

Abstract
Type 2 diabetes mellitus (DM) appears to be a significant risk factor for Alzheimer disease (AD). Insulin and insulin-like growth factor-1 (IGF-1) also have intense effects in the central nervous system (CNS), regulating key processes such as neuronal survival and longevity, as well as learning and memory. Hyperglycaemia induces increased peripheral utilization of insulin, resulting in reduced insulin transport into the brain. Whereas the density of brain insulin receptor decreases during age, IGF-1 receptor increases, suggesting that specific insulin-mediated signals is involved in aging and possibly in cognitive decline. Molecular mechanisms that protect CNS neurons against β-amyloid-derived-diffusible ligands (ADDL), responsible for synaptic deterioration underlying AD memory failure, have been identified. The protection mechanism does not involve simple competition between ADDLs and insulin, but rather it is signalling dependent down-regulation of ADDL-binding sites. Defective insulin signalling make neurons energy deficient and vulnerable to oxidizing or other metabolic insults and impairs synaptic plasticity. In fact, destruction of mitochondria, by oxidation of a dynamic-like transporter protein, may cause synapse loss in AD. Moreover, interaction between Aβ and τ proteins could be cause of neuronal loss. Hyperinsulinaemia as well as complete lack of insulin result in increased τ phosphorylation, leading to an imbalance of insulin-regulated τ kinases and phosphatates. However, amyloid peptides accumulation is currently seen as a key step in the pathogenesis of AD. Inflammation interacts with processing and deposit of β-amyloid. Chronic hyperinsulinemia may exacerbate inflammatory responses and increase markers of oxidative stress. In addition, insulin appears to act as ‘neuromodulator’, influencing release and reuptake of neurotransmitters, and improving learning and memory. Thus, experimental and clinical evidence show that insulin action influences cerebral functions. In this paper, we reviewed several mechanisms by which insulin may affect pathophysiology in AD.
Keywords: Alzheimer’s disease, dementia and diabetes mellitus, insulin therapy, insulin receptor, insulin resistance
Introduction
Alzheimer’s disease (AD) is a neurological disorder characterized by profound memory loss and progressive dementia. The pathological and histological hallmarks of AD include amyloid plaques, neurofibrillary tangles and amyloidal angiopathy, accompanied by diffuse loss of neurons and synapses [1]. Environmental and genetic factors interact in the development of disease. Type 2 diabetes mellitus (DM) appears to be a significant risk factor for vascular dementia and AD in several epidemiological studies [2, 3]. Recent longitudinal studies have shown that AD and disorder of glucose metabolism are related [4, 5]. One explanation could be that vascular complications of diabetes result in neurodegenerative disease [6]. On the other hand, in addition to its peripheral metabolic effects, insulin also appears have important outcome on brain functions. A recent commentary offers two models of the link between Type 2 DM and AD: (1) ‘central insulin resistance’ and (2) inflammation. Both mechanisms influence insulin sensitivity in the brain, finally leading to β-amiloid accumulation and, consequently, to AD [7]. Complex molecular mechanisms, referring to insulin and/or insulin-like growth factor-1 (IGF-1) signalling could link DM and AD [8]. In fact, there is evidence that altered insulin and/or IGF-1 signalling to brain cells is probably responsible of amyloid accumulation in AD [9] and several independent effects of insulin on brain functions and cognitive performance have been described [10]. Insulin resistance with associated hyperinsulinemia are the mechanisms suggested to explain the increased AD risk in diabetes [11]. Subsequent investigations demonstrated reduced blood glucose levels and increased insulin levels in patients with late onset AD compared to aged controls or patients with vascular dementia. Although the authors concluded that these findings did not support an association between diabetes and AD [12], the same data were reinterpreted as an increased prevalence of insulin resistance in AD. The latter conclusion contradicts the finding that glucose administration could both increase plasma insulin levels and improve cognition in AD. Working under the assumption that increased insulin rather than glucose was responsible for the improvement in memory, further studies were used to demonstrate that the administration of insulin significantly improved memory performance in AD [8, 13]. Hyperinsulinemic euglycaemic clamp studies in humans showed improvement of attention in AD patients and neuroelectric changes in evoked potential induced by insulin [14]. In contrast, increases in plasma glucose that were not accompanied by increases in insulin levels did not influence cognitive performances [15]. The Rotterdam Study was one of the first epidemiology surveys to provide convincing evidence on a relationship between DM and dementia based on a significantly higher prevalence of dementia in patients with insulin-dependent (Type 1) DM compared to non-diabetic aged controls [3]. In addition, the possible association between DM-insulin resistance and degree of hippocampal and amygdala atrophy was investigated in vivo by magnetic resonance imaging [16]. The study showed that: (1) Individuals with DM had greater degree of hippocampal and amygdala atrophy compared with patients who did not have DM and (2) severity of insulin resistance