obtained by solving the equation dRm/dαm = 0 for αm = α1 = α*, which yields
| 3.7 |
Therefore, even without any link between transmission and virulence, there is a non-zero optimal level of virulence if β > μ + 1/c, where parameter c captures the intensity of super-infection (the greater the c, the higher the optimal-level virulence).
In its simplest form, the super-infection function introduces a strong nonlinearity in the system: more virulent strains always take over a host, and less virulent strains are always ousted. Nowak & May [15] showed that, because of this, super-infection can lead to the persistence of a high number of strains with complicated (and potentially chaotic) dynamics. This is also discussed in box 1.
Box 1. Evolutionary branching and super-infections.
Multiple infections can lead to evolutionary branching in the parasite population [43–46]. Most studies have focused on how the shape of the transmission–virulence trade-off affects the branching [43]. However, it is not necessary to assume such a trade-off to obtain branching and varying the super-infection function itself is sufficient.
To find the singular strategies, we need to derive the invasion fitness of a rare mutant (given in equation (3.4)) with respect to αm, which yields, assuming that β and α are independent,
| 3.8 |
For simplicity, we assume a constant population size, whose density is normalized to 1 (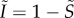 ). Furthermore, we know from equation (3.2) that
). Furthermore, we know from equation (3.2) that 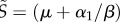 .
.
As shown in the electronic supplementary material, appendix D, the secondary derivatives, which we need to assess evolutionary stability and convergence stability, can be written as follows if αm→α1:
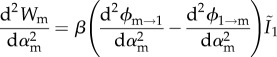 |
3.9a |
and
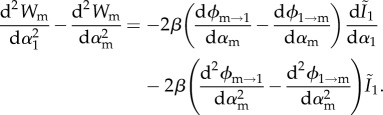 |
3.9b |
The first condition tells us about the evolutionary stability of a singular strategy: if this second derivative is negative, then the strategy cannot be invaded by any other mutant strategy. On the contrary, if it is positive, any mutant strategy can invade. The second condition tells us about convergence stability. If it is positive, the singularity is convergence stable, which means that, in a monomorphic population, the trait value evolves towards it. For further details, see [47].
We investigate the effect that the super-infection function (ϕ) has on the evolutionary dynamics. In the ‘classical’ super-infection model, ϕi →j = cαi. Therefore, the singularity is bound to be convergent stable (equation (3.9b) is always positive). Note that condition (3.9a) is equal to zero, which means that mutants have neither an advantage nor a disadvantage. This echoes Nowak & May's [15] result with the coexistence of an infinite number of strategies. In an adaptive dynamics model, because mutants are rare, we expect them to go extinct by drift and to see a distribution of traits next to the ESS, as in figure 2a.
Figure 2.
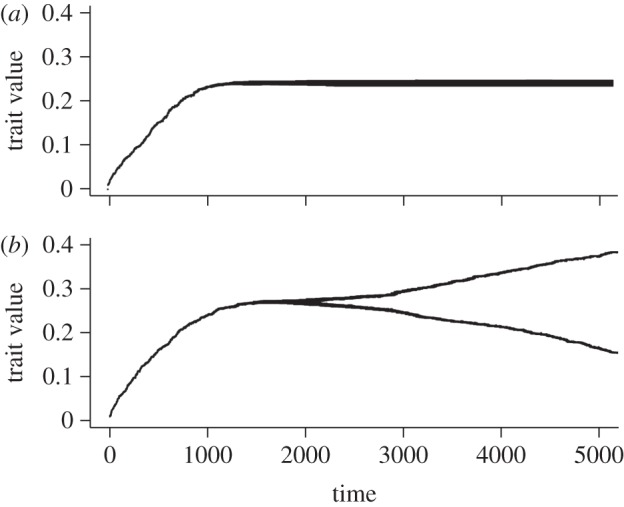
Virulence evolution in the presence of super-infection (box 1). (a) The super-infection function is ϕi→j = 4αi and in (b) 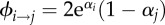 . Other parameter values are β = 0.5 and μ = 0.01. Simulations are coded as in [48]: population dynamics processes are deterministic and mutation events are stochastic.
. Other parameter values are β = 0.5 and μ = 0.01. Simulations are coded as in [48]: population dynamics processes are deterministic and mutation events are stochastic.
From equations (3.9a) and (3.9b), we see that there are two conditions to be met for the singular strategy to be a branching point. As shown in the electronic supplementary material, appendix D, in the limit where αm→α1 these can be written as
| 3.10a |
and
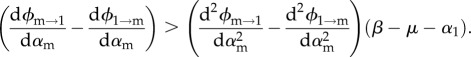 |
3.10b |
In order to find a super-infection that satisfies these conditions, it helps to simplify the super-infection process itself by breaking it down into two terms describing the ability of the invading strain to take over the host (f) and the ability of the resident strain to resist super-infection (g). One can then see the super-infection function at the product of two terms that depend on the virulence of each of the strains (mathematically, ϕi→j = f(αi)g(αj)). That functions f and g differ can be justified biologically by the fact that one of the strains is always rare, whereas the other is already established within the host.
If we choose the functions so that the first derivative of f is much greater than the first derivative of g and also such that the second derivative of f is slightly greater than that of g, we can find a super-infection function that satisfies the branching conditions. For instance, if
| 3.11 |
where β > μ + 1/c. Note that in this model α∈[0, 1].
Biologically, this means that increasing virulence increases the competitive ability exponentially but that the ability to resist invasion does not increase that rapidly (it does increase with virulence though).
With such a function, we obtain evolutionary branching, as shown in figure 2b. At first, the trait value converges towards a singular strategy. The difference from the other scenario is that this strategy is a fitness minimum and therefore any mutant with a lower or a larger fitness can invade. As there is no transmission–virulence trade-off in this model, we expect one of the branches to go to complete avirulence and the other to go to high virulence. As explained in the main text, the branching can be interpreted as parasite specialization to infecting susceptible hosts versus taking over already infected hosts.
Another important result is that the presence of super-infection can alter the effect that variations in host life history have on trait evolution. For instance, increased host background mortality is known to select for increased levels of virulence, because the parasite has only limited time to exploit the same amount of host resources. Under high levels of super-infection, the opposite result is reached, and virulence decreases with increased host mortality [49].
Few evolutionary models have ventured into incorporating super-infection (or more generally multiple infections) in a spatial context. Caraco et al. [50] provide an exception. They show that, as expected, increasing the ‘viscosity’ of the space counter-selects more virulent strains and that one of the effects of super-infection is to favour coexistence between strains.
So far, we have only mentioned studies on the direction of trait evolution or on the maintenance of diversity. One recurrent question in evolutionary ecology is how diversity is generated. The presence of super-infection in epidemiological models has been shown to have the potential to lead to evolutionary branching. Gandon et al. [49] found such branching but their model made an extra assumption, which was that the host population was diverse, with one resistant and one sensitive genotype. More recently, it was shown using a critical function analysis that super-infection can lead to evolutionary branching for some carefully chosen transmission–virulence trade-off functions [43]. However, this branching cannot occur if there is constant population size or logistic growth of the host. Note that, in this model, the super-infection function only depends on the difference between the virulence of the two strains. In box 1, I show that this branching is possible without assuming a transmission–virulence trade-off and only by varying the super-infection function. Because this function stems from within-host dynamics, assessing its exact shape could be more feasible than for the transmission–virulence trade-off, which is likely to be blurred by small parameter variations [51].
