

Abstract
FE65 is a cytosolic adapter protein and an important binding partner of amyloid precursor protein. Dependent on Thr668 phosphorylation in amyloid precursor protein, which influences amyloidogenic amyloid precursor protein processing, FE65 undergoes nuclear translocation, thereby transmitting a signal from the cell membrane to the nucleus. As this translocation may be relevant in Alzheimer disease, and as FE65 consists of three protein–protein interaction domains able to bind and affect a variety of other proteins and downstream signaling pathways, the identification of the FE65 interactome is of central interest in Alzheimer disease research. In this study, we identified 121 proteins as new potential FE65 interacting proteins in a pulldown/mass spectrometry approach using human post-mortem brain samples as protein pools for recombinantly expressed FE65. Co-immunoprecipitation assays further validated the interaction of FE65 with the candidates SV2A and SERCA2. In parallel, we investigated the whole cell proteome of primary hippocampal neurons from FE65/FE65L1 double knockout mice. Notably, the validated FE65 binding proteins were also found to be differentially abundant in neurons derived from the FE65 knockout mice relative to wild-type control neurons. SERCA2 is an important player in cellular calcium homeostasis, which was found to be up-regulated in double knockout neurons. Indeed, knock-down of FE65 in HEK293T cells also evoked an elevated sensitivity to thapsigargin, a stressor specifically targeting the activity of SERCA2. Thus, our results suggest that FE65 is involved in the regulation of intracellular calcium homeostasis. Whereas transfection of FE65 alone caused a typical dot-like phenotype in the nucleus, co-transfection of SV2A significantly reduced the percentage of FE65 dot-positive cells, pointing to a possible role for SV2A in the modulation of FE65 intracellular targeting. Given that SV2A has a signaling function at the presynapse, its effect on FE65 intracellular localization suggests that the SV2A/FE65 interaction might play a role in synaptic signal transduction.
FE65 is an intracellular adapter protein consisting of three protein–protein interaction domains, two phosphotyrosine binding and one WW group II domain (1, 2). The protein is of central interest in Alzheimer disease (AD)1 because it binds amyloid precursor protein (APP) (3, 4), which plays a central role in AD pathology (5). The binding of APP to FE65 is responsible for APP-dependent cell signaling (6), APP cleavage and secretion (7, 8), apoptosis (9), and neurite outgrowth (10). In addition to APP, many other proteins are reported to bind FE65, linking the adapter to a variety of central cellular mechanisms in different subcellular compartments, which might be of relevance in AD as well.
For example, its interaction with the mammalian homolog of Drosophila enabled, which binds the WW domain of FE65 via its PPLP amino acid motif (1), points to a neuronal function of the adapter protein in regulating actin dynamics in lamellipodia (11, 12). Hints regarding its neuronal function were also derived from the interaction of FE65 with the P2X receptor subunit, raising the possibility that FE65 modulates receptor function at excitatory synapses (13). The finding that the PTB1 domain is also able to bind to the microtubule-associated protein Tau (14) is another observation characterizing the adapter protein as potentially relevant for neuronal cell viability.
Next to APP, FE65 has several other interacting proteins (mostly receptors) at the cell membrane. Its PTB1 domain mediates binding to the lipoprotein receptor-related protein (LRP) (isoform not specified) (15), the apolipoprotein E receptor APOER2 (LRP8) (16), and the very low-density lipoprotein receptor (17). This group of interacting proteins is completed by Megalin, another LRP family member (18), and the estrogen receptor alpha (19), for which the FE65 interacting domain is not yet known. Notably, the phosphorylation-dependent (20, 21) LRP–FE65 binding affects the secretion of the secreted domain of APP α and amyloid β (8), and it is not hard to imagine that other FE65 interactions in this cellular compartment affect the cleavage of APP and putatively also of other transmembrane proteins mentioned above.
Potentially most fascinating is the identification of FE65 interacting proteins within the nucleus, pointing to a signal-transduction mechanism from the cell membrane to DNA-associated processes. The WW domain in FE65 is responsible for the nuclear translocation of FE65, whereas the interaction with APP anchors the protein in the cytosol (22). The binding of APP to the PTB2 domain of FE65 was described as diminished upon T668 phosphorylation of APP (23). However, Chang et al. reported that T668 phosphorylation is essential for FE65 binding to AICD (the APP intracellular domain corresponding to the last 50 amino acids of APP (C-terminal)) and its nuclear translocation (24). Structural examinations of both FE65 PTB domains also point to the relevance of phosphorylation for protein interaction (25, 26). In addition, AICD phosphorylation at Y682, which is significantly increased in AD patient brains, was shown to inhibit the interaction between APP and FE65 and thus control nuclear translocation of the adapter protein (27, 28). In the nucleus, FE65 has several other interaction partners. For example, α-globin transcription factor CP2/late SV40 factor/leader binding protein 1, for which an association between a noncoding polymorphism and sporadic AD was suggested (29), was identified as binding to the PTB1 domain in FE65 (30). Other ligands include the nucleosome assembly factor SET (31) and Teashirt, which recruits histone deacetylases, producing a gene-silencing complex able to target caspase-4 (32). Finally, a complex consisting of AICD and FE65 was shown to induce transcriptional activation of a reporter construct upon interaction with a third PTB1-binding protein, the histone acetyltransferase TIP60 (6). However, the biological significance of this observation is still unclear. Although several putative AICD transcriptional target genes have been reported, the findings are controversial (31, 33–36). Aside from gene expression changes, a possible role of FE65 in DNA damage or repair was inferred from higher levels of the histone γ-H2AX in FE65 knockout fibroblasts exposed to DNA-damaging agents (37).
These data indicate that FE65 functions in a variety of different pathways. Several of these interactions suggest a role for FE65 proteins in brain development and possibly neurodegeneration. Given the possibility that not all FE65 binding proteins have been identified, we performed an FE65 interaction study using recombinantly expressed FE65 and a protein pool prepared from human post-mortem brain. Interesting new candidates were identified via state-of-the-art mass spectrometry and subsequently validated in independent experiments. In parallel, we assessed the proteome of primary hippocampal neurons from FE65/FE65L1 double knockout (DKO) mice (38) versus neurons from wild-type (WT) control animals. Notably, some of the FE65 interacting proteins identified were also differentially abundant in WT and DKO neurons, indicating that FE65 protein deficiency alters the levels of some of its binding proteins.
MATERIALS AND METHODS
Expression of Recombinant FE65 in Escherichia coli
After transforming the E. coli BL21 Star™ strain (Invitrogen) with the previously cloned vector pGEX-4T-1-FE65, we established a culture. The synthesis of the GST-fusion protein was started by adding isopropyl β-D-1-thiogalactopyranoside.
The E. coli pellet was resuspended in 10 volumes of lysis buffer (1 mm DTT, 50 μg/ml Lysozym, 25 μg/ml DNase I, 1 mm PMSF, dissolved in PBS) with a Potter-Elvehjem homogenizer. Cell disruption was achieved with a high-pressure homogenizer (EmulsiFlex-C5, Avestin, Canada). After a safety centrifugation to remove unlysed cells, the supernatant was centrifuged at 4 °C and 23,428 × g for 1 h.
Preparation of Human Brain Lysates
First, 30.7 g of frozen human brain tissue (occipital cortex of a control case, post-mortem time < 4 h, obtained from the Brain Bank Center Munich) was solubilized in 90 ml of immunoprecipitation buffer (10 mm Tris (pH 7.0), 50 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, and Complete Mini Protease Inhibitor Mixture (Roche Diagnostics, Switzerland)) and homogenized with a dispersing instrument (Ultra-Turrax, IKA, Staufen, Germany). After a further homogenization step with a Potter-Elvehjem, the sample was centrifuged at 4 °C and 700 × g for 10 min. The supernatant was sonicated four times with a probe sonicator on ice for 30 s. Another centrifugation step was performed at 4 °C and 13,000 × g for 10 min. The resulting supernatant was divided into six equal portions. One milliliter of Protino® Glutathione Agarose 4 (Macherey-Nagel, Düren Germany) was added to every portion and incubated on a multi-axle rotating mixer at 4 °C for 1 h. The sample was centrifuged at 4 °C and 600 × g for 5 min. The supernatant was used as the human brain lysate. An ethical vote for the work with human brain samples is available (No. 2875, ethical commission of Ruhr-University Bochum).
Brain Pulldown Assay Using Recombinant FE65
The supernatants of the FE65 and control lysates (the secreted domain of APP was used as the control) were divided into three equal parts each and incubated separately with three preparations of the human brain lysate on a multi-axle rotating mixer at 4 °C for 2 h. Three hundred microliters of Protino® Glutathione Agarose 4 (Macherey-Nagel, Germany) were added to every sample and incubated while being mixed at 4 °C for another 2.5 h. The samples were centrifuged at 4 °C and 600 × g for 5 min. The supernatant was discarded, and the resulting sediment was resuspended in 5 ml of PBS. This wash step was repeated three times, with the addition of 1% Triton X-100 in the second wash step. The glutathione beads were incubated with 300 μl of PBS and 10 units of thrombin while being mixed at 4 °C overnight.
On the next day, the thrombin-digested samples were centrifuged at 4 °C and 600 × g for 1 min. The supernatant was used for one-dimensional SDS-PAGE.
Hippocampal Neuronal Cultures
Hippocampi were removed from brains of E15 WT control or FE65/FE65L1 DKO embryos (38) and gently triturated with a series of fire-polished Pasteur pipettes of different pore sizes. In total, we used WT and DKO hippocampi from three animals for the study. Suspended dissociated cells were plated on six-well plates (6 × 105 cells/well for cortical neurons and 4 × 105 cells/well for hippocampal neurons) precoated with poly-d-lysine (0.1 mg/ml) and laminin (4 μg/ml). Cells were plated and maintained in neuronal-selection medium consisting of Neurobasal medium, B27 supplement, 2 mm glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). 25 μm glutamate was added to the initial plating medium. Half the neuronal culture medium (1 ml) was replaced every three to four days. Neurons were harvested for proteomic analysis at 13 days in vitro.