associated with degree of amygdala atrophy. The inability to convincingly demonstrate a correlation between DM and AD, or find evidence that DM causes neuropathology, led to the alternative hypothesis that diabetes may serve as a cofactor in the pathogenesis of dementia and possibly AD. In this regard, epidemiological studies showed that hyperinsulinemia in patients with APO E4− genotype was correlated with AD-type dementia, whereas in the absence of diabetes, APO E4+ genotype was also correlated with AD [17], suggesting that APO E4 genotype and DM contribute independently to the pathogenesis of AD. Correspondingly, post-mortem studies have shown that individuals with DM and APO E4 genotype had significantly more abundant Aβ deposits and neurofibrillary tangles compared with diabetics who did not have an APO E4 allele [18]. In this review, we will summarize current evidence supporting the association between insulin action, insulin receptors, IGF-1 and AD, and we will describe the underlying mechanisms.
Insulin, IGF-1 in the brain
Insulin is almost exclusively synthesized and secreted into the plasma by pancreatic β cells and has important role in metabolic homeostasis. IGF-1 is synthesized by liver in response to pituitary growth hormone (GH) and affects growth processes; however, many other tissue, including the brain, are also able to synthesize IGF1 locally, out of GH control [19]. Although accumulated evidence indicate that insulin is derived from peripheral insulin and transferred by a transporter regulated way through the blood–brain barrier (BBB) [20, 21], there is also evidence consistent with local synthesis of insulin in the brain. In fact, Schechter et al. demonstrated that insulin can be produced locally in rabbit neuronal cells from culture [22]. Besides, Devaskar et al. revealed localization of insulin expressing neurons involved in associative areas of limbic system and areas regulating olfaction [23]. On the other hand, it is now generally thought that insulin synthesis in the brain is restricted is not synthesized to any significant amount in adult developed brain [21]. It may be possible that insulin could be synthesized during a specific period of brain development. In the larva of fruit fly Drosophila, insulin-producing cells exist in brain and secrete insulin into the circulatory system; ablation of these cells caused developmental delay, growth retardation and elevated carbohydrate levels, which are characteristics of human DM [24]. Over the past few years, it has become clear that insulin and IGF-1 also have intense effects in the central nervous system (CNS), regulating key processes such as energy homeostasis, neuronal survival, longevity, as well as learning and memory. Insulin and IGF-1 bind to tyrosine kinase receptors, the insulin receptor (IR) and IGF-1 receptor (IGF-1R), which share a high degree of identity in their structure and function. Insulin and IR are abundant but selectively distributed in the brain. Rodent studies have shown that insulin binding is highest in the olfactory bulb, cerebral cortex, hippocampus, hypothalamus, amygdala and septum [25, 26]. In the adult mammalian brain, two types of IR were found: a peripheral type and a neuron-specific type [27]. Insulin signalling within the cell is mediated, in general, by two functional cascades, one acting through the phosphatidylinositol-3 (PI3) kinase pathway, and other acting through the mitogen-activated protein kinase pathway [28]. Binding of insulin or IGF-1 induces a conformational change of the receptor and activates tyrosine-kinase which leads to auto-phosphorylation of the intracellular β-subunit [29]. Tyrosine-phosphorylate IR and IGF-1R β-subunits recruit and subsequently phosphorylate tyrosine residues of the intracellular insulin receptor substrates (IRS). The IRS protein family has at least four members, IRS-1 to -4 [30]. Giovannone et al. revealed that IRS proteins are homologue in structure and function but show distinct tissue distribution: IRS-1 and IRS-2 are widely distributed throughout different tissues and the brain, whereas IRS-3 is only expressed in rodent adipose tissue, and IRS-4 is predominantly localized in hypothalamus, thymus, skeletal muscle, heart, kidney and liver [31]. IR and IGF-1R are also expressed on brain capillaries and mediate the high-efficiency translocation of insulin and IGF-1 into the brain across the BBB [32, 33]. Insulin and IGF-1 interact in regulation of glucose disposal [34] and IGF-1 mimic insulin signalling in the brain [35], suggesting a partial overlapping. Several studies have shown the highest IR density in olfactory bulb, hippocampal formation, hypothalamus and cerebral cortex [36]. In post-mortem studies in adult humans, Adem et al. showed the highest IGF-1R density in hippocampus, amygdala and parahippocampal gyrus [37]. Whereas the density of brain IR decreases during age, IGF-1R increases, suggesting that specific insulin mediated signals are involved in aging and possibly cause age associated cognitive decline [38, 39]. Insulin has been shown to cross the BBB by different mechanisms: extracellular pathways, non-saturable transmembrane diffusion or saturable active transport [40]. Currently, the majority of studies suggest that the largest proportion of insulin crosses the BBB by receptor-mediated transport [41]. In contrast, insulin IGF-1 is formed within the CNS during the development and, to a lesser extent, in the mature brain [42]. However, besides the local expression of IGF-1, recent studies suggest that IGF-1 might cross the BBB via an analogous mechanism such as insulin [43].