At first, the evolutionary branching can seem surprising (why would we switch from directional to disruptive selection?) but it is easy to interpret with an adaptive dynamics perspective [47]. Contrary to most population genetics models, in which fitness is an absolute value associated with a genotype, in adaptive dynamics models fitness is defined as a function of the environment. In a game theory manner, the same mutant strain can invade or not depending on which resident it competes with. Therefore, the fitness landscape changes as the parasite population evolves and what appeared to be a peak from the distance (the singularity) can, in fact, turn out to be a valley once it is reached. The parasite population can thus evolve from a monomorphic state to a dimorphic state, where very virulent parasites (adapted to infecting already-infected hosts) coexist with moderately virulent parasites (adapted to infecting susceptible hosts). See also box 1 for further details.
4. Co-infections frameworks
The super-infection hypothesis simplifies calculations and so allows strong analytical predictions. However, the assumption that upon infection an invading strain immediately replaces the resident strain in a host is unsatisfying. Co-infection models introduce additional complexity by including a co-infected host class and, as already pointed out, this complexity can be daunting at first. For instance, if there are n strains in the population, following the dynamics of each strain requires of the order of n2 equations. Furthermore, if we want to account for the order of infection, this means that the number of parameters also explodes. And, of course, this is assuming that there can be at most only two strains per host (an assumption most models make).
Contrary to super-infection models, there exists a great diversity of co-infection models. I first present a co-infection model, which is technically incorrect in evolutionary epidemiology but nevertheless is used widely. Then, I present (correct) models that capture co-infection by closely related parasites or by parasites from different species. I conclude the section by discussing models permitting larger numbers of co-infecting strains per host.
4.1. The incorrect co-infection model
Several co-infection models [16,52–54] consist of four host classes: uninfected hosts (of density S), hosts infected by a resident strain (I1), hosts infected by a mutant strain (Im) and doubly infected hosts (D1m), as shown in figure 1b. It is difficult to trace the origin of this type of model, because it is often erroneously attributed to Dietz [55], who uses a different formalism: he does consider four host classes but they are based on a host immune criterion (‘Is a host susceptible to a strain?’) instead of an infection criterion (‘Which strains are present in a host?’).
Although seemingly intuitive, such a four-class model based on infection status should not be used in evolutionary epidemiology. To understand why, let us perform a classical invasion analysis [39,40,56] and perturbate a resident system with strain 1 at equilibrium (equation (3.2)) by adding a second (rare) mutant strain. Mathematically, this results in the addition of two host categories (Im and D1m), the dynamics of which are governed by the following equations:
 |
4.1a |
and
| 4.1b |
where βij is the transmission rate of strain i in a host co-infected by strains i and j. For the sake of simplicity, we will assume here that the order of infection (resident then mutant or mutant then resident) does not affect the value of the epidemiological parameters βm1, β1m and α1m and therefore that there is only one type of doubly infected host.
As shown in the electronic supplementary material, appendix B, the expression for the invasion fitness of this rare mutant emerging in a population infected by a resident strain (or Rm) is
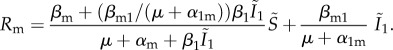 |
4.2 |
Let us now consider what happens in the neutral case, i.e. when the resident strain is identical to the mutant strain such that α1 = αm = α and β1 = βm = β. As explained before, we expect to find Rm = 1, because the mutant has no advantage over the resident strain. After some calculations involving equation (3.2) shown in the electronic supplementary material, appendix C, we find that there is neutrality if and only if
| 4.3 |
Equation (4.3) implies that the transmission rate of a mutant strain from a co-infected host (βm1) is a linear function of the overall virulence in this co-infected host (α1m), which makes sense biologically. The problem is that this linear relationship involves host densities. What would make sense biologically would be that within-host quantities shape the relationship. In other words, there is no reason why epidemiological parameters of a host should be shaped by host densities.
Overall, with this model, the invasion fitness of a neutral mutant strain is likely to be positive for realistic biological assumptions. The mutant's fitness advantage stems from its rarity: it can infect susceptible hosts and hosts infected by the resident strain, whereas the resident strain can infect susceptible hosts only (hosts infected by the mutant strain are also rare). As the density of the mutant increases, this negative frequency-dependent effect vanishes. For additional discussion on this effect, see [57,58].
4.2. Co-infections by the same species
The first (unbiased) co-infection model in evolutionary epidemiology was devised by van Baalen & Sabelis [17]. Note that another co-infection model was published the same year by May & Nowak [59] but it is discussed later on (in the section about co-infection with n strains), partly because it somehow exhibits the limitation raised in §4.1.
Van Baalen & Sabelis's [17] model can seem counterintuitive since, as shown in figure 1c, it accounts co-infections by the same (resident) strain (D11). As they argue (and as I show in the electronic supplementary material, appendix C), this solves the neutrality problem and avoids giving a frequency-dependent advantage to the mutant. One could wonder what the biological basis for these hosts infected by the same strain is. If we are dealing with macro-parasites, the interpretation is intuitive as D11 can be seen as hosts with a double parasite load. The reasoning is the same for micro-parasites but, given their underlying biology, D11 hosts resemble singly infected host.
In this model, the dynamics of the resident strain are governed by the equations
| 4.4a |
| 4.4b |
| 4.4c |
where λ1 = β1I1 + β11D11 is the force of infection of strain 1 as defined by [16], and σS and σI capture the vulnerability of S and I hosts to a new infection. Whether two co-infecting strains share host resources (i.e. β11 = β1) or whether co-infected hosts have a doubled parasite burden (i.e. β11 = 2β1) needs to be specified in the model hypothesis as discussed later. Note that the vulnerability parameters (σS and σI) were not present in the original model. They were added later on because they allow the composition of the resident population to be varied by increasing or decreasing the fraction of co-infected hosts [33]. These parameters are assumed to be independent of the nature of the strain but, as in super-infection models, one could envisage a situation where these σ would vary.
Equilibrium densities of the resident system can be expressed as a function of the equilibrium density of singly infected hosts ( ),
),
| 4.5 |
As the vulnerability of infected hosts to further infection (σI) increases, the fraction of co-infected hosts increases. The fraction of susceptible hosts decreases as they become more vulnerable (σS increases) and it increases as infected hosts become more vulnerable.
When the mutant strain emerges, it adds at least three new host classes: hosts infected by the mutant strain (Im), hosts co-infected by the resident and the mutant strains (D1m) and hosts co-infected twice by the mutant (Dmm). The last hosts can be neglected in an invasion analysis, because the mutant is assumed to be rare and this is a second-order term (it might not be the case if co-transmission is allowed though [60]). In the end, the system of ODEs capturing the mutant dynamics is very similar to previous system (4.1) except that there are hosts co-infected by the resident (D11). The invasion analysis, derived in [17,33], also looks similar
| 4.6 |
The neutrality condition is checked in the electronic supplementary material, appendix C. I find that if αm = α1, αm1 = α11, βm = β1 and βm1 = β11, then we always have Rm = 1.