Spectral Counting–based Label-free Proteomics
The eluted fractions from the brain pulldown assay and the hippocampal neuron lysates were separated via SDS-PAGE on a 12% Bis-Tris gel for HPLC analysis. Each lane of the gel was cut into 10 pieces. The resulting gel pieces were washed alternately with 10 mm ammonium hydrogen carbonate (NH4HCO3) and 50% (v/v) 10 mm ammonium hydrogen carbonate, 50% (v/v) acetonitrile (ACN) and afterward were digested with trypsin in 10 mm HCl and 50 mm ammonium hydrogen carbonate at 37 °C overnight. The resulting peptides were extracted from the gel slices twice by means of sonication in 50% (v/v) ACN, 50% (v/v) 0.1% trifluoroacetic acid (TFA). After the extracts were merged, the ACN was removed in a centrifugal vacuum concentrator. The final sample volumes were adjusted to 40 μl with 0.1% TFA.
Nano-HPLC-MS/MS was carried out on an UltiMate 3000 RSLCnano LC system (Dionex Sunnyvale, CA). After the samples had been loaded on a trap column (75 μm × 2 cm, particle size = 3 μm, pore size = 100 Å; Dionex) and washed with 0.1% TFA (flow rate: 10 μl/min), the trap column was connected with an analytical C18 column (75 μm × 25 cm, particle size = 2 μm, pore size = 100 Å; Dionex). The following system of solvents was used for separating the peptides with a flow rate of 400 nl/min: 95% ACN, 0.1% formic acid (A); and 80% ACN, 0.1% formic acid (B). First, a gradient from 0% to 40% B (95 min) was performed, followed by a second gradient from 40% to 95% B within 2 min. Finally, 95% B was used for 5 min, followed by equilibration with buffer A. The HPLC system was connected to a nanoelectrospray ionization source (Thermo Fisher Scientific). Electrospray ionization MS/MS was performed on an LTQ Orbitrap Velos (Thermo Fisher Scientific). MS spectra were scanned between 300 and 2000 m/z with a resolution of 30,000 and a maximal acquisition time of 500 ms. The m/z values initiating MS/MS were set on a dynamic exclusion list for 35 s. Lock mass polydimethylcyclosiloxane (m/z 445.120) was used for internal recalibration. The 20 most intensive ions (charge > 1) were selected for MS/MS fragmentation in the ion trap. Fragments were generated via low-energy collision-induced dissociation on isolated ions with a collision energy of 35% and a maximal acquisition time of 50 ms.
Raw files were transformed to *.mgf files using ProteomeDiscoverer 1.3 software (Thermo Fisher Scientific). Mascot generic files were imported into ProteinScape™ 2.1 (Bruker Daltonics, Bremen, Germany) and analyzed using MASCOT (version 2.2.0, Matrix Science, London, UK) with a peptide mass tolerance of 10 ppm and a fragment mass tolerance of 0.5 Da. One missed cleavage site after tryptic digestion was allowed in the database search. Carbamidomethylation (C), oxidation (M), and phosphorylation (S, T, Y) were regarded as variable modifications. All data were searched against the UniProt/Swiss-Prot database (downloaded on June 1, 2011; containing 529,056 entries, plus one shuffled decoy entry for each target entry for false discovery rate estimation with taxonomy restriction for human (resulting in 40,620 target and decoy entries)).
After identification of the peptides, an algorithm, using a minimal peptide score and a minimal peptide count per protein, was applied by performing the following steps.
First, the Mascot ion scores of the peptides, which had a score of at least the minimum peptide score, were summed up for all corresponding protein groups (a group consisted of all entries in the database containing the same set of identified peptides). If two peptides were equal except for the score, only the higher score was used. The second step was to report the highest scoring protein group, which had at least the minimum number of peptides not yet flagged as used up, and flag all the peptides of the reported proteins as used up. The third step was to repeat the second step until no more protein groups were reported.
A local false discovery rate was calculated for each protein group, with a group regarded as decoy if it contained only decoy proteins. With this strategy the minimal peptide score was calculated, which yielded the list with the most target (opposed to decoy) groups beneath a false discovery rate of 5%. For the given data, the minimum number of peptides per protein was set at 2 to exclude “one-hit wonders,” which yielded a minimal peptide score of 21 for the longest list. From among the proteins in this list (supplemental Table S7), every peptide spectrum match was extracted.
The peptide spectrum matches were used for creating a pivot table with Microsoft Excel. As it is possible for a peptide to belong to more than one protein, all non-unique peptides were excluded, and the spectral counts of peptides belonging to one distinct protein only were used. Label-free quantification was based on the spectral index, which is calculated by adding all spectral counts of the peptides belonging to a certain protein. For the identification of significant proteins between samples and controls, the ratio index and Student's t test of the normalized spectral indices of every protein were calculated. The significance threshold was defined as a p value of 0.05. This approach has been successfully used in other proteomic studies by our group (39–44).
Immunoblotting
Eluted proteins from the pulldown assay (three independent replicates from FE65 and three from the control pulldown) were separated via SDS-PAGE using 12% Bis-Tris gels and transferred to nitrocellulose membranes. The membranes were incubated with the following antibodies: anti-SV2A antibody (dilution factor: 1:250, catalog no.: HPA007863, Sigma-Aldrich), anti-VGLUT1 antibody (dilution factor: 1:500, catalog no.: V0389, Sigma-Aldrich), anti-RYR3 antibody (dilution factor: 1:500, catalog no.: ab56000, Abcam, Cambridge, UK), and anti-SERCA2 antibody (dilution factor: 1:500, catalog no.: NB100–237, Novus Biologicals Littleton, CO). The antibodies were diluted in StartingBlockTM TBS Blocking Buffer (Thermo Fisher Scientific). The secondary IRDyeTM 800CW antibody (dilution factor: 1:15,000, catalog no.: 926–32211, LI-COR Biosciences Lincoln, NE) was applied. The Odyssey Infrared Imaging System and Odyssey Application Software version 3.0.21 (LI-COR Biosciences) were used for protein detection.
Immunofluorescence Staining
HEK293T or SH-SY5Y cells were transfected with equal amounts of pEGFP-FE65 and the empty backbone (pcDNA3.1zeo) for the pSV2A clone, pEGFP-FE65 plus pSV2A or pEGFP-FE65/pSV2A/pTIP60 DNAs. The pSV2A expression vector was kindly provided by Professor Bajjalieh, Department of Pharmacology, University of Washington (45). The resulting FE65/SV2A-overexpressing cells were fixed, permeabilized, and blocked. Afterward they were incubated with anti-SV2A antibody (dilution factor: 1:100, catalog no.: SC11939, Santa Cruz Biotechnology, Santa Cruz, CA) and subsequently with a TRITC-conjugated anti-rabbit secondary antibody (dilution factor: 1:200, catalog no.: T5268, Sigma-Aldrich). The visualization of dot-positive cells was performed using an IX51 fluorescence microscope (Olympus Microscopy, Hamburg, Germany). At least three fields of vision were evaluated to calculate the percentage of dot-positive cells. Experiments were performed in triplicate.
Co-immunoprecipitation Assays
Co-immunoprecipitation assays were used to verify protein–protein interactions. In brief, magnetic beads (MB) (Invitrogen, Oslo, Norway) were covalently bound to either anti-mouse-Ig or anti-rabbit-Ig directed against the species-specific Fc part of the primary antibody. The following primary antibodies were used: FE65 (05–759, Millipore), GFP (sc 9996, Santa Cruz Biotechnology Darmstadt, Germany), SV2A (HPA007863, Sigma), and SERCA2 (NB-100–237, Novus Biologicals).
Cell extracts (500 μg of total protein) and 100 μl of MB were incubated together with a primary antibody (0.7 μg of antibody/50 μl of MB) for 48 h at 4 °C. The concentration of MB for all immunoprecipitations was 6.5 × 108 MB/ml. Following incubation of the bead slurry with the cell extracts, the MB were washed three times with ice-cold immunoprecipitation buffer (10 mm Tris, 50 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% (v/v) Triton X-100, 1 tablet/50 ml Complete Mini Protease Inhibitor) and the bead precipitates were denaturared in SDS-PAGE sample buffer at 90 °C. After brief centrifugation, the supernatants were loaded on a 12% SDS gel for subsequent immunoblot analysis. Experiments were performed in duplicate.
Dissociated Dorsal Root Ganglia Cell Culture, Microinjection, and Confocal Laser Scanning Microscopy
Insertion of pEGFP-FE65 and SV2A vectors into single neurons of dissociated dorsal root ganglia cultures was performed using the microinjection technique described by Meller (46). Briefly, sterile glass capillaries (Femtotips, Eppendorf, Hamburg, Germany) were back-filled with 2 μl of vector suspension in distilled water. Microinjection was accomplished under visual control on an inverse microscope equipped with phase-contrast optics (Zeiss, Jena, Germany). The pressure injection tool (Eppendorf) maintained a constant pressure of 5 to 15 hPa on the tip prior to the injection, which was achieved with up to 15 to 30 hPa of increased pressure over 0.2 s. During microinjection, cultures were kept at 37 °C on a heatable stage, and they were rinsed in fresh medium following the treatment. A postincubation time of ∼24 h was sufficient to demonstrate the expression of FE65 and SV2A throughout the prolongations of dorsal root ganglia cells. Therefore, cell cultures were fixed in 4% paraformaldehyde followed by a 0.1% Triton-X-100 incubation in PBS for 10 min. Thereafter cultures were rinsed with PBS and incubated with the primary anti-SV2A antibody (SC11939, Santa Cruz Biotechnology, 1:100 in PBS) overnight at 4 °C. After intensive washing with PBS and blocking of nonspecific binding components with 10% goat serum (G9023, Sigma-Aldrich) for 30 min, samples were incubated with secondary anti-rabbit IgG TRITC antibody (T5268, Sigma-Aldrich; 1:200 in PBS) for 1.5 h at room temperature. Finally, cell cultures were rinsed in PBS and coverslipped in mounting medium (S302380-2, Dako Hamburg, Germany).