Glucose metabolism and AD
Some of the earliest work on senile dementia, which probably corresponded to AD, vascular dementia or a combination of both, documented the development of altered brain metabolism soon after the onset of clinical symptoms [44, 45]. The metabolic abnormalities consisted of impaired glucose utilization and energy metabolism, with features that resemble Type 2 DM [44]. In addition, several studies confirmed that cerebral metabolism declined before the deterioration of cognitive functions, suggesting that energy failure is one of the earliest reversible hallmarks of AD. These observations led to the hypothesis that AD-associated abnormalities in energy metabolism are caused by IR action in the brain, that is brain diabetes [45]. Plasma glucose is transported across the BBB by several glucose transporter (GLUT) isoforms: GLUT 1 (expressed on BBB endothelial cells and cortical membranes), GLUT3 (expressed on neurons), GLUT4 and GLUT8 (expressed in intracellular compartments of neurons and translated to cell membranes in response to insulin) [46]. Rats express GLUT4 transporters in cerebellum, sensorimotor cortex, hippocampus, pituitary and hypothalamus [47]. Similarly, GLUT8 transporters are present in hippocampus and hypothalamus [48]. Therefore, overlapping distributions of insulin, IR and insulin-sensitive GLUT isoforms constitute a platform for insulin-stimulated glucose uptake in selective brain regions, such as the hippocampus, a structure that supports memory [47].
The insulin and IGF-1 system in AD
Type 2 diabetic patients are insulin resistant and have chronic hyperinsulinemia. In normal physiology, insulin facilitates memory as demonstrated when administration at optimal doses and in contrast of sufficient glucose availability [15]. The peripheral utilization of insulin reduces insulin transport into the brain, ultimately producing brain insulin deficiency [49], and abrogating the beneficial influences of insulin on the brain functions [15]. Different insulin levels have been observed in different brain regions [32, 50], probably linked to multiple insulin actions in CNS. Studies on Type 2 DM animal models have shown a reduced uptake of insulin into the brain. It was observed that obese diabetic Zucker rats have a decreased insulin transport into the brain, reduced brain levels of insulin and peripheral hyperglycaemia [32, 40, 50]. Recent studies linked diabetes with AD [8, 9, 18] and suggested that the brain may be influenced by changes in insulin levels and sensitivity. The observations that insulin, insulin receptors and C-peptide levels in cerebrospinal fluid (CSF) appears to be reduced in aging [51], along with the finding that AD patients have lower levels of insulin in the CSF, suggest impaired transport of insulin into the brain [52]. However, the salutary effect of insulin on brain functions are reserved under conditions that impair its functioning, such as insulin resistance [53]. Frolich et al. found that neuronal tyrosine-kinase activity is decreased in AD patients compared to age-matched controls [39]. The overall expression of IGF-1R is reduced in AD brains dependent on the severity of the disease. Brain IGF-1 mRNA levels diminish in severe AD, whereas IGF-1 serum levels are increased in early stages of the disease, suggesting that IGF-1 resistance plays a role in the pathogenesis of AD [39]. IRS-1/2 protein expression is reduced in AD brains, and inactivating Serine-phosphorylation of IRS-312 and Ser616 is improved, leading to impaired insulin resistance and IGF-1R signalling [54]. Given that IRS are widely expressed in the hippocampus, the most studied brain region for learning and memory, it seems to be plausible that decline of insulin resistance signalling leads to cognitive impairment [55]. Experiments with adult mice lacking liver IGF-1 production with an up of 85% reduction in circulating IGF-1 showed impaired spatial memory in the Morris water maze task compared to wild-type litter mates [55]. These findings might explain the reduction of