The major insight provided by this co-infection model was to highlight the feedback loop between epidemiological dynamics and trait evolution. Van Baalen and Sabelis show that the evolutionarily stable level of virulence increases with the force of the infection (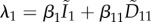 ). This is a direct evolutionary effect: the greater the force of the infection, the more co-infections there are and the more virulent strains are favoured (because they are assumed to have a competitive advantage). However, the force of the infection decreases as a function of the virulence of the resident strain (α1), which creates an epidemiological feedback. The optimal level of virulence is found by combining these two forces.
). This is a direct evolutionary effect: the greater the force of the infection, the more co-infections there are and the more virulent strains are favoured (because they are assumed to have a competitive advantage). However, the force of the infection decreases as a function of the virulence of the resident strain (α1), which creates an epidemiological feedback. The optimal level of virulence is found by combining these two forces.
This model also explored other biological questions in a pioneering way. For instance, van Baalen and Sabelis allow the possibility for parasites to plastically change their strategy in response to co-infection. At the time, there was little evidence for such behaviour, which made the parametrization of their model difficult (but see [61] for a recent review on plastic responses in parasites).
This model seems appropriate to describe co-infections by strains from the same species because there is only one resident strain [11]. Considering two species would imply that what we called the ‘mutant’ strain is in fact a strain from a different species. In such a situation, we would not be studying the evolution of a parasite species but rather the replacement of one species by another.
4.3. Co-infections by different species
Choisy & de Roode [23] developed a model that relaxes one of the main limitations of the van Baalen and Sabelis model by considering co-infections by different species (or very different strains). In this case, the resident state is dimorphic (figure 1d), and the dynamics of the resident strains of each of the two species are governed by the following equations:
| 4.7a |
| 4.7b |
| 4.7c |
| 4.7d |
where the forces of infection of each species are 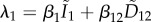 and
and 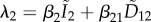 . As before, βij is the transmission rate of strain i from a host co-infected by strains i and j. (For the sake of simplicity, the vulnerability parameters are not shown in these equations but they could apply as in system (4.4).) Unfortunately, the dimension of the resident system of ODEs makes it impossible to obtain an analytical expression for the equilibrium densities, even if the total host population size is constant. This is probably the main drawback of this model, although it could be addressed using the implicit function theorem or numerical approaches.
. As before, βij is the transmission rate of strain i from a host co-infected by strains i and j. (For the sake of simplicity, the vulnerability parameters are not shown in these equations but they could apply as in system (4.4).) Unfortunately, the dimension of the resident system of ODEs makes it impossible to obtain an analytical expression for the equilibrium densities, even if the total host population size is constant. This is probably the main drawback of this model, although it could be addressed using the implicit function theorem or numerical approaches.
Let us consider the fate of a rare mutant of species 1. Its dynamics are governed by the two following equations:
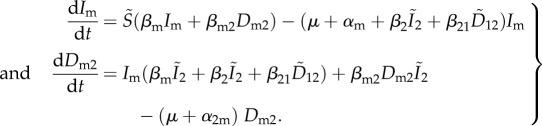 |
4.8 |
Note that second-order terms (e.g. products between Im and D2m) can be neglected, because the mutant is rare.
After some calculations described in [23] and in the electronic supplementary material, appendix B, one can show that the invasion fitness of the mutant is:
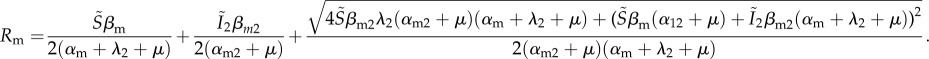 |
4.9 |
As in the previous case, if we assume that the mutant and the corresponding resident strains are identical (i.e. αm = α1, αm1 = α11, αm2 = α12, βm = β1, βm2 = β12 and βm1 = β11), then we find that R = 1. I show in the electronic supplementary material, appendix C, that the neutrality condition is independent of the input rate of susceptible hosts (ρ), of the definition of the overall virulence (α12) and of the transmission from co-infected hosts (β12 and β21).
An important feature of this model is that the mutant strain cannot co-infect hosts with its corresponding resident strain but only with the other resident strain (an assumption that is relaxed in §4.4.). This is why this model seems well suited to a scenario where co-infections only occur between unrelated parasites. In the electronic supplementary material, appendix A, I remove this assumption and allow both for within- and between-species co-infections. The corresponding model is shown in figure 1e. I also find that the neutrality condition is satisfied. This more complicated model could be used to follow parasite evolution after evolutionary branching.
In their study, Choisy & de Roode [23] vary the intensity of within-host competition by varying a single parameter in the definition of the overall virulence (i.e. the virulence expressed by co-infected hosts) and of the transmission rate from co-infected hosts. They show that the more intense the competition, the higher the virulence. They also study the effect of parasite plasticity, which they define as the ability for a parasite to be more (or less) virulent in co-infections than in single infections (a limitation being that their plasticity is a fixed scaling parameter). This plasticity leads to lower ESS values. Finally, contrary to the simplified picture shown in figure 1d, they also include partial recovery in the model. By varying the scaling parameter of the recovery rate, they show that, as the efficiency of the immune response decreases in co-infections (for instance owing to immune impairment), co-ESS levels decrease.
Note that all the effects studied by Choisy and de Roode could also be modelled in the framework for co-infections by the same species [17]. However, their model can capture differences between parasite species. As discussed below, one can assume that the two species have different transmission routes or that their virulences are constrained by different trade-offs [60]. This opens many perspectives to include biological details in co-infection models.
4.4. n-Infections
The co-infection models described until now share the same limitation that they only allow up to two parasite strains per host. This is not the case for kin selection models, which can easily be applied for a variable number of strains per host n [10] but lack an epidemiological dimension [33].
May & Nowak [59] developed a co-infection model that allows hosts to be infected by an arbitrary number of strains, while maintaining an epidemiological setting. To achieve this, they had to make two simplifying assumptions. First, the overall virulence, i.e. the virulence expressed by co-infected hosts, is equal to the virulence of the most virulent strain infecting this host. Second, transmission rates are unaffected by the presence of other strains. The combination of these two assumptions means that their model bears many similarities to single infection models. In fact, with these two assumptions, the epidemiology of the most virulent strain is identical to what would be observed with single infections only. Less virulent strains are affected because they can share hosts with more virulent strains and this decreases the duration of the infection (which is set by the most virulent strain). The model allows for strain coexistence, because there is no limitation in susceptible hosts: each strain always has access to the same number of susceptible hosts (the ones it has not already infected). One of the advantages of the model is that it is possible to approximate the average level of virulence in the population ( ) for a case without transmission–virulence trade-off by
) for a case without transmission–virulence trade-off by
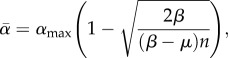 |
4.10 |
where n is the number of parasite strains circulating in the host population, which have been assigned random virulence values in [0,1]. αmax is the virulence of the most virulent strain. The larger the n, the closer the average virulence is to that of the most virulent strain. The authors do not provide any intuitive explanation as to why this should occur. One possibility to interpret this result is to bear in mind that the spread of the most virulent strain is completely unaffected by the other strains. On the contrary, any strain with intermediate virulence will see its spread hampered by more virulent strains. As the number of strains increases, so does the risk that a strain will share its host with a very virulent strain that will kill it rapidly. In the end, the average level of virulence will follow that of the most virulent strains because their spread is less affected by co-infections and because there are more of them as n increases. Overall, there is indeed a link between the number of strains and the evolution of virulence, but one can wonder to what extent this is a sampling effect: the more strains there are, the more likely it is that very virulent strains will be found.