Immunostained dorsal root ganglia samples (n = 10) were evaluated via confocal laser scanning microscopy using a Zeiss LSM 510 (Zeiss, Jena, Germany) in the multitracking sequential mode set for the detection of GFP (green, 488 nm) and TRITC (red, 543 nm) with the appropriate emission band-pass filters.
FE65 Knockdown and MTT Assay
The FE65 knockdown was established by means of transfection with a GIPZ lentiviral shRNAmir vector (Open Biosystems Darmstadt, Germany) in HEK293T cells. For the control state, a nonsilencing GIPZ lentiviral shRNAmir vector (Open Biosystems) was used. Successful knockdown was validated by means of quantitative real-time PCR and immunoblotting (not shown). Primer and knockdown shRNAmir sequences can be given upon request. The MTT assay is a colorimetric assay measuring the reduction of MTT (Sigma-Aldrich, Germany) to a purple formazan dye in living cells, enabling assessment of cell viability. Cells were seeded into 96-well plates and treated with the noncompetitive inhibitor of SERCA, thapsigargin (3 and 8 μm; Alexis, Lörrach Germany), for 24 h or with the proteasome inhibitor epoxomicin (50 nm; Biotrend, Cologne, Germany) or MG132 (10 μm; Calbiochem, Germany) for 48 h. An equal volume of dimethyl sulfoxide was used as a negative control. After incubation with MTT for 3 to 4 h, the insoluble formazan dye was dissolved with isopropanol/HCl (1:1) and the absorbance of the solution was measured and quantified in a photometer at 560 nm. Reduced MTT activity (formazan build-up) correlates with decreased cell viability. Six replicates were studied for each condition.
RESULTS
Identification of FE65 Interacting Proteins
The central goal of this study was the identification of novel FE65 interacting proteins from human brain. A human post-mortem occipital cortex lysate was used as the protein pool from which candidate FE65 binding proteins were identified from a pulldown assay using a GST–FE65 fusion protein. The applied workflow is shown in Fig. 1. As shown in Fig. 1, GST–FE65 fusion protein was present in the lanes corresponding to isopropyl β-D-1-thiogalactopyranoside–treated pGEX-4T-1-FE65 transformed E. coli (F1–F3). As a control we used a protein of similar size lacking the protein–protein interaction domains found in FE65 that was cloned in the same GST fusion vector. Thus, the control lanes (C1–C3) show a signal with a molecular weight similar to that of the GST–FE65 fusion protein. The proteins recovered from the pulldown experiment were identified from gel slices via mass spectrometry analysis. For data analysis we used a label-free approach that had been successfully applied before in a variety of studies (41, 44, 47). Proteins identified as significantly enriched (p < 0.05 in at least one sample of each group with more than 0 (spectral index)) using FE65 as bait are listed in Table I. In addition, some protein candidates were identified solely in the FE65 pulldown samples (F1–F3), and never in the controls. Such candidate proteins, termed “black/white proteins” (spectral index in at least two samples of F1–F3 with more than 0), are listed in supplemental Table S1. In total, we identified 35 significantly enriched novel proteins and 86 novel black/white proteins as potential candidates for FE65 interacting proteins.
Fig. 1.

Workflow for the identification of novel FE65 interacting proteins using human brain tissue lysate. Human FE65 cDNA was cloned into the bacterial pGEX-4T-1 expression vector. Following isopropyl β-D-1-thiogalactopyranoside induction, a strong GST-FE65 signal was evident on SDS-PAGE, a finding supported by mass spectrometry data. The GST-FE65 expressing E. coli lysate was incubated with a human occipital cortex lysate. FE65 interacting proteins were isolated using glutathione agarose beads. Following various wash steps and thrombin cleavage to release FE65 polypeptides from the agarose beads, the eluted fractions were analyzed via SDS-PAGE. The same procedure was performed using a control protein of similar size. Each lane was cut into 10 parts, and peptides were extracted after tryptic digestion of each gel piece. Experiments were performed in triplicate for FE65 (F1, F2, F3) and the control (C1, C2, C3). Mass spectrometry was performed on an LTQ Orbitrap Velos instrument. For quantification, we used an in-house label-free quantification method used previously in several studies.
Table I. Novel FE65 interacting protein candidates (significant candidates) (corresponding table in color can be found in supplemental Table S1).
| Accession | Protein name | Mean counts, FE65 eluate | Mean counts, control eluate | Ratio | t test |
|---|---|---|---|---|---|
| Q9P2U7a | Vesicular glutamate transporter 1 (SLC17A7, VGLUT1) | 43.8 | 3.9 | 11.2 | 0.0001 |
| P50993b | Sodium/potassium-transporting ATPase subunit α-2 (ATP1A2) | 86.2 | 30.2 | 2.9 | 0.0002 |
| O00213 | Amyloid β A4 precursor protein-binding family B member 1 (FE65) | 3629.5 | 714.4 | 5.1 | 0.0003 |
| Q7L0J3b | Synaptic vesicle glycoprotein 2A (SV2A) | 21.4 | 5.9 | 3.6 | 0.0008 |
| P34932 | Heat shock 70 kDa protein 4 (HSPA4) | 47.6 | 7.5 | 6.4 | 0.0018 |
| P23634b | Plasma membrane calcium-transporting ATPase 4 (ATP2B4) | 6.2 | 0.9 | 7.1 | 0.0032 |
| P20020b | Plasma membrane calcium-transporting ATPase 1 (ATP2B1) | 12.8 | 5.4 | 2.4 | 0.0039 |
| P78559b | Microtubule-associated protein 1A (MAP1A) | 6.7 | 1.8 | 3.7 | 0.0065 |
| P43003b | Excitatory amino acid transporter 1 (SLC1A3) | 22.0 | 12.1 | 1.8 | 0.0072 |
| P00505 | Aspartate aminotransferase, mitochondrial (GOT2) | 3.8 | 0.3 | 14.7 | 0.0094 |
| P07954b | Fumarate hydratase, mitochondrial (FH) | 21.4 | 11.7 | 1.8 | 0.0101 |
| P08195 | 4F2 cell-surface antigen heavy chain (SLC3A2) | 12.3 | 3.2 | 3.8 | 0.0102 |
| P32004b | Neural cell adhesion molecule L1 (L1CAM) | 6.8 | 2.0 | 3.4 | 0.0120 |
| P14415 | Sodium/potassium-transporting ATPase subunit β-2 (ATP1B2) | 7.5 | 1.7 | 4.3 | 0.0131 |
| P11216b | Glycogen phosphorylase, brain form (PYGB) | 37.9 | 17.6 | 2.1 | 0.0135 |
| Q15413 | Ryanodine receptor 3 (RYR3) | 12.2 | 1.8 | 6.7 | 0.0164 |
| Q92598b | Heat shock protein 105 kDa (HSPH1) | 39.3 | 11.3 | 3.5 | 0.0169 |
| Q6PCE3b | Glucose 1,6-bisphosphate synthase (PG2L1) | 16.5 | 1.5 | 11.1 | 0.0190 |
| P16615b | Sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (ATP2A2, SERCA2) | 4.4 | 0.7 | 6.3 | 0.0194 |
| P00488 | Coagulation factor XIII A chain (F13A1) | 13.1 | 3.1 | 4.2 | 0.0232 |
| Q15904 | V-type proton ATPase subunit S1 (ATP6AP1) | 2.7 | 0.9 | 3.1 | 0.0235 |
| Q8IX18 | Probable ATP-dependent RNA helicase DHX40 (HX40) | 9.8 | 2.7 | 3.6 | 0.0240 |
| P22695b | Cytochrome b-c1 complex subunit 2, mitochondrial (UQCRC2) | 18.1 | 6.6 | 2.7 | 0.0241 |
| P54803 | Galactocerebrosidase (GALC) | 2.4 | 0.6 | 3.9 | 0.0270 |
| P11021 | 78 kDa glucose-regulated protein (HSPA5) | 108.0 | 68.2 | 1.6 | 0.0280 |
| Q9NQC3b | Reticulon-4 (RTN4) | 3.0 | 0.8 | 3.8 | 0.0304 |
| Q9HDC9 | Adipocyte plasma membrane-associated protein (APMAP) | 6.6 | 1.3 | 5.0 | 0.0308 |
| Q9NS86b | LanC-like protein 2 (LANCL2) | 5.3 | 0.4 | 12.4 | 0.0329 |
| O15394 | Neural cell adhesion molecule 2 (NCAM2) | 5.8 | 2.2 | 2.7 | 0.0344 |
| P63096 | Guanine nucleotide-binding protein G(i) subunit alpha-1 (GNAI1) | 2.3 | 0.9 | 2.7 | 0.0386 |
| P08107 | Heat shock 70 kDa protein 1A/1B (HSPA1A) | 63.9 | 21.8 | 2.9 | 0.0412 |
| O43813 | LanC-like protein 1 (LANCL1) | 6.7 | 3.2 | 2.1 | 0.0450 |
| P43004 | Excitatory amino acid transporter 2 (SLC1A2) | 16.6 | 6.5 | 2.6 | 0.0474 |
| P35579b | Myosin-9 (MYH9) | 4.2 | 1.0 | 4.5 | 0.0489 |
| P55290b | Cadherin-13 (CDH13) | 4.2 | 1.6 | 2.7 | 0.0498 |
a Similar protein regulated in DKO study (significant or black/white).
b Regulated in DKO study (significant or black/white).