cognitive functions during aging, because IGF-1 serum levels diminish under physiological conditions [56]. Unpredictably, studies in neuronal-IR-knockout mice (NIRKO) did not provide evidence for impairment in learning and memory, proposing that insulin resistance alone is not a key feature in dementia and neurodegeneration [17]. The revelation of a down-regulation of brain insulin signalling in obesity and diabetes leads to the proposal brain insulin resistance [57]. The down-regulation of neural insulin action results associated to change of hippocampal electrical activity [58] and to modulation of GABA [59], AMPA receptors [60] and N-L calcium channels [61]. Moreover, there are conflicting findings regarding the effects of antidiabetic therapy on clinical and neuropathology of AD. The Honolulu-Asia Aging Study demonstrated improvement of cognitive function and memory following induced hyperinsulinemia in patients with AD, independently of diabetes and blood glucose levels [2]. Conversely, the Rotterdam Study [3] observed increased risk of dementia in patients with diabetes treated with insulin. In fact, in this prospective study, 6370 non-demented and non-diabetic elderly patients were evaluated. At the end of follow-up, 126 patients became demented, of whom 89 had AD. DM almost doubled the risk of dementia [relative risk (RR) 1.9 (1.2–3.1)] and patients treated with insulin were at higher risk of dementia [RR 4.3 (1.7–10.5)]. In opposition, recent studies suggest that the combination of insulinic therapy with other diabetes medications is associated to a lower neuritic plaques [63] and to slower cognitive decline in patients with AD [13]. Besides, studies in animals have revealed the beneficial effects of peripheral and cerebroventricular injections of insulin on memory and learning [63]. Several studies have recognized that increasing plasma glucose levels improves memory in patients with AD [14, 15, 32]. Increasing plasma glucose levels also increases endogenous insulin levels, raising the query whether memory improvement is because of changes in insulin, independently of hyperglycaemia [14], although the exact mechanism remains unclear. Dense IR distributions have been documented in the dentate gyros, CA1 and CA3 fields of the hippocampus [64]. These regions are known to play a role in declarative memory and they are affected earlier and most severely by the neuropathologic changes of AD [65]. Increased plasma insulin levels result in amplified insulin binding in hippocampus. In turn, increased brain insulin levels results in enlarged glucose utilization in the entorhinal cortex [66]. Johnstone et al. observed that activation of the PI3 kinase pathway triggers GLUT4 translocation and consequently cellular glucose uptake in peripheral insulin-sensitive tissue as well as in some brain regions [28]. In contrast to the traditional notion that the brain is not an insulin-sensitive organ, insulin-promoted glucose utilization also results in glycolytic production of acetyl-CoA and subsequent increase in acetylcholine [67], a neurotransmitter closely linked to memory function and severely reduced in AD. Craft et al. confirm that elevated insulin without hyperglycaemia enhances memory in adults with AD, when endogenous insulin was suppressed by concomitant infusion of somatostatin analogues [14]. Moreover, the beneficial effect of insulin appears to be reduced when insulin resistance is present [17]. Craft et al. showed acute effect of hyperinsulinemia in older adults and in patients with AD using a hyperinsulinemic–euglycaemic clamps [15]. Low doses of insulin that result in plasma insulin levels of 10–20 μU/ml improve memory in normal patients. AD patients with insulin resistance required higher insulin doses (60–85 μU/ml) to obtain memory improvement. AD patients in this subgroup were not carriers of APO E4 allele. To date, no genetic risk factors have been identified for these patients, raising the possibility that factors relating to insulin resistance may be important for AD pathogenesis [15].