Furthermore, it is important to stress that May and Nowak's model rests on existing diversity. If we analyse their model for n = 2, where the second strain is a mutant strain of the first (resident strain), then it is clear that we are again faced with the bias described in §4.1: the second strain can infect any host types, whereas the resident strain can infect only susceptible hosts. Therefore, this model also confers a problematic frequency-dependent advantage to rare neutral strains.
For almost 20 years, there has not really been any new n-infection epidemiological model (i.e. a model that allows for an arbitrary number of strains). However, recently, Lion [25] generalized the van Baalen and Sabelis co-infection model [17] to an arbitrary number of strains. What is particularly satisfying compared with the previous model [59] is that Lion's model can account for within-host interactions that shape overall virulence and transmission rate from co-infected hosts. The mutant invasion fitness he finds is
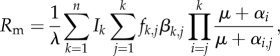 |
4.11 |
where λ is the force of infection of the resident strain, Ik is the density of hosts that are infected by k strains and fk,j is a term that captures the within-host competitiveness of the mutant strain (it corresponds to the fraction of infections caused by a co-infected host that transmit the focal/mutant strain). The notations are slightly different from that used above, because here βk,j is the transmission rate of a mutant strain in a host co-infected by k other strains and where the mutant was the jth strain to infect. As in the van Baalen and Sabelis model, this model captures co-infections by strains from the same species and Rm gives the fitness of a mutant strain that emerges in a population, where a resident strain is already present and at equilibrium. Furthermore, contrary to May and Nowak's model, this model does not include a frequency-dependent advantage.
With Lion's model, it is possible to express the evolutionary stable level of virulence as a function of the average number of strains per host. Depending on the within-host interactions (captured by the function f), these can select for increasing or decreasing level of virulence. Lion shows that his model can be interpreted in a kin selection framework. He thus recovers classical results from kin selection models [27] with the important difference that he allows for epidemiological feedbacks. In other words, the relatedness between co-infecting parasites, which typically is a constant parameter in kin selection models, is replaced by a demographic average of the number of strains per host.
4.5. Multiple infections and parasite transmission modes
Overall, it is striking to see that, from an epidemiological point of view, multiple infection evolutionary models are simple and usually involve ‘susceptible–infected’ (SI) or ‘susceptible–infected–recovered’ (SIR) settings (and, as discussed below, recovery is not always included). There are some exceptions, which are described below.
The past few years have seen a marked interest in using multiple infections to control vector-borne infectious diseases. For instance, a strain of the vertically transmitted symbiont Wolbachia has been shown to protect Aedes aegypti mosquitoes against dengue, chikungunya and Plasmodium [62]. The problem, however, is that such intervention strategies are not evolutionarily neutral and they can have unexpected consequences [63].
Some have attempted to predict the parasite's evolutionary response to the spread of a protective vertically transmitted symbiont in the host population. This requires a model with co-infections by different species, where one of the species is transmitted vertically (from parent to offspring) and the other horizontally; these parasites are denoted vertically transmitted parasite (VTP) and horizontally transmitted parasite (HTP), respectively. In their model, Jones et al. [64] show that the invasion fitness of a mutant for the HTP is slightly simpler than equation (4.9) because one of the parasites is transmitted vertically,
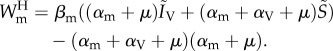 |
4.12 |
The authors find that the virulence of the VTP (αV) and the feminization it causes (which increases the density of hosts infected by the VTP, 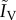 ) can both select for more virulent parasites. Of course, the VTP can evolve as well, leading to co-evolutionary dynamics. Note that some models consider co-infections by VTPs [65], but, because they exclude horizontal transmission, they do not fall into our definition of epidemiological models.
) can both select for more virulent parasites. Of course, the VTP can evolve as well, leading to co-evolutionary dynamics. Note that some models consider co-infections by VTPs [65], but, because they exclude horizontal transmission, they do not fall into our definition of epidemiological models.
Most models assume that virulence, i.e. the decrease in host fitness owing to the infection, is expressed in terms of increased mortality. However, decreasing host fecundity also decreases host fitness. The reason why fecundity is ignored in most models is that it usually does not enter the expression of parasite fitness. VTPs are an exception because obviously castrating the host decreases the parasite's reproductive success. There are other situations where host fecundity can affect parasite fitness; for instance, if there is spatial structure in the model or if the hosts are allowed to co-evolve with the parasite. However, few (if any) models have combined multiple infections with these settings.
The model used by Sorrell et al. [66] stands out in terms of parasite life cycle. They study the evolution of covert infection, which means evolving from an SIR to an SEIR system, where the E stands for ‘exposed’ hosts, who are infected but not infectious. One of the explanations the authors offer for the evolution of this additional host state is that being exposed could protect the host against super-infection by potentially more virulent strains. Interestingly, they allow virulence to act on both host survival or fecundity.
Another biological process that can affect pathogen evolution is co-transmission, which is the fact that more than one parasite can be transmitted upon a single infection event [60]. This of course requires a co-infection framework (with co-infections either by the same species or by different species) in which transitions from the susceptible state to the co-infected state are allowed. I showed that allowing for co-transmission modifies the selective pressures on the parasite strains (or species) in that the more they tend to co-transmit, the more their interests are aligned. This can lead to unexpected evolutionary outcomes. For instance, for very high co-transmission rates between two different species, a species that is less virulent in a system with only single infections can evolve to be more virulent than another species, which is more virulent in single infections. Furthermore, one of the outcomes of this model is that the prevalence of co-infections can be a poor predictor of virulence evolution because if this prevalence is due to co-transmission we expect different trait evolution than if it is due to increased susceptibility of singly infected hosts.
5. Short-term evolutionary dynamics
The epidemiology Price equation framework [22,24] allows study of the effect super-infections can exert in the short term. This framework assumes that the parasite population is diverse: n parasite strains circulate in the population and each strain can have different epidemiological parameters (denoted by a subscript i). The method tracks the density of susceptible hosts (S), the total density of infected hosts (IT) and the average trait values. As explained in detail in the electronic supplementary material, appendix E, and in [67], an SI model with super-infections can be written as
| 5.1a |
| 5.1b |
| 5.1c |
and
| 5.1d |
where notations are identical to those used in the super-infection model (ϕ is the super-infection rate, β is the transmission rate and α is the virulence). Lines above a variable indicate average values, the superscript [j] indicates an average over all strains, and var and cov indicate genetic variances and covariances between two infection life-history traits. For instance, if the covariance term cov(βi, αi) is strictly positive, it means that strains with a high transmission rate (βi) also tend to have a high virulence (αi).
Equations (5.1c) and (5.1d) capture variations in the average value of virulence and transmission rate over time but, more generally, the dynamics of any infection life-history trait x are given by
| 5.2 |
Note that, in all these equations, I assumed for simplicity that there is no mutational bias [67].