In addition to the identification of novel FE65 interacting protein candidates, our study identified previously reported FE65 interacting proteins such as APP (3), LRP1 (pro-low-density LRP 1) (15), glycogen synthase kinase 3 β (48), microtubule-associated protein tau (14), and 14–3-3γ (49) (supplemental Table S2). α2-macroglobulin, which is an LRP ligand, was also identified in our study. In total, we were able to identify 33% of the currently known FE65 interacting proteins that are known to be expressed in brain tissue (supplemental Table S2). The lack of identification of other known FE65 binding proteins might have been due to several factors—for example, the possibility of protein degradation in the human post-mortem brain sample; poor recovery of proteins, especially of nuclear proteins and membrane proteins; large protein complexes in our human brain sample; limited accessibility via mass spectrometry detection; and the choice of unique peptides for the data analysis.
FE65 Interacting Protein Candidates Are Differentially Abundant in FE65/FE65L1 DKO and WT Hippocampal Neurons
In order to validate proteins identified in the FE65 pulldown assay and to select interacting proteins for further study, we identified those proteins that differed between primary hippocampal neurons cultured from FE65/FE65L1 knockout mice (38) and WT controls. We reasoned that the stability of a subset of FE65 interacting proteins might be affected in primary neuron cultures lacking FE65 and FE65L1, being aware that the abundance of a protein will not necessarily be associated with protein interaction levels, which are often regulated by additional mechanisms such as phosphorylation. The complete results of the FE65/FE65L1 knockout study are given in supplemental Table S3. All proteins that were differentially expressed in FE65/FE65L1 DKO hippocampal neurons are listed in supplemental Table S4. Proteins highlighted in yellow correspond to proteins that were identified in the pulldown study and differentially expressed (significantly regulated or black/white protein) in hippocampal neurons from WT and FE65/FE65L1 DKO mice. Proteins highlighted in green were also identified in the pulldown experiment and regulated in the hippocampal neuron comparison, but they only share similar functions as members of the same protein family. Proteins highlighted in orange were identified as black/white proteins in the interaction study and as regulated in the hippocampal neuron comparison. Finally, proteins highlighted in blue share similar functions with those differentially expressed in the FE65/FE65L1 DKO and WT hippocampal neurons.
In brief, a yellow mark corresponds to identical proteins (19 candidates) that were identified as significantly regulated in the pulldown study and regulated (significantly regulated or black/white protein) in the DKO mice study. Proteins in green (12 candidates) were also identified as significantly regulated in the pulldown study and regulated in the DKO study and might be of similar function (members of the same protein family). Proteins in orange (13 candidates) were identified as black/white proteins in the interaction study and as regulated in the DKO study. Finally, proteins labeled in blue (five candidates) were also identified as black/white proteins in the interaction study and are similar (i.e. might be of similar function) to corresponding proteins in the FE65/FE65L1 DKO study. Notably, 53% of proteins identified as significantly regulated in the pulldown experiment were also identified as regulated in the DKO study (supplemental Table S6).
Validation Experiments Revealed SV2A, VGLUT1, RYR3, and SERCA2 as Novel FE65 Interacting Proteins
The proteins SV2A, VGLUT1, RYR3, and SERCA2 were selected for further experiments based on their statistical significance in the FE65 pulldown experiment and their co-identification in the FE65/FE65L1 DKO and control neuron comparison. Initially, significantly regulated proteins with a ratio of >3 and a p value of <0.001 in the pulldown experiment awaked special interest for subsequent analysis (SV2A, VGLUT1). In a second step we considered proteins with a ratio of >6 and a p value of <0.02. From that list we excluded the 70-kDA heat shock protein 4 and glucose 1,6-bisphosphate synthase, as such proteins (or related proteins) are often found in proteomic experiments for unknown reasons (50). In order to validate our findings, we immunoblotted the eluted brain fractions from three FE65 and three control pulldown samples (Fig. 2). The initial brain lysate was used to assign the correct molecular weight for each of the FE65 interacting proteins. A HEK293 cell lysate was included to assess the expression of the candidate proteins in these cells in order to use them for subsequent experiments. Validation experiments for SV2A are shown in Fig. 2A. A prominent signal was evident in all FE65 samples (F1–F3), but never in controls (C1–C3). The brain lysate sample revealed a strong SV2A signal in the range of 70–100 kDa, consistent with the molecular weight of the full-length protein of 82.6 kDa, and another signal at about 60 kDa, potentially corresponding to a cleavage product. SV2A signals corresponding to both the full-length and the 60-kDa species were evident in the FE65 pulldowns but absent in the control pulldowns (Fig. 2A). These data are consistent with the mass spectrometry data showing that SV2A was identified in two different gel pieces corresponding to the correct protein size (supplemental Table S5). Notably, this cleavage product seemed to be significantly more pulled down by FE65 than the full-length protein, which might have been caused by structural changes of SV2A upon its cleavage. HEK293 cells also revealed SV2A positive full-length and 60-kDa signals in immunoblots, indicating that SV2A is expressed in HEK293 cells. This is in agreement with data in the TIGER database showing that SV2A is expressed in kidney (51). However, we were not able to identify the endogenous SV2A protein in HEK293T cells using mass spectrometry. Immunoblotting for VGLUT1 revealed prominent signals in the FE65 pulldown samples with lower signals in the three controls (Fig. 2B). In the brain lysate sample, the VGLUT1 antibody recognized a signal at a molecular weight of about 80 kDa that was identical to the signal in the FE65 pulldown samples (arrow in Fig. 2B). However, the signal at 62 kDa, which corresponded to the VGLUT1 full-length size, did not show any difference between the FE65 pulldown samples and the controls. In contrast, potential VGLUT1 cleavage products (signals at ∼30 and 36 kDa) were significantly more pulled down by FE65 in contrast to the controls. These findings are supported by the MS data showing that VGLUT1 peptides were identified in three different gel pieces of the corresponding molecular weight (supplemental Table S5). In HEK293 cells, the VGLUT1 immunoblotting demonstrated a strong signal at about or ∼, but not both ∼27 kDa, putatively a cleavage product of VGLUT1. Similar to SV2A, the VGLUT1 protein was not detected by mass spectrometry of HEK293T cellular proteins. These data point to a VGLUT1 isoform 80 kDa in size that is able to interact with FE65 as well as its cleavage products, whereas the 62-kDa isoform is not binding to FE65. Next, we studied the candidate FE65 interacting protein RYR3, which is a very large protein of 4870 amino acids in size with a calculated molecular weight of 536 kDa. As expected, we were not able to identify such a large protein via the standard SDS-PAGE procedure. Mass spectrometry identified RYR3 in gel pieces corresponding to 15–30 kDa (supplemental Table S5), suggesting a possible cleavage product of the receptor. A corresponding signal was also evident in immunoblotting of the FE65 samples, as well as in the brain and HEK293 lysates (Fig. 2C). RYR3 was previously reported to be expressed in HEK293 cells (52). Again, we were not able to detect the protein in HEK293T cells via mass spectrometry. Among all the candidates, the calcium transporter SERCA2 was identified with the lowest mean counts (supplemental Table S5) in gel pieces corresponding to proteins ranging from 30 to 55 kDa in size, again suggesting cleavage of the full-length protein in the post-mortem brain sample. Immunoblot validation experiments demonstrated a very low-intensity SERCA2 signal at 75 kDa (Fig. 2D), which may also correspond to a larger proteolytic fragment of the 109-kDa SERCA2 protein. A longer exposure of the blot slightly enhanced the signal (Fig. 2D, zoom-in box marked by arrow). In order to validate these findings, an additional immunoblot was performed, again demonstrating slight signals in the FE65 samples (box marked by asterisk in Fig. 2). An additional SERCA2 signal was evident at about 50 kDa, which is consistent with the identification of SERCA2 in 30–55 kDa slices via MS. SERCA2 is highly expressed in HEK293 cells, as demonstrated by immunoblotting (full-length signal as well as potential cleavage product; Fig. 2D) and mass spectrometry (not shown). As demonstrated below, we were able to validate the interaction of SV2A with FE65 and SERCA2 with FE65 using co-immunoprecipitation. In contrast, this was not true for the candidate proteins RYR3 and VGLUT1.
Fig. 2.

SV2A, VGLUT1, RYR3, and SERCA2 are novel FE65 interacting proteins. Candidates chosen for further validation from the interaction study were examined in three FE65 (F1–F3) and three control (C1–C3) pulldown assays. A, immunoblotting using the anti-SV2A antibody revealed strong signals corresponding to the full-length (FL) SV2A protein in all F samples that were absent in controls. In addition, a putative SV2A cleavage product (CP) signal was evident in both pulldown eluates and in the brain lysate (BL) used as starting material for the pulldown experiments. B, similarly, VGLUT1 immunoblotting revealed a specific signal in the FE65 precipitates, but only a weak signal in controls (a second brain lysate preparation (BL2) was used in addition to BL). C, a putative RYR3 cleavage product was specifically identified in the F samples, but not in controls. D, finally, immunoblotting for SERCA2 validated our initial findings. A longer exposure (box marked by arrow) as well as an independent experiment (box marked by asterisk) revealed weak signals in the F samples that were not present in the control lanes.
SV2A Modulates the Intracellular Localization of FE65 and Co-immunoprecipitation Experiments Confirm FE65–SV2A Interaction in HEK293 Cells
FE65/SV2A-overexpressing HEK293 cells were used for further validation of this interaction because endogenous SV2A was not detected in HEK293 cells via mass spectrometry, and its abundance as identified via immunoblotting might not correspond to levels convenient for subsequent experiments. Overexpression of an FE65-EGFP vector alone induced a characteristic nuclear dot-like phenotype in 15.9% of HEK293 cells (Fig. 3A), pointing to moderate endogenous expression of the FE65 interacting protein TIP60, which is necessary for the observed phenotype. In order to evaluate a hypothetical role for SV2A in the intracellular localization of FE65, we co-expressed FE65 and SV2A in HEK293 cells. Quantitation of the number of dot-positive cells in FE65 and FE65/SV2A expressing cells revealed a reduced number of dot-positive cells in FE65/SV2A expressing cells (Figs. 3B and 3C, p < 10−4). As the number of dot-positive cells significantly depends on the co-expression of the FE65 interacting protein TIP60 (53), we next assessed the percentage of dot-positive cells in FE65/TIP60 versus FE65/TIP60/SV2A transfected HEK293 cells (Figs. 3D and 3E). Quantitation of the number of dot-positive cells revealed no difference (Fig. 3F). These data suggest that TIP60 expression sequesters FE65 in the nucleus away from SV2A, which is localized in the cytosol (see Fig. 4B), and prevents SV2A-dependent relocalization of FE65 (Fig. 3F). Similar experiments repeated in the neuroblastoma cell line, SH-SY5Y, revealed comparable results: a significant reduction in the percentage of dot-positive cells caused by FE65 expression in SV2A co-expressing cells (Figs. 3G–3I, p < 10−3). Again, the co-transfection of TIP60 abolished the influence of SV2A on dot-positive cells (Figs. 3J–3L).