The insulin and oxidative stress in AD
Insulin promotes cell membrane expression of N-methyl-D-aspartate (NMDA) receptors, with increased neuronal Ca2+ influx [69]. Ca2+ influx presumably activates Ca2+-dependent enzymes, including α-dependent enzymes and strengthens neuronal synaptic association [10]. Besides, a recent study identified a molecular mechanism that protects CNS neurons against β-amyloid-derived-diffusible ligands (ADDL), responsible for synaptic deterioration underlying AD memory failure. The authors found ADDL binding to particular synaptic sites, and the resulting oxidative stress on synapses loss are markedly decreased by the presence of insulin. The protection mechanism does not involve simple competition between ADDLs and insulin, but rather is signalling dependent down-regulation of ADDL-binding sites [69]. Another metabolic disturbance of emerging importance in AD involves insulin signalling in the brain. Levels of insulin receptors, glucose-transport proteins and other insulin pathway components are reduced in some studies of AD brain (central resistance) [32]. Han et al. proposed a central insulin resistance together with decreased brain insulin levels might lead to accumulation of β-amyloid and consequently AD [7]. Insulin and brain-derived IGF-1 instigate signals in the brain by activating the PI3 kinase–Akt pathway and the mitogen-activated protein kinase-extracellular signal-regulated kinase pathway [70], but it is unclear whether signalling is up-regulated (compensatory) or down-regulated (pathologic) in AD. Aging and life span are also influenced by insulin. Resistance to insulin signalling makes neurons energy deficient and vulnerable to oxidizing or other metabolic insults and impairs synaptic plasticity [71]. Both in AD and in normal aging process mtDNA sustains high levels of oxidative damage [72] (Fig. 1). In fact, it was observed the accumulation of Aβ within structural damaged mitochondria isolated from the brains of AD patients [72, 73] and transgenic brains [74], which impair critical mitochondrial enzymes. Dysfunctional mitochondria release oxidizing free radicals, with peroxidation of membrane lipids and output of toxic aldehydes that cause considerable oxidative stress in AD and in normal aging brains [75]. Other essential proteins resulted oxidized, yielding carbonyl and nitrated derivatives, in neuronal cytoplasm in cerebral regions of neurodegeneration, in human brain affected by AD [76]. Subsequently, increased membrane permeability to calcium, and impaired glucose transport aggravate the energy imbalance [77]. Experimental model show that markers of oxidative damage precede pathological changes [78]. Destruction of mitochondria by the oxidation of a dynamic-like transporter protein may cause synapse loss in AD [79]. The ‘receptor for advanced glycation end products’ (RAGE) mediates Aβ’s pro-oxidant effects on neural, microglial and cerebrovascular cells [80]. The RAGE receptor is a multi-ligand receptor, and one of its ligands is Aβ [80]. RAGE regulates several intracellular pathways [81], such stimulates expression of b-site amyloid precursor protein (APP)-cleaving enzyme 1 (BACE1) [82], an enzyme that is necessary for Aβ production. Moreover, RAGE seems to negatively affect the long-term potentiation (LTP) synaptic process of learning and memory [83]. Moreover, RAGE can induce its own expression through activation of the transcription factor NF-κB [81]. RAGE also exists in a soluble form, structured by alternative splicing [84] or proteolytic cleavage by the metalloprotease 10 (ADAM 10) [85]. Soluble RAGE (sRAGE) contains the ligand-binding site, but does not have the signalling properties of full-length RAGE (flRAGE). Soluble RAGE should also have an increased propensity to scavenge Aβ, thus increased protective properties. Nevertheless it was observed that flRAGE is engaged in positive feedback mechanisms, enhancing its own production, and limiting sRAGE proposed protective actions. This notion is supported by the finding that flRAGE expression is increased in AD brains [86]. Indeed, studies have shown that sRAGE can inhibit the accumulation and aggregation of Aβ in mice brains [87]. In addition, it has been shown that sRAGE is present at lower levels in the blood and brain of AD patients [88]. Abnormal expression of RAGE forms in AD brain suggests that it is relevant to the pathogenesis of neuronal dysfunction and death.
Fig 1.