Let us focus on the equation governing the dynamics of the average virulence (equation (5.1c)). The first term is proportional to the density of susceptible hosts and indicates that higher levels of virulence can be selected if they increase the parasite transmission rate (which is also known as the transmission–virulence trade-off). The second term goes in the opposite direction and it captures the fact that virulence is always counter-selected because it decreases infection duration (as long as virulence is variable in the parasite population, −var(αi) will be negative). The last two terms are due to super-infection, and it is not a coincidence that they are proportional to the density of infected hosts (IT). In short, more virulent strains are favoured if they are more capable of taking over hosts already infected (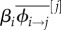 ) and they are counter-selected if they are more susceptible to super-infection (
) and they are counter-selected if they are more susceptible to super-infection (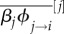 ). Note that the averaging over all the other strains j in the population is slightly different when taking over new hosts and when being super-infected as in the former case there is only one transmission rate (that of the focal strain). The exact same reasoning can be applied to the evolutionary dynamics of the transmission rate (equation (5.1d)) or any infection life-history trait (equation (5.2)).
). Note that the averaging over all the other strains j in the population is slightly different when taking over new hosts and when being super-infected as in the former case there is only one transmission rate (that of the focal strain). The exact same reasoning can be applied to the evolutionary dynamics of the transmission rate (equation (5.1d)) or any infection life-history trait (equation (5.2)).
The advantage of this formulation is that it tracks changes in average trait value as the total densities of susceptible hosts or infected hosts vary. Therefore, we directly see how epidemiology feeds back to affect trait evolution. We also see that transmission and super-infection rates are both included in some covariance terms, which implies that it could be very difficult to tease apart effects owing to super-infection from effects owing to transmission rate [42].
Day & Gandon [68] recently extended this framework to study the consequences multiple infections can have on the evolution of two traits that are determined by a single locus. A typical example is multi-locus drug resistance. The Price equation approach unravels the importance of feedbacks between the epidemiological and evolutionary dynamics. In particular, the rate at which hosts meet affects possibilities for recombination, which determines how fast multi-locus resistance spreads. Note that their framework can also be used to study the role of recombination in the evolution of drug resistance over the course of an infection, e.g. in the case of human immunodeficiency virus.
6. Nested models with multiple infections
Within-host models of co-infections were briefly presented in §2, but some studies have worked on nesting these models into evolutionary epidemiology models. One of the critiques against these so-called nested models is that they sometimes can be unnecessary, as analyses can be performed at the within-host level first and then at the between-host level [37]. If co-infections are allowed, nesting becomes more relevant, because the time at which a new strain can co-infect a host depends on the epidemiological state of the population.
Coombs et al.'s [69] study raises these questions, although their model could be argued not to be a real co-infection model because all infected hosts are co-infected (what varies is the ratio of each of the two strains within each host). The system they analyse can be written as follows:
| 6.1a |
| 6.1b |
| 6.1c |
where  is the transmission rate that creates new infections with strain mix
is the transmission rate that creates new infections with strain mix 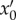 from individuals that were initially infected with a mix x0, I(t, a, x0) is the density of hosts infected a units of time ago by an initial mix x0 and α(a, x0) is the virulence of these infected hosts.
from individuals that were initially infected with a mix x0, I(t, a, x0) is the density of hosts infected a units of time ago by an initial mix x0 and α(a, x0) is the virulence of these infected hosts.
One limitation of their model is that the infection always needs to be seeded by a fraction of the two viruses. This way, the authors only need to follow the composition of the initial dose that seeded the infection. This makes the approach similar to Price equation models [24], where one follows the average value of an infection life-history trait (e.g. virulence over time). From a more biological point of view, the fact that the two strains are always transmitted together bears many similarities to a co-transmission model [60]. This raises specific issues, because, in this situation, the fitness of the two parasites are linked.
Alizon & van Baalen [44] incorporated within-host dynamics in the co-infection model developed in [17]. As indicated above, one of the issues raised by multiple infections is that time scales overlap. In this model, this is materialized by the fact that, in a co-infected host, the newly arrived strain might not have the time to replace the resident strain before the end of the infection. They approximate the growth of the second strain using the following function:
| 6.2 |
where x is the density of the second strain, x0 is its initial dose and Δr is the difference between the within-host fitness of the second and the first strain. The reproductive output, BI, the second strain can achieve from a co-infection is obtained by weighting this density with a survival function,
| 6.3 |
In the ‘classical’ co-infection model by van Baalen & Sabelis [17], the singular strategy (where the fitness gradient is zero) is always evolutionarily stable. Therefore, the pathogen population eventually reaches a state where all the strains have the same virulence, which corresponds to an ESS. One of the consequences of the overlap between the within-host and the between-host dynamics is that the evolutionary stability of the singular strategy can change. What was an ESS then becomes a branching point (see §3 and box 1 for further details).
In some cases, the nesting is less essential as the within-host and the between-host dynamics are studied separately. However, it can still help to formulate biologically relevant hypotheses. For instance, Alizon & Lion [33] analysed an earlier kin selection model for within-host public goods production [30] and included an explicit within-host model to derive epidemiological parameters (virulence and transmission rate). In the same vein, Boldin & Diekmann [70] analysed a nested model with super-infections. The originality of their approach is that it relies on a branching process to evaluate the success of the super-infection. They consider three possibilities for the super-infection: (i) a discontinuous function (it only takes values 0 or 1), (ii) continuous function that is not differentiable in 0 (arguably the most biological case) and (iii) a fully continuous function. Their goal is to see whether within-host and between-host selective pressures coincide or not. They show that the assumption made on the super-infection function has important consequences for whether the continuously stable strategy at the within-host and at the between-host level match or not. They also show that super-infection can lead to evolutionary branching depending on the shape of the transmission–virulence trade-off function (see also box 1 and [71] for further details).
7. Discussion
There is a growing interest in multiple infections not only because of their clinical implications [6,7] but also because they can radically modify selective pressures acting on pathogen evolution [9,10,13]. However, considering what happens in a co-infected host is not sufficient to accurately predict virulence evolution because of epidemiological feedbacks, hence the necessity for evolutionary epidemiology approaches [11,33].
The super-infection framework allows for analytical approaches but this comes at the expense of biological realism. Furthermore, its biological relevance is criticized even for the case of bacteriophage lambda, for which recent results show that coexistence within the same bacterium is possible owing to multiple integration sites [72]. The co-infection framework is more realistic in this respect. However, it raises many biological and technical questions. Three co-infection models currently coexist. I show that one of these models is inherently biased because it is not neutral: it generates a frequency-dependent advantage, so that a rare strain will grow in the population almost independently of its trait value. This model is widely used in epidemiology [16,53,54] and this is perfectly fine as long as one does not use it to perform an invasion analysis. One way to address this problem is to consider co-infections twice by the same strain [17]. Another way is to consider a system with two resident strains [23]. Note that this neutrality problem is not restricted to virulence evolution models but that it occurs for any invasion analysis [58].