Fig. 3.
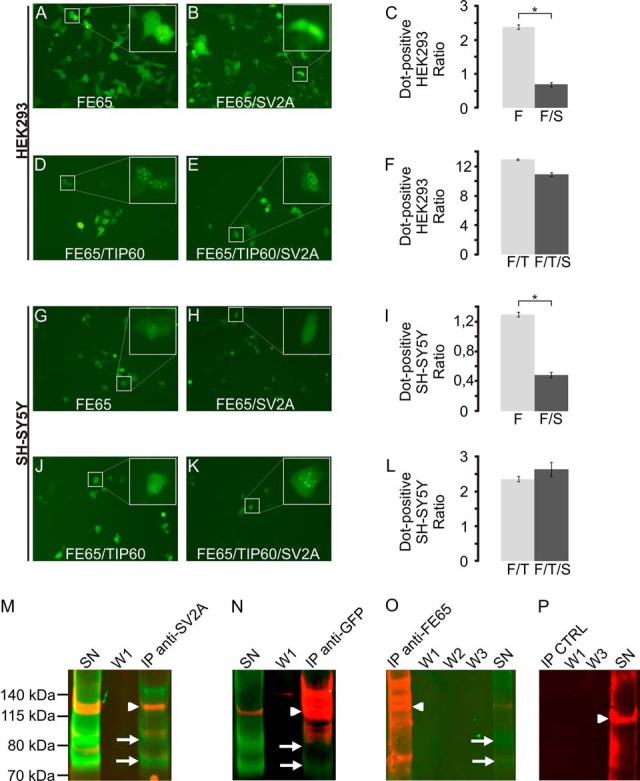
SV2A modulates the localization of FE65 and interacts with the adapter protein in co-immunoprecipitation assays. A–C, co-transfection of SV2A significantly modified the localization of FE65 in HEK293 cells, but not if TIP60 was co-transfected as well (D–F). Similar results were obtained with co-transfection experiments in SH-SY5Y (G–L). Co-immunoprecipitation assays validated the interaction of SV2A with FE65. In SV2A precipitates (M), both FE65 (arrowhead) and SV2A (arrows, two isoforms) could be identified. The co-immunoprecipitation supernatant (SN) still contained both proteins, whereas no signal was evident in the first washing step (W1). Conversely, immunoprecipitation of FE65-EGFP using an anti-GFP antibody (N) revealed the co-immunoprecipitation of SV2A (arrow). Immunoprecipitation using an anti-FE65 WW antibody (O) did not co-precipitate SV2A. However, SV2A was present in the immunoprecipitation supernatant. Control experiments using beads without antibody (P) showed no SV2A or FE65 signal.
Fig. 4.
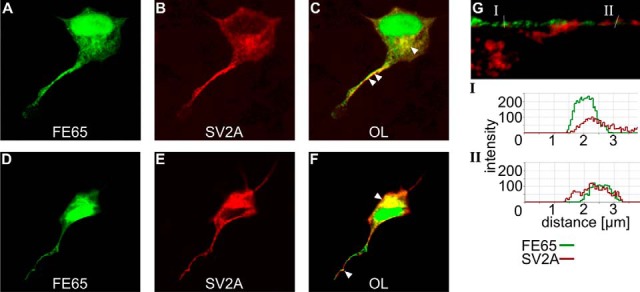
SV2A and FE65 co-localize in the cell body and neurites of chicken dorsal root ganglion cells. Following microinjection of SV2A and FE65 expression vectors, intracellular localization of SV2A (B, E) was visualized by means of immunofluorescence staining and confocal microscopy. FE65 was expressed as an FE65–EGFP fusion protein (A, D). The overlay of both images (C, F) revealed co-localization in the perinuclear region and in neurites (loci of co-localization are indicated by arrowheads). Analysis of the signal intensity of FE65 and SV2A staining in neurite cross sections (G) supported the co-localization of both proteins in neurites (intensity shown for two profiles).
Further validation of the FE65–SV2A interaction was obtained using co-immunoprecipitation assays. Following co-transfection of FE65 and SV2A in HEK293 cells, SV2A complexes were precipitated using an anti-SV2A antibody and subsequently analyzed via immunoblotting for FE65 (red) and SV2A (green) in parallel (Fig. 3M). Significant co-precipitation of FE65 was obtained (lane “IP anti-SV2A,” FE65 in red (arrowhead), SV2A in green (arrows)). As expected, the co-immunprecipitation supernatant still contained both proteins (SN), whereas no signal was evident in the first washing step (W1). Similar results were obtained when an anti-GFP antibody was used for immunoprecipitation (Fig. 3N). EGFP–FE65 complexes were recovered using an anti-GFP antibody. Prominent SV2A (green, arrow) and FE65 (red, arrowhead) signals were evident in the anti-GFP immunoprecipitation lane. In contrast, SV2A was not recovered in FE65 complexes obtained with an anti-WW FE65 antibody (Fig. 3O), possibly because of competitive binding of the antibody and SV2A for FE65. Controls using unloaded beads revealed neither an FE65 nor an SV2A signal unspecifically bound to the beads (Fig. 3P, IP CTRL).
FE65 Co-localizes with SV2A in Neurites of Chicken Dorsal Root Ganglion Cells
Confocal microscopy was used to examine FE65 and SV2A staining in pFE65–EGFP and pSV2A double-transfected dorsal root ganglion cells (Fig. 4). FE65 expression localized to the nucleus, the perinuclear region, and neurites (Figs. 4A and 4D). In contrast, SV2A was absent from the nucleus (Figs. 4B and 4E) but was present in the perinuclear region and in neurites. The overlay (Figs. 4C and 4F) revealed putative intracellular regions of co-localization. A prominent co-localization was evident in perinuclear regions, potentially the endoplasmic reticulum. Additionally, fluorescent intensity analysis of neurite cross sections pointed to a co-localization of both signals in regions of the neurite (Fig. 4G).
FE65 and SERCA2 Co-immunoprecipitate from HEK293 Cells
Co-immunoprecipitation experiments were performed to validate the interaction of FE65 with SERCA2 in HEK293 cells overexpressing both proteins. Using an anti-GFP antibody or an anti-WW FE65 antibody, complexes were precipitated and analyzed via immunoblotting for SERCA2 and FE65 in parallel (Fig. 5A). SERCA2 (green) was detected in immune complexes obtained using a GFP antibody or an anti-WW FE65 antibody. Conversely, FE65 (red) was detected in SERCA2 immune complexes. The control experiment without antibody revealed neither an FE65 nor a SERCA2 signal.
Fig. 5.

FE65 and SERCA2 co-immunoprecipitate and FE65 knockdown sensitizes cells to the endoplasmic reticulum stressor thapsigargin. A, co-immunoprecipitation assays for FE65 (red) revealed the precipitation of SERCA2 (green) using antibodies against EGFP (IP GFP) or FE65 (IP FE65). FE65 was identified on the SV2A immunoprecipitates (IP SERCA2). Lanes labeled with FE65 and SERCA2 correspond to lysates of cells expressing the individual protein. Lane SN corresponds to the supernatant after immunoprecipitation of a control (without primary antibody) demonstrating the presence of both proteins in the supernatant. B–D, the knockdown of FE65 in HEK293 cells resulted in less cell viability following 8 μm thapsigargin (Tha) stimulation after 24 h (B) and following 3 μm and 8 μm Tha stimulation after 48 h (C). The proteasomal inhibitors epoxomicin (Epoxo) and MG132 revealed no differences in cell viability in FE65 knockdown cells versus controls (D).
Knockdown of FE65 Sensitizes Cells to the Endoplasmic Reticulum Stressor Thapsigargin
SERCA2 and RYR3 are critical players in cellular calcium homeostasis (54, 55). In order to test the hypothesis that FE65 affects intracellular calcium levels, we generated a stable FE65 knockdown cell line and measured cell viability following thapsigargin treatment. Thapsigargin is a noncompetitive inhibitor of SERCA (56). In the presence of 3 μm thapsigargin, no significant difference in MTT activity was observed at 24 h (Fig. 5B). However, application of 8 μm thapsigargin for 24 h revealed significantly less MTT activity in FE65 knockdown cells than in controls. Significant MTT activity differences were observed in cells treated for 48 h with 3 and 8 μm thapsigargin (Fig. 5C). In order to test the specificity of the cellular stress response, we examined cell viability following treatment with the proteasomal inhibitors epoxomicin and MG132 (Fig. 5D). In contrast to thapsigargin stimulation, no significant differences in cell viability were observed between FE65 knockdown and control cells treated with these proteasome inhibitors. However, both MG132 and epoxomycin treatment reduced cell viability to the same extent in control and FE65 knockdown cells (Fig. 5D). These data suggest that the interaction of FE65 and SERCA2 plays a pivotal role in determining the viability of cells treated with the endoplasmic reticulum stressor thapsigargin.