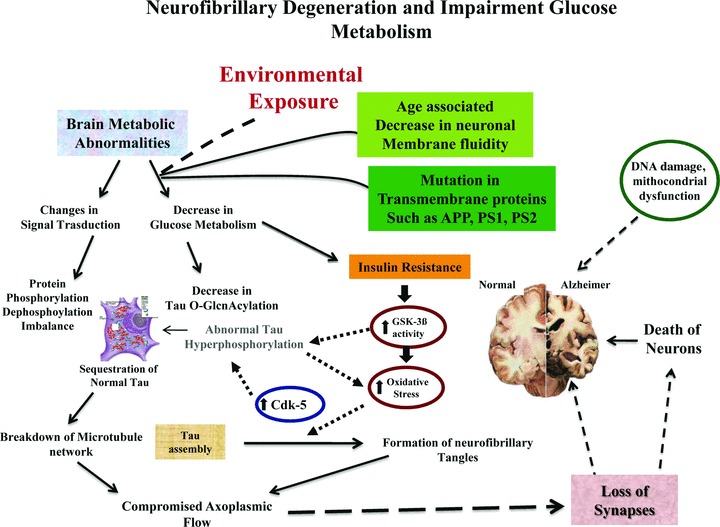
Protein phosphorylation/dephosphorylation imbalance is generated, at least in part, by a decrease in the activities of τ phosphatases and increase the activities of τ kinase (i.e. cdk5, GSK-3, etc.) affected by insulin. Impaired insulin signalling stimulates GSK-3β activity that increases oxidative stress and τ hyper-phosphorylation, by Cdk-5. Severe or sustained oxidative injury leads to mithocondrial DNA damage, mithocondrial dysfunction, apoptosis and the attendant cell loss and impaired neuronal function lead to dementia. Age reduces membrane fluidity inducing mutations in transmembrane proteins (i.e. PS1, PS2, APP), and vulnerability of the cell membrane to variation in pathological signal transduction.
The insulin and IGF-1R system and τ phosphorylation
Neurofibrillary tangles are intracellular polymers of τ proteins, observed in cytoplasm of neurons [89] in AD and in other neurodegenerative disorders, such as frontotemporal dementia, Pick’s disease, corticobasal degeneration, supranuclear palsy. Normally, soluble τ proteins assemble with tubulin to constitute cross bridge between adjacent microtubule and promote stability of microtubules and vesicle transport [89]. Neurofibrillary tangles are hyperphosphorylated and aggregated form of τ proteins. Hyperphosphorylation of τ induces abnormal insoluble τ protein, formation of helical filaments and lack of affinity for microtubules [90]. Studies in humans [91] and in mice [92] supposed that the interaction between Aβ and τ proteins is necessary to cause neuronal loss. When hyperphosphorylated, τ aggregates and interferes with intraneuronal metabolism and transport, leading to neurodegeneration. Protein phosphatases 2A (PP2A) is the major phosphatases with 70% activity in human brains [93]. This implies a protective role of PP2A in neurodegeneration, which is consistent with the finding that PP2A activity is reduced in AD brains [94]. In vitro experiments demonstrated that τ phosphorylation is normally regulated by insulin and IGF-1 [95]. The phosphorylation of τ is mainly promoted by GSK-3β and cyclin-dependent-kinase5 (Cdk5). GSK-3β is functionally important for regulating glycogen metabolism, cell cycle kinetics, proliferation survival and cell migration. These effects are mediated by growth factor stimulating phosphorylation and attending inhibition of GSK-3β activity [90] (Fig. 1). GSK-3β is a serine/threonine kinase, modulated by insulin-IGF-1 signalling. However, PP2A dephosphorylates GSK-3β [96], which then phosphorylates τ at several sites leading to an equilibrium of phosphorylation and dephosphorylation of τ. Thus, impaired insulin resistance-IGF-1R signalling might lead to hyperphosphorylation of τ protein and increased formation of neurofibrillary tangles. In IGF-1 knockout mice, a substantial increase of site-specific τ phosphorylation at Ser396 and Ser202 could be demonstrated whereas τ mRNA as well as τ protein levels remained unchanged [95]. This suggests a protective role of IGF-1 to prevent τ hyperphosphorylation. Hyperphosphorylation of τ at Thr231 was found in NIRKO mice brain [58]. Furthermore, in IRS-2 knockout mice hyperphosphorylation of τ at Ser202 was demonstrated [97]. The pattern of τ phosphorylation in NIRKO mice is different from IRS-2 deficient mice, suggesting that not only insulin resistance, occurring in both mouse models, but also other factors might be involved, for example hyperinsulinemia to compensate insulin resistance. Freud et al. showed that the brain is a direct target of peripheral circulating insulin where it leads to intraneuronal insulin receptor signalling inducing presumably unfavourable τ phosphorylation, predisposing for tangle formation [98]. In fact, authors demonstrated a significant increase of τ phosphorylation at Ser202 in the brain within 10 min. after 1-mU insulin injection, indicating a correlation in vivo between τ phosphorylation and peripheral insulin levels [98]. Thus, insulin-stimulated τ phosphorylation provides a mechanism