The model with co-infections twice by the same strain has the advantage that its resident state (i.e. with only one strain in the system) can be derived analytically, which greatly increases the tractability of the model. However, a potential limitation can come from the biological interpretation of the hosts co-infected by the same strain. They have been considered to be hosts with a double pathogen load [58] or to be hosts identical in all points to singly infected hosts [17]. The most appropriate assumption depends on the host–parasite interaction studied, but data are likely to be lacking because, by definition, the pathogens from the first and from the second infection event are undistinguishable, which makes it difficult to know if a host is singly or doubly infected by the same strain. Overall, the main limitation of this model is that testing it with data is complicated. One possibility could be to consider scenarios similar to some plant fungal pathogens, where each infection is restricted to an organ (a leaf).
Another experimental problem is that it seems difficult to compare a system with or without co-infections when dealing with closely related parasite strains. One way to circumvent this problem is to vary host background mortality rate, the idea being that with higher rates there should be fewer multiple infections. This was performed by Ebert & Mangin [73], although their motivation was not to study multiple infections in the first place. In addition, Day & Gandon [22] show that short-term evolution and demographic feedbacks can further complicate the results.
The model introduced by Choisy & de Roode [23] solves many of the issues related to biological over-simplications. Their model is best suited to describe the case of co-infections by different species, a scenario for which there are more biological data [9]. Indeed, it is simpler to perform evolutionary experiments with or without co-infection by different species [11,74]. The main limitation of this model is technical, because the resident equilibrium state cannot be solved analytically.
In the electronic supplementary material, appendix A, I derive a model that alleviates the assumption made by Choisy and de Roode [23] that co-infections can only occur between different species (and not between a mutant and a resident strain of the same species). This model could be used to study what happens after an evolutionary branching in a population, i.e. a parasite population with two resident strains that diverged recently.
Few models allow for the host to be infected by more than two strains. May & Nowak [59] develop a model to study virulence evolution in which they allow for a potentially infinite number of strains to coexist within a host. However, this is not an invasion analysis as all the strains are present in the population initially, and the authors look at the equilibrium distribution of strains. It also exhibits the negative frequency-dependent bias mentioned above. Recently, Lion [25] developed an extension of the van Baalen and Sabelis co-infection model to allow for more than two strains per host. One particularly insightful aspect of Lion's results is that they can be analysed in a kin selection perspective, thus allowing comparisons with earlier results to evaluate the role of epidemiological feedbacks. Finally, the newly developed Price equation framework in epidemiology combines an explicit description of the genetic diversity of the population, which is inherent to population genetics models, with the dynamic features of epidemiological models.
Several of the implications of multiple infection models for experimental evolution are discussed in [11]. Regarding theoretical approaches, one challenge is to manage to incorporate data on within-host competition between different parasite strains into an epidemiological setting. One possibility for this would be to use a Price equation framework, as done by Day et al. [75] for single infections. The problem is that this framework cannot yet deal with co-infections and has to rely on a super-infection assumption.
Another topic that has received little attention to date is the role of partial recovery, i.e. when hosts only clear one of the co-infecting parasites but not all of them. Most models assume full recovery that occurs at a constant rate and completely cures the host. Allowing for partial recovery (only one parasite strain is removed) would complicate the transitions between host classes but could prove to be essential if one of the parasites causes short/acute infections. Related to this idea, accounting for the immune status of the host is also a largely open question. More generally, allowing for cross-immunity between parasite strains/species would be interesting in that it would allow parasites to interact through the immune system of a host without requiring that the two are present in the host at the same time. Capturing the exact shape of cross-immunity functions could prove to be difficult but, on the other hand, not requiring parasite coexistence in the host would greatly improve the biological insights of super-infection models.
Acknowledgements
I thank S. Lion for his help in showing the neutrality in two co-infection models and for helpful comments. I also thank A. Morozov for inviting me to contribute to this Theme Issue. Finally, this review was greatly improved by the comments and suggestions made by O. Diekmann, M. Sofonea and T. Caraco.
Funding statement
This study is supported by the CNRS, the IRD and by an ATIP Avenir from the CNRS and the INSERM.
References
- 1.Sharp GB, Kawaoka Y, Jones DJ, Bean WJ, Pryor SP, Hinshaw V, Webster RG. 1997. Coinfection of wild ducks by influenza A viruses: distribution patterns and biological significance. J. Virol. 71, 6128–6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petney TN, Andrews RH. 1998. Multiparasite communities in animals and humans: frequency, structure and pathogenic significance. Int. J. Parasitol. 28, 377–393. ( 10.1016/S0020-7519(97)00189-6) [DOI] [PubMed] [Google Scholar]
- 3.Lord CC, Barnard B, Day K, Hargrove JW, McNamara JJ, Paul REL, Trenholme K, Woolhouse MEJ. 1999. Aggregation and distribution of strains in microparasites. Phil. Trans. R. Soc. Lond. B 354, 799–807. ( 10.1098/rstb.1999.0432) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cox FEG. 2001. Concomitant infections, parasites and immune responses. Parasitology 122, S23–S38. ( 10.1017/S003118200001698X) [DOI] [PubMed] [Google Scholar]
- 5.Telfer S, Lambin X, Birtles R, Beldomenico P, Burthe S, Paterson S, Begon M. 2010. Species interactions in a parasite community drive infection risk in a wildlife population. Science 330, 243–246. ( 10.1126/science.1190333) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brogden KA, Guthmiller JM, Taylor CE. 2005. Human polymicrobial infections. Lancet 365, 253–255. ( 10.1016/S0140-6736(05)17745-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balmer O, Tanner M. 2011. Prevalence and implications of multiple-strain infections. Lancet Infect. Dis. 11, 868–878. ( 10.1016/S1473-3099(11)70241-9) [DOI] [PubMed] [Google Scholar]