DISCUSSION
The FE65 adapter protein is relevant to AD because of its interaction with AICD (57, 58) and its effect on β amyloid generation (59). Furthermore, FE65 protein family interactions with APP and the amyloid precursor-like proteins play a role in brain development (38). Therefore, identification of the FE65 interactome is a necessary prerequisite for understanding the biological functions of FE65 in the central nervous system. Among the many new protein candidates identified as potential FE65 interacting proteins in this work, RYR3, SERCA2, VGLUT1, and SV2A were selected for further study, as these candidates were also co-identified in the FE65/FE65L1 DKO and control neuron comparison. The binding of SV2A and SERCA2 with FE65 was validated using co-immunoprecipitation in HEK293 cells. Additionally, the generation of the characteristic nuclear FE65 phenotype depends on the co-transfection of SV2A showing fewer nuclear dots in FE65/SV2A co-transfected cells. Both proteins co-localize in neurons in the perinuclear region and in neurites.
Our pulldown assays identified known FE65 interactors such as APP and LRP, and these interactions have been extensively studied for effects on APP processing (23, 59). However, for other reported interactors such as Tau, 14-3-3γ, and glycogen synthase kinase 3 β, ours is the first report confirming these interactions. The identification of α2-macroglobulin, a secreted protein, with FE65, a cytosolic protein, suggests that its recovery in FE65 pulldown assays results from its interaction with LRP (60), a shared binding partner. The lack of identification of other known FE65 binding proteins such as the amyloid precursor-like proteins and TIP60 might be due to several factors. These include the possibility of protein degradation in the human post-mortem brain sample used (4 h post-mortem time); poor recovery of proteins, especially of nuclear proteins and membrane proteins; large protein complexes in our human brain sample; limited accessibility to mass spectrometry detection; and the choice of unique peptides for data analysis. Despite these limitations, our approach revealed novel interacting proteins. Our new findings, as well as the present knowledge on the FE65 interactome, are summarized in Fig. 6.
Fig. 6.
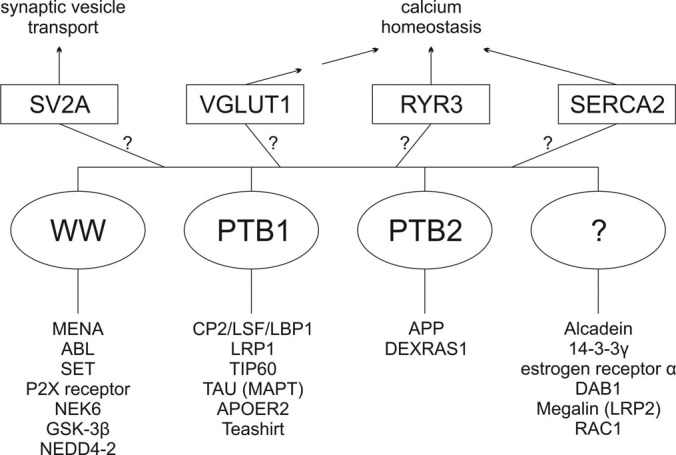
The FE65 interactome. Diagram summarizing known and novel FE65 interactors. Known FE65 interactors are shown below FE65, and the interactors identified and validated in this study, along with the cellular processes that are or may be altered by their interaction with FE65, are shown above FE65.
SV2A and a variety of members of the solute-like carrier proteins, of which VGLUT1 is a member, were significantly more abundant in the FE65/FE65L1 DKO hippocampal neurons than in control neurons. VGLUT1 (SLC17A7) was not identified in the hippocampal neuron proteomic study comparing FE65/FE65L1 knockout and control neurons, possibly because of the limited sensitivity of the mass spectrometry analysis. The regulation of SV2A protein levels in FE65/FE65L1 DKO neurons suggests a role for this protein–protein interaction in neurons. SV2A is a presynaptic vesicle protein and a molecular target for antiepileptic drugs (61). The association of SV2A with AD has not been described, but some disorientation in AD is associated with epileptiform activity and might be prevented with antiepileptic drugs (62). In contrast, VGLUT1, the vesicular glutamate transporter in neurons, has been found to be differentially expressed in different AD mouse models (63, 64). The observation that FE65 is associated with synaptic plasticity (65) might in part be due to the interaction of FE65 with SV2A and VGLUT1. FE65 interactions with APP localize to Rab5-containing synaptic organelles, but not to synaptic vesicles (12). A function for FE65 at the synapse has not been established. However, up-regulation of FE65 proteins in Mint/X11-deficient neurons suggests that these proteins are partially functionally redundant (66). Mint/X11 proteins, which also bind the APP NPXY motif, are regulators of presynaptic neurotransmitter release. The identification of FE65 binding to SV2A and VGLUT1, proteins that also affect neurotransmitter release, suggests that the functional redundancy of FE65 and Mint/X11 proteins might be independent of their binding to APP.
AD is characterized by an altered glutamatergic activation, and glutamate can promote calcium dyshomeostasis, another feature of the disease (67). SERCA2 is a protein that affects cellular calcium homeostasis (54, 68) and was identified as an FE65 interacting protein in this study. SERCA2 was initially described as an intracellular pump located in the sarcoplasmic or endoplasmic reticula of muscle cells. This enzyme catalyzes the hydrolysis of ATP coupled with the translocation of calcium from the cytosol to the sarcoplasmic reticulum lumen. Our data show that SERCA2 protein levels were higher in primary hippocampal neuron cultures of FE65/FE65L1 DKO mice. Thus, the interplay of FE65 and SERCA2 might have an effect on cellular calcium homeostasis. Calcium release from intracellular stores in the endoplasmic reticulum occurs through the activation of two types of calcium receptors: inositol 1,4,5-trisphosphate and ryanodine receptors (69). RYR3 was also identified as an FE65 interacting protein in this study, again pointing to the relevance of FE65 in regulating cellular calcium homeostasis. In good agreement with this hypothesis, FE65 knockdown cells are more sensitive to thapsigargin, an inhibitor of SERCA activity, than controls. Notably, a role for the APP intracellular domain in regulating phosphoinositide-mediated calcium signaling has been reported (70). In addition, presenilin, the catalytic subunit of γ-secretase, has been shown to regulate the activity of the SERCA pump (71). This might be caused by elevated liberation of AICD upon APP cleavage by the presenilin complex, binding of AICD to FE65 (or conformational change of FE65), and subsequent further interaction of the complex with the FE65 interacting proteins SERCA2 or RYR3.
Supplementary Material
Acknowledgments
The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD000437 and DOI 10.6019/PXD000437.
Footnotes
Author contributions: T. Mueller designed research; F.M.N., M.H.N., A.S., T. Mastalski, S.S., C.L., F.E., J.S., S.Y.G., N.R., D.K., C.T., and T. Mueller performed research; F.M.N., M.H.N., A.P., S.H., R.E., J.U., M.E., J.S., S.Y.G., and T. Mueller analyzed data; H.E.M., S.Y.G., K.M., and T. Mueller wrote the paper.
* This work was funded by FoRUM (Forschungsförderung Ruhr-Universität Bochum Medizinische Fakultät) AZ-F616-2008 and AD F680-2009. We thank the federal state North Rhine-Westphalia for funding within the project PURE. S.Y.G. and J.S. were funded by the National Institutes of Health (AG15903).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- AD
- Alzheimer disease
- AICD
- amyloid precursor protein intracellular domain
- APP
- amyloid precursor protein
- ACN
- acetonitrile
- Bis-Tris
- bis(2-hydroxyethyl)iminotris(hydroxymethyl)methane
- DKO
- double knockout
- EGFP
- enhanced green fluorescent protein
- FE65L1
- FE65-like protein
- GFP
- green fluorescent protein
- GST
- glutathione S-transferase
- HEK293T
- human embryonic kidney 293 cells
- LRP
- lipoprotein receptor-related protein
- MB
- magnetic beads
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PBS
- phosphate-buffered saline
- PTB
- phosphotyrosine-binding domain
- RYR
- ryanodine receptor
- SDS-PAGE
- sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SERCA2
- sarco/endoplasmic reticulum Ca2+-ATPase
- SV2A
- synaptic vesicle glycoprotein 2A
- TIP60
- K(lysine) acetyltransferase 5
- VGLUT1
- vesicular glutamate transporter 1
- WT
- wild type.
REFERENCES
- 1. Ermekova K. S., Zambrano N., Linn H., Minopoli G., Gertler F., Russo T., Sudol M. (1997) The WW domain of neural protein FE65 interacts with proline-rich motifs in MENA, the mammalian homolog of Drosophila enabled. J. Biol. Chem. 272, 32869–32877 [DOI] [PubMed] [Google Scholar]
- 2. Espanel X., Sudol M. (1999) A single point mutation in a group I WW domain shifts its specificity to that of group II WW domains. J. Biol. Chem. 274, 17284–17289 [DOI] [PubMed] [Google Scholar]
- 3. Borg J. P., Ooi J., Levy E., Margolis B. (1996) The phosphotyrosine interaction domains of X11 and FE65 bind to distinct sites on the YENPTY motif of amyloid precursor protein. Mol. Cell. Biol. 16, 6229–6241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bressler S. L., Gray M. D., Sopher B. L., Hu Q., Hearn M. G., Pham D. G., Dinulos M. B., Fukuchi K., Sisodia S. S., Miller M. A., Disteche C. M., Martin G. M. (1996) cDNA cloning and chromosome mapping of the human Fe65 gene: interaction of the conserved cytoplasmic domains of the human beta-amyloid precursor protein and its homologues with the mouse Fe65 protein. Hum. Mol. Genet. 5, 1589–1598 [DOI] [PubMed] [Google Scholar]
- 5. Selkoe D. J. (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 81, 741–766 [DOI] [PubMed] [Google Scholar]
- 6. Cao X., Sudhof T. C. (2001) A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293, 115–120 [DOI] [PubMed] [Google Scholar]
- 7. Santiard-Baron D., Langui D., Delehedde M., Delatour B., Schombert B., Touchet N., Tremp G., Paul M. F., Blanchard V., Sergeant N., Delacourte A., Duyckaerts C., Pradier L., Mercken L. (2005) Expression of human FE65 in amyloid precursor protein transgenic mice is associated with a reduction in beta-amyloid load. J. Neurochem. 93, 330–338 [DOI] [PubMed] [Google Scholar]
- 8. Pietrzik C. U., Yoon I. S., Jaeger S., Busse T., Weggen S., Koo E. H. (2004) FE65 constitutes the functional link between the low-density lipoprotein receptor-related protein and the amyloid precursor protein. J. Neurosci. 24, 4259–4265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kinoshita A., Whelan C. M., Berezovska O., Hyman B. T. (2002) The gamma secretase-generated carboxyl-terminal domain of the amyloid precursor protein induces apoptosis via Tip60 in H4 cells. J. Biol. Chem. 277, 28530–28536 [DOI] [PubMed] [Google Scholar]
- 10. Ikin A. F., Sabo S. L., Lanier L. M., Buxbaum J. D. (2007) A macromolecular complex involving the amyloid precursor protein (APP) and the cytosolic adapter FE65 is a negative regulator of axon branching. Mol. Cell. Neurosci. 35, 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sabo S. L., Ikin A. F., Buxbaum J. D., Greengard P. (2001) The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. J. Cell Biol. 153, 1403–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sabo S. L., Ikin A. F., Buxbaum J. D., Greengard P. (2003) The amyloid precursor protein and its regulatory protein, FE65, in growth cones and synapses in vitro and in vivo. J. Neurosci. 23, 5407–5415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Masin M., Kerschensteiner D., Dumke K., Rubio M. E., Soto F. (2006) Fe65 interacts with P2X2 subunits at excitatory synapses and modulates receptor function. J. Biol. Chem. 281, 4100–4108 [DOI] [PubMed] [Google Scholar]
- 14. Barbato C., Canu N., Zambrano N., Serafino A., Minopoli G., Ciotti M. T., Amadoro G., Russo T., Calissano P. (2005) Interaction of Tau with Fe65 links tau to APP. Neurobiol. Dis. 18, 399–408 [DOI] [PubMed] [Google Scholar]
- 15. Kinoshita A., Whelan C. M., Smith C. J., Mikhailenko I., Rebeck G. W., Strickland D. K., Hyman B. T. (2001) Demonstration by fluorescence resonance energy transfer of two sites of interaction between the low-density lipoprotein receptor-related protein and the amyloid precursor protein: role of the intracellular adapter protein Fe65. J. Neurosci. 21, 8354–8361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hoe H. S., Magill L. A., Guenette S., Fu Z., Vicini S., Rebeck G. W. (2006) FE65 interaction with the ApoE receptor ApoEr2. J. Biol. Chem. 281, 24521–24530 [DOI] [PubMed] [Google Scholar]
- 17. Dumanis S. B., Chamberlain K. A., Sohn Y. J., Lee Y. J., Guenette S. Y., Suzuki T., Mathews P. M., Pak D. T., Rebeck G. W., Suh Y. H., Park H. S., Hoe H. S. (2012) FE65 as a link between VLDLR and APP to regulate their trafficking and processing. Mol. Neurodegener. 7, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alvira-Botero X., Perez-Gonzalez R., Spuch C., Vargas T., Antequera D., Garzon M., Bermejo-Pareja F., Carro E. (2010) Megalin interacts with APP and the intracellular adapter protein FE65 in neurons. Mol. Cell Neurosci. 45, 306–315 [DOI] [PubMed] [Google Scholar]
- 19. Bao J., Cao C., Zhang X., Jiang F., Nicosia S. V., Bai W. (2007) Suppression of beta-amyloid precursor protein signaling into the nucleus by estrogens mediated through complex formation between the estrogen receptor and Fe65. Mol. Cell. Biol. 27, 1321–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klug W., Dietl A., Simon B., Sinning I., Wild K. (2011) Phosphorylation of LRP1 regulates the interaction with Fe65. FEBS Lett. 585, 3229–3235 [DOI] [PubMed] [Google Scholar]
- 21. Mulvihill M. M., Guttman M., Komives E. A. (2011) Protein interactions among Fe65, the low-density lipoprotein receptor-related protein, and the amyloid precursor protein. Biochemistry 50, 6208–6216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Minopoli G., de Candia P., Bonetti A., Faraonio R., Zambrano N., Russo T. (2001) The beta-amyloid precursor protein functions as a cytosolic anchoring site that prevents Fe65 nuclear translocation. J. Biol. Chem. 276, 6545–6550 [DOI] [PubMed] [Google Scholar]
- 23. Ando K., Iijima K. I., Elliott J. I., Kirino Y., Suzuki T. (2001) Phosphorylation-dependent regulation of the interaction of amyloid precursor protein with Fe65 affects the production of beta-amyloid. J. Biol. Chem. 276, 40353–40361 [DOI] [PubMed] [Google Scholar]
- 24. Chang K. A., Kim H. S., Ha T. Y., Ha J. W., Shin K. Y., Jeong Y. H., Lee J. P., Park C. H., Kim S., Baik T. K., Suh Y. H. (2006) Phosphorylation of amyloid precursor protein (APP) at Thr668 regulates the nuclear translocation of the APP intracellular domain and induces neurodegeneration. Mol. Cell. Biol. 26, 4327–4338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Radzimanowski J., Simon B., Sattler M., Beyreuther K., Sinning I., Wild K. (2008) Structure of the intracellular domain of the amyloid precursor protein in complex with Fe65-PTB2. EMBO Rep. 9, 1134–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Radzimanowski J., Ravaud S., Schlesinger S., Koch J., Beyreuther K., Sinning I., Wild K. (2008) Crystal structure of the human Fe65-PTB1 domain. J. Biol. Chem. 283, 23113–23120 [DOI] [PubMed] [Google Scholar]
- 27. Zhou D., Zambrano N., Russo T., D'Adamio L. (2009) Phosphorylation of a tyrosine in the amyloid-beta protein precursor intracellular domain inhibits Fe65 binding and signaling. J. Alzheimers Dis. 16, 301–307 [DOI] [PubMed] [Google Scholar]
- 28. Barbagallo A. P., Weldon R., Tamayev R., Zhou D., Giliberto L., Foreman O., D'Adamio L. (2010) Tyr(682) in the intracellular domain of APP regulates amyloidogenic APP processing in vivo. PLoS One 5, e15503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lambert J. C., Goumidi L., Vrieze F. W., Frigard B., Harris J. M., Cummings A., Coates J., Pasquier F., Cottel D., Gaillac M., St C. D., Mann D. M., Hardy J., Lendon C. L., Amouyel P., Chartier-Harlin M. C. (2000) The transcriptional factor LBP-1c/CP2/LSF gene on chromosome 12 is a genetic determinant of Alzheimer's disease. Hum. Mol. Genet. 9, 2275–2280 [DOI] [PubMed] [Google Scholar]
- 30. Zambrano N., Minopoli G., de Candia P., Russo T. (1998) The Fe65 adaptor protein interacts through its PID1 domain with the transcription factor CP2/LSF/LBP1. J. Biol. Chem. 273, 20128–20133 [DOI] [PubMed] [Google Scholar]
- 31. Telese F., Bruni P., Donizetti A., Gianni D., D'Ambrosio C., Scaloni A., Zambrano N., Rosenfeld M. G., Russo T. (2005) Transcription regulation by the adaptor protein Fe65 and the nucleosome assembly factor SET. EMBO Rep. 6, 77–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kajiwara Y., Akram A., Katsel P., Haroutunian V., Schmeidler J., Beecham G., Haines J. L., Pericak-Vance M. A., Buxbaum J. D. (2009) FE65 binds Teashirt, inhibiting expression of the primate-specific caspase-4. PLoS One 4, e5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baek S. H., Ohgi K. A., Rose D. W., Koo E. H., Glass C. K., Rosenfeld M. G. (2002) Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-kappa B and beta-amyloid precursor protein. Cell 110, 55–67 [DOI] [PubMed] [Google Scholar]
- 34. Bimonte M., Gianni D., Allegra D., Russo T., Zambrano N. (2004) Mutation of the feh-1 gene, the Caenorhabditis elegans orthologue of mammalian Fe65, decreases the expression of two acetylcholinesterase genes. Eur. J. Neurosci. 20, 1483–1488 [DOI] [PubMed] [Google Scholar]
- 35. von Rotz R. C., Kohli B. M., Bosset J., Meier M., Suzuki T., Nitsch R. M., Konietzko U. (2004) The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J. Cell Sci. 117, 4435–4448 [DOI] [PubMed] [Google Scholar]
- 36. Muller T., Concannon C. G., Ward M. W., Walsh C. M., Tirniceriu A. L., Tribl F., Kogel D., Prehn J. H., Egensperger R. (2007) Modulation of gene expression and cytoskeletal dynamics by the amyloid precursor protein intracellular domain (AICD). Mol. Biol. Cell 18, 201–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Minopoli G., Stante M., Napolitano F., Telese F., Aloia L., De F. M., Di L. R., Pacelli R., Brunetti A., Zambrano N., Russo T. (2007) Essential roles for Fe65, Alzheimer amyloid precursor-binding protein, in the cellular response to DNA damage. J. Biol. Chem. 282, 831–835 [DOI] [PubMed] [Google Scholar]