- 8.Griffiths EC, Pedersen AB, Fenton A, Petchey OL. 2011. The nature and consequences of coinfection in humans. J. Infect. 63, 200–206. ( 10.1016/j.jinf.2011.06.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Read AF, Taylor LH. 2001. The ecology of genetically diverse infections. Science 292, 1099–1102. ( 10.1126/science.1059410) [DOI] [PubMed] [Google Scholar]
- 10.Brown SP, Hochberg ME, Grenfell BT. 2002. Does multiple infection select for raised virulence? Trends Microbiol. 10, 401–405. ( 10.1016/S0966-842X(02)02413-7) [DOI] [PubMed] [Google Scholar]
- 11.Alizon S, de Roode JC, Michalakis Y. 2013. Multiple infections and the evolution of virulence. Ecol. Lett. 16, 556–567. ( 10.1111/ele.12076) [DOI] [PubMed] [Google Scholar]
- 12.Brown SP, West SA, Diggle SP, Griffin AS. 2009. Social evolution in micro-organisms and a Trojan horse approach to medical intervention strategies. Phil. Trans. R. Soc. B 364, 3157–3168. ( 10.1098/rstb.2009.0055) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rigaud T, Perrot-Minnot M-J, Brown MJF. 2010. Parasite and host assemblages: embracing the reality will improve our knowledge of parasite transmission and virulence. Proc. R. Soc. B 277, 3693–3702. ( 10.1098/rspb.2010.1163) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levin S, Pimentel D. 1981. Selection of intermediate rates of increase in parasite-host systems. Am. Nat. 117, 308–315. ( 10.1086/283708) [DOI] [Google Scholar]
- 15.Nowak MA, May RM. 1994. Superinfection and the evolution of parasite virulence. Proc. R. Soc. Lond. B 255, 81–89. ( 10.1098/rspb.1994.0012) [DOI] [PubMed] [Google Scholar]
- 16.Anderson RM, May RM. 1991. Infectious diseases of humans. Dynamics and control. Oxford, UK: Oxford University Press. [Google Scholar]
- 17.van Baalen M, Sabelis MW. 1995. The dynamics of multiple infection and the evolution of virulence. Am. Nat. 146, 881–910. ( 10.1086/285830) [DOI] [Google Scholar]
- 18.Mideo N, Day T. 2008. The evolution of reproductive restraint in malaria. Proc. R. Soc. B 275, 1217–1224. ( 10.1098/rspb.2007.1545) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reece SE, Drew DR, Gardner A. 2008. Sex ratio adjustment and kin discrimination in malaria parasites. Nature 453, 609–614. ( 10.1038/nature06954) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koelle K, Cobey S, Grenfell B, Pascual M. 2006. Epochal evolution shapes the phylodynamics of interpandemic influenza A (H3N2) in humans. Science 314, 1898–1903. ( 10.1126/science.1132745) [DOI] [PubMed] [Google Scholar]
- 21.Abu-Raddad LJ, Patnaik P, Kublin JG. 2006. Dual infection with HIV and malaria fuels the spread of both diseases in sub-Saharan Africa. Science 314, 1603–1606. ( 10.1126/science.1132338) [DOI] [PubMed] [Google Scholar]
- 22.Day T, Gandon S. 2007. Applying population-genetic models in theoretical evolutionary epidemiology. Ecol. Lett. 10, 876–888. ( 10.1111/j.1461-0248.2007.01091.x) [DOI] [PubMed] [Google Scholar]
- 23.Choisy M, de Roode JC. 2010. Mixed infections and the evolution of virulence: effects of resource competition, parasite plasticity and impaired host immunity. Am. Nat. 175, E105–E118. ( 10.1086/651587) [DOI] [PubMed] [Google Scholar]
- 24.Day T, Proulx SR. 2004. A general theory for the evolutionary dynamics of virulence. Am. Nat. 163, E40–E63. ( 10.1086/382548) [DOI] [PubMed] [Google Scholar]
- 25.Lion S. 2013. Multiple infections, kin selection and the evolutionary epidemiology of parasite traits. J. Evol. Biol. 26, 2107–2122. ( 10.1111/jeb.12207) [DOI] [PubMed] [Google Scholar]
- 26.Hamilton WD. 1972. Altruism and related phenomena, mainly in social insects. Annu. Rev. Ecol. Syst. 3, 193–232. ( 10.1146/annurev.es.03.110172.001205) [DOI] [Google Scholar]
- 27.Frank SA. 1992. A kin selection model for the evolution of virulence. Proc. R. Soc. Lond. B 250, 195–197. ( 10.1098/rspb.1992.0149) [DOI] [PubMed] [Google Scholar]
- 28.Frank SA. 1996. Models of parasite virulence. Q. Rev. Biol. 71, 37–78. ( 10.1086/419267) [DOI] [PubMed] [Google Scholar]
- 29.Brown SP. 2001. Collective action in RNA virus. J. Evol. Biol. 14, 821–828. ( 10.1046/j.1420-9101.2001.00317.x) [DOI] [Google Scholar]
- 30.West SA, Buckling A. 2003. Cooperation, virulence and siderophore production in bacterial parasites. Proc. R. Soc. Lond. B 270, 37–44. ( 10.1098/rspb.2002.2209) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gardner A, West SA, Buckling A. 2004. Bacteriocins, spite and virulence. Proc. R. Soc. Lond. B 271, 1529–1535. ( 10.1098/rspb.2004.2756) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor PD, Frank SA. 1996. How to make a kin selection model. J. Theor. Biol. 180, 27–37. ( 10.1006/jtbi.1996.0075) [DOI] [PubMed] [Google Scholar]
- 33.Alizon S, Lion S. 2011. Within-host parasite cooperation and the evolution of virulence. Proc. R. Soc. B 278, 3738–3747. ( 10.1098/rspb.2011.0471) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eshel I. 1977. On the founder effect and the evolution of altruistic traits: an ecogenetical approach. Theor. Popul. Biol. 11, 410–424. ( 10.1016/0040-5809(77)90020-X) [DOI] [PubMed] [Google Scholar]
- 35.Sabelis MW, Janssen A, Diekmann O, Jansen VA, van Gool E, van Baalen M. 2005. Global persistence despite local extinction in acarine predator–prey systems: lessons from experimental and mathematical exercises. Adv. Ecol. Res. 37, 183–220. ( 10.1016/S0065-2504(04)37006-6) [DOI] [Google Scholar]
- 36.Bremermann HJ, Pickering J. 1983. A game-theoretical model of parasite virulence. J. Theor. Biol. 100, 411–426. ( 10.1016/0022-5193(83)90438-1) [DOI] [PubMed] [Google Scholar]
- 37.Mideo N, Alizon S, Day T. 2008. Linking within- and between-host dynamics in the evolutionary epidemiology of infectious diseases. Trends Ecol. Evol. 23, 511–517. ( 10.1016/j.tree.2008.05.009). [DOI] [PubMed] [Google Scholar]
- 38.Dieckmann U. 2002. Adaptive dynamics of pathogen-host interactions. In Adaptive dynamics of infectious diseases: in pursuit of virulence management (eds Dieckmann U, Metz JAJ, Sabelis MW, Sigmund K.), pp. 39–59. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 39.Diekmann O, Heesterbeek J. 2000. Mathematical epidemiology of infectious diseases: model building, analysis, and interpretation. New York, NY: Wiley. [Google Scholar]
- 40.Hurford A, Cownden D, Day T. 2010. Next-generation tools for evolutionary invasion analyses. J. R. Soc. Interface 7, 561–571. ( 10.1098/rsif.2009.0448) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Roode JC, et al. 2005. Virulence and competitive ability in genetically diverse malaria infections. Proc. Natl Acad. Sci. USA 102, 7624–7628. (doi:10.1073pnas.0500078102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alizon S, Michalakis Y. 2011. The transmission–virulence trade-off and superinfection: comments to Smith. Evolution 65, 3633–3638. ( 10.1111/j.1558-5646.2011.01432.x) [DOI] [PubMed] [Google Scholar]
- 43.Boldin B, Geritz S, Kisdi E. 2009. Superinfections and adaptive dynamics of pathogen virulence revisited: a critical function analysis. Evol. Ecol. Res. 11, 153–175. [Google Scholar]
- 44.Alizon S, van Baalen M. 2008. Multiple infections, immune dynamics and virulence evolution. Am. Nat. 172, E150–E158. ( 10.1086/590958) [DOI] [PubMed] [Google Scholar]
- 45.Gandon S, Mackinnon MJ, Nee S, Read AF. 2001. Imperfect vaccines and the evolution of pathogen virulence. Nature 414, 751–756. ( 10.1038/414751a) [DOI] [PubMed] [Google Scholar]
- 46.Gandon S. 2004. Evolution of multihost parasites. Evolution 58, 455–469. ( 10.1111/j.0014-3820.2004.tb01669.x) [DOI] [PubMed] [Google Scholar]
- 47.Geritz SAH, Kisdi E, Meszéna G, Metz JAJ. 1998. Evolutionarily singular strategies and the adaptive growth and branching of the evolutionary tree. Evol. Ecol. 4, 1–79. ( 10.1023/A:1006554906681) [DOI] [Google Scholar]
- 48.Luciani F, Alizon S. 2009. The evolutionary dynamics of a rapidly mutating virus within and between hosts: the case of hepatitis C virus. PLoS Comput. Biol. 5, e1000565 ( 10.1371/journal.pcbi.1000565) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gandon S, van Baalen M, Jansen VAA. 2002. The evolution of parasite virulence, superinfection and host resistance. Am. Nat. 159, 658–669. ( 10.1086/339993) [DOI] [PubMed] [Google Scholar]
- 50.Caraco T, Glavanakov S, Li S, Maniatty W, Szymanski BK. 2006. Spatially structured superinfection and the evolution of disease virulence. Theor. Popul. Biol. 69, 367–384. ( 10.1016/j.tpb.2005.12.004) [DOI] [PubMed] [Google Scholar]
- 51.Alizon S, van Baalen M. 2005. Emergence of a convex trade-off between transmission and virulence. Am. Nat. 165, E155–E167. ( 10.1086/430053) [DOI] [PubMed] [Google Scholar]
- 52.Adler FR, Brunet RC. 1991. The dynamics of simultaneous infections with altered susceptibilities. Theor. Popul. Biol. 40, 369–410. ( 10.1016/0040-5809(91)90061-J) [DOI] [PubMed] [Google Scholar]
- 53.Lipsitch M. 1997. Transmission rates and HIV virulence: comments to Massad. Evolution 51, 319–320. ( 10.2307/2410988) [DOI] [PubMed] [Google Scholar]
- 54.Mosquera J, Adler FR. 1998. Evolution of virulence: a unified framework for coinfection and superinfection. J. Theor. Biol. 195, 293–313. ( 10.1006/jtbi.1998.0793) [DOI] [PubMed] [Google Scholar]
- 55.Dietz K. 1979. Epidemiologic interference of virus populations. J. Math. Biol. 8, 291–300. ( 10.1007/BF00276314) [DOI] [PubMed] [Google Scholar]
- 56.Otto S, Day T. 2007. A biologist's guide to mathematical modeling in ecology and evolution. Princeton, NJ: Princeton University Press. [Google Scholar]
- 57.Alizon S. 2008. Decreased overall virulence in co-infected hosts leads to the persistence of virulent parasites. Am. Nat. 172, E67–E79. ( 10.1086/588077) [DOI] [PubMed] [Google Scholar]
- 58.Lipsitch M, Colijn C, Cohen T, Hanage WP, Fraser C. 2009. No coexistence for free: neutral null models for multistrain pathogens. Epidemics 1, 2–13. ( 10.1016/j.epidem.2008.07.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.May RM, Nowak MA. 1995. Coinfection and the evolution of parasite virulence. Proc. R. Soc. Lond. B 261, 209–215. ( 10.1098/rspb.1995.0138) [DOI] [PubMed] [Google Scholar]
- 60.Alizon S. 2013. Parasite co-transmission and the evolutionary epidemiology of virulence. Evolution 67, 921–933. ( 10.1111/j.1558-5646.2012.01827.x) [DOI] [PubMed] [Google Scholar]
- 61.Mideo N, Reece SE. 2012. Plasticity in parasite phenotypes: evolutionary and ecological implications for disease. Future Microbiol. 7, 17–24. ( 10.2217/fmb.11.134) [DOI] [PubMed] [Google Scholar]
- 62.Moreira LA, et al. 2009. A Wolbachia symbiont in Aedes aegypti limits infection with dengue, chikungunya, and Plasmodium. Cell 139, 1268–1278. ( 10.1016/j.cell.2009.11.042) [DOI] [PubMed] [Google Scholar]
- 63.Michalakis Y, Renaud F. 2009. Malaria: evolution in vector control. Nature 462, 298–300. ( 10.1038/462298a) [DOI] [PubMed] [Google Scholar]
- 64.Jones E, White A, Boots M. 2010. The evolutionary implications of conflict between parasites with different transmission modes. Evolution 64, 2408–2416. ( 10.1111/j.1558-5646.2010.00992.x) [DOI] [PubMed] [Google Scholar]
- 65.Vautrin E, Charles S, Genieys S, Vavre F. 2007. Evolution and invasion dynamics of multiple infections with Wolbachia investigated using matrix based models. J. Theor. Biol. 245, 197–209. ( 10.1016/j.jtbi.2006.09.035) [DOI] [PubMed] [Google Scholar]
- 66.Sorrell I, White A, Pedersen AB, Hails RS, Boots M. 2009. The evolution of covert, silent infection as a parasite strategy. Proc. R. Soc. B 276, 2217–2226. ( 10.1098/rspb.2008.1915) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Day T, Gandon S. 2006. Insights from Price's equation into evolutionary epidemiology. In Disease evolution: models, concepts, and data analyses (eds Feng Z, Dieckmann U, Levin SA.), pp. 23–44. DIMACS Series in Discrete Mathematics and Theoretical Computer Science, vol. 7. Providence, RI: American Mathematical Society. [Google Scholar]
- 68.Day T, Gandon S. 2012. The evolutionary epidemiology of multilocus drug resistance. Evolution 66, 1582–1597. ( 10.1111/j.1558-5646.2011.01533.x) [DOI] [PubMed] [Google Scholar]
- 69.Coombs D, Gilchrist MA, Ball CL. 2007. Evaluating the importance of within- and between-host selection pressures on the evolution of chronic pathogens. Theor. Popul. Biol. 72, 576–591. ( 10.1016/j.tpb.2007.08.005) [DOI] [PubMed] [Google Scholar]
- 70.Boldin B, Diekmann O. 2008. Superinfections can induce evolutionarily stable coexistence of pathogens. J. Math. Biol. 56, 635–672. ( 10.1007/s00285-007-0135-1) [DOI] [PubMed] [Google Scholar]
- 71.Boldin B, Diekmann O. In press. An extension of the classification of evolutionarily singular strategies in adaptive dynamics. J. Math. Biol. ( 10.1007/s00285-013-0725-z) [DOI] [PubMed] [Google Scholar]
- 72.Fogg PCM, Allison HE, Saunders JR, McCarthy AJ. 2010. Bacteriophage lambda: a paradigm revisited. J. Virol. 84, 6876–6879. ( 10.1128/JVI.02177-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ebert D, Mangin K. 1997. The influence of host demography on the evolution of virulence of microsporidian gut parasite. Evolution 51, 1828–1837. ( 10.2307/2411005) [DOI] [PubMed] [Google Scholar]
- 74.Garbutt J, Bonsall MB, Wright DJ, Raymond B. 2011. Antagonistic competition moderates virulence in Bacillus thuringiensis. Ecol. Lett. 14, 765–772. ( 10.1111/j.1461-0248.2011.01638.x) [DOI] [PubMed] [Google Scholar]
- 75.Day T, Alizon S, Mideo N. 2011. Bridging scales in the evolution of infectious disease life histories: theory. Evolution 65, 3448–3461. ( 10.1111/j.1558-5646.2011.01394.x) [DOI] [PubMed] [Google Scholar]