- 38. Guenette S., Chang Y., Hiesberger T., Richardson J. A., Eckman C. B., Eckman E. A., Hammer R. E., Herz J. (2006) Essential roles for the FE65 amyloid precursor protein-interacting proteins in brain development. EMBO J. 25, 420–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maerkens A., Kley R. A., Olive M., Theis V., van der Ven P. F., Reimann J., Milting H., Schreiner A., Uszkoreit J., Eisenacher M., Barkovits K., Guttsches A. K., Tonillo J., Kuhlmann K., Meyer H. E., Schroder R., Tegenthoff M., Furst D. O., Muller T., Goldfarb L. G., Vorgerd M., Marcus K. (2013) Differential proteomic analysis of abnormal intramyoplasmic aggregates in desminopathy. J. Proteomics, 90, 14–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kley R. A., Maerkens A., Leber Y., Theis V., Schreiner A., van der Ven P. F., Uszkoreit J., Stephan C., Eulitz S., Euler N., Kirschner J., Muller K., Meyer H. E., Tegenthoff M., Furst D. O., Vorgerd M., Muller T., Marcus K. (2013) A combined laser microdissection and mass spectrometry approach reveals new disease relevant proteins accumulating in aggregates of filaminopathy patients. Mol. Cell. Proteomics 12, 215–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schrotter A., Pfeiffer K., El M. F., Platta H., Erdmann R., Meyer H. E., Egensperger R., Marcus K., Muller T. (2012) The APP family members are key players in S-adenosylmethionine formation by MAT2A and modify BACE1 and PSEN1 gene expression—relevance for Alzheimer's disease. Mol. Cell. Proteomics 11, 1274–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Muller T., Schrotter A., Loosse C., Pfeiffer K., Theiss C., Kauth M., Meyer H. E., Marcus K. (2013) A ternary complex consisting of AICD, FE65, and TIP60 down-regulates Stathmin1. Biochim. Biophys. Acta 1834, 387–394 [DOI] [PubMed] [Google Scholar]
- 43. Muller T., Schrotter A., Loosse C., Helling S., Stephan C., Ahrens M., Uszkoreit J., Eisenacher M., Meyer H. E., Marcus K. (2011) Sense and nonsense of pathway analysis software in proteomics. J. Proteome Res. 10, 5398–5408 [DOI] [PubMed] [Google Scholar]
- 44. Muller T., Loosse C., Schrotter A., Schnabel A., Helling S., Egensperger R., Marcus K. (2011) The AICD interacting protein DAB1 is up-regulated in Alzheimer frontal cortex brain samples and causes deregulation of proteins involved in gene expression changes. Curr. Alzheimer Res. 8, 573–582 [DOI] [PubMed] [Google Scholar]
- 45. Nowack A., Malarkey E. B., Yao J., Bleckert A., Hill J., Bajjalieh S. M. (2011) Levetiracetam reverses synaptic deficits produced by overexpression of SV2A. PLoS One 6, e29560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Meller K. (1992) Axoplasmic transport of horseradish peroxidase in single neurons of the dorsal root ganglion studied in vitro by microinjection. Cell Tissue Res. 270, 139–148 [DOI] [PubMed] [Google Scholar]
- 47. Spitzer P., Klafki H. W., Blennow K., Buee L., Esselmann H., Herruka S. K., Jimenez C., Klivenyi P., Lewczuk P., Maler J. M., Markus K., Meyer H. E., Morris C., Muller T., Otto M., Parnetti L., Soininen H., Schraen S., Teunissen C., Vecsei L., Zetterberg H., Wiltfang J. (2010) cNEUPRO: novel biomarkers for neurodegenerative diseases. Int. J. Alzheimers Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee E. J., Hyun S., Chun J., Shin S. H., Lee K. E., Yeon K. H., Park T. Y., Kang S. S. (2008) The PPLA motif of glycogen synthase kinase 3beta is required for interaction with Fe65. Mol. Cells 26, 100–105 [PubMed] [Google Scholar]
- 49. Sumioka A., Nagaishi S., Yoshida T., Lin A., Miura M., Suzuki T. (2005) Role of 14-3-3gamma in FE65-dependent gene transactivation mediated by the amyloid beta-protein precursor cytoplasmic fragment. J. Biol. Chem. 280, 42364–42374 [DOI] [PubMed] [Google Scholar]
- 50. Petrak J., Ivanek R., Toman O., Cmejla R., Cmejlova J., Vyoral D., Zivny J., Vulpe C. D. (2008) Deja vu in proteomics. A hit parade of repeatedly identified differentially expressed proteins. Proteomics 8, 1744–1749 [DOI] [PubMed] [Google Scholar]
- 51. Liu X., Yu X., Zack D. J., Zhu H., Qian J. (2008) TiGER: a database for tissue-specific gene expression and regulation. BMC Bioinformatics 9, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yamaguchi N., Xu L., Pasek D. A., Evans K. E., Chen S. R., Meissner G. (2005) Calmodulin regulation and identification of calmodulin binding region of type-3 ryanodine receptor calcium release channel. Biochemistry 44, 15074–15081 [DOI] [PubMed] [Google Scholar]
- 53. Schroetter A., Mastalsik T., Nensa F. M., Neumann M., Loosse C., Pfeiffer K., El Magraoui F., Platta H., Erdmann R., Theiss C., Uszkoreit J., Eisenacher M., Meyer H. E., Marcus K., Mueller T. (2013) FE65 regulates and interacts with the Bloom syndrome protein in dynamic nuclear spheres—potential relevance to Alzheimer's disease. J. Cell Sci. 126, 2480–2492 [DOI] [PubMed] [Google Scholar]
- 54. Arbabian A., Brouland J. P., Gelebart P., Kovacs T., Bobe R., Enouf J., Papp B. (2011) Endoplasmic reticulum calcium pumps and cancer. Biofactors 37, 139–149 [DOI] [PubMed] [Google Scholar]
- 55. Perez C. F., Lopez J. R., Allen P. D. (2005) Expression levels of RyR1 and RyR3 control resting free Ca2+ in skeletal muscle. Am. J. Physiol. Cell Physiol. 288, C640–C649 [DOI] [PubMed] [Google Scholar]
- 56. Rishi A. K., Yu M., Tsai-Wu J. J., Belani C. P., Fontana J. A., Baker D. L., Periasamy M., Hussain A. (1998) Gene amplification and transcriptional upregulation of the sarco/endoplasmic reticulum Ca2+ transport ATPase in thapsigargin-resistant hamster smooth muscle cells. Nucleic Acids Res. 26, 4529–4537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Goodger Z. V., Rajendran L., Trutzel A., Kohli B. M., Nitsch R. M., Konietzko U. (2009) Nuclear signaling by the APP intracellular domain occurs predominantly through the amyloidogenic processing pathway. J. Cell Sci. 122, 3703–3714 [DOI] [PubMed] [Google Scholar]
- 58. Ghosal K., Vogt D. L., Liang M., Shen Y., Lamb B. T., Pimplikar S. W. (2009) Alzheimer's disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc. Natl. Acad. Sci. U.S.A. 106, 18367–18372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sabo S. L., Lanier L. M., Ikin A. F., Khorkova O., Sahasrabudhe S., Greengard P., Buxbaum J. D. (1999) Regulation of beta-amyloid secretion by FE65, an amyloid protein precursor-binding protein. J. Biol. Chem. 274, 7952–7957 [DOI] [PubMed] [Google Scholar]
- 60. Trommsdorff M., Borg J. P., Margolis B., Herz J. (1998) Interaction of cytosolic adaptor proteins with neuronal apolipoprotein E receptors and the amyloid precursor protein. J. Biol. Chem. 273, 33556–33560 [DOI] [PubMed] [Google Scholar]
- 61. Rogawski M. A., Bazil C. W. (2008) New molecular targets for antiepileptic drugs: alpha(2)delta, SV2A, and K (v) 7/KCNQ/M potassium channels. Curr. Neurol. Neurosci. Rep. 8, 345–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Palop J. J., Mucke L. (2009) Epilepsy and cognitive impairments in Alzheimer disease. Arch. Neurol. 66, 435–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schallier A., Smolders I., Van D. D., Loyens E., De Deyn P. P., Michotte A., Michotte Y., Massie A. (2011) Region- and age-specific changes in glutamate transport in the AbetaPP23 mouse model for Alzheimer's disease. J. Alzheimers Dis. 24, 287–300 [DOI] [PubMed] [Google Scholar]
- 64. Cassano T., Serviddio G., Gaetani S., Romano A., Dipasquale P., Cianci S., Bellanti F., Laconca L., Romano A. D., Padalino I., LaFerla F. M., Nicoletti F., Cuomo V., Vendemiale G. (2012) Glutamatergic alterations and mitochondrial impairment in a murine model of Alzheimer disease. Neurobiol. Aging 33, 1121. [DOI] [PubMed] [Google Scholar]
- 65. Wang Y., Zhang M., Moon C., Hu Q., Wang B., Martin G., Sun Z., Wang H. (2009) The APP-interacting protein FE65 is required for hippocampus-dependent learning and long-term potentiation. Learn. Mem. 16, 537–544 [DOI] [PubMed] [Google Scholar]
- 66. Ho A., Morishita W., Atasoy D., Liu X., Tabuchi K., Hammer R. E., Malenka R. C., Sudhof T. C. (2006) Genetic analysis of Mint/X11 proteins: essential presynaptic functions of a neuronal adaptor protein family. J. Neurosci. 26, 13089–13101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bezprozvanny I., Mattson M. P. (2008) Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 31, 454–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Luo D., Sun H., Xiao R. P., Han Q. (2005) Caffeine induced Ca2+ release and capacitative Ca2+ entry in human embryonic kidney (HEK293) cells. Eur. J. Pharmacol. 509, 109–115 [DOI] [PubMed] [Google Scholar]
- 69. Corona C., Pensalfini A., Frazzini V., Sensi S. L. (2011) New therapeutic targets in Alzheimer's disease: brain deregulation of calcium and zinc. Cell Death Dis. 2, e176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Leissring M. A., Murphy M. P., Mead T. R., Akbari Y., Sugarman M. C., Jannatipour M., Anliker B., Muller U., Saftig P., De Strooper B., Wolfe M. S., Golde T. E., LaFerla F. M. (2002) A physiologic signaling role for the gamma-secretase-derived intracellular fragment of APP. Proc. Natl. Acad. Sci. U.S.A. 99, 4697–4702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Green K. N., Demuro A., Akbari Y., Hitt B. D., Smith I. F., Parker I., LaFerla F. M. (2008) SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J. Cell Biol. 181, 1107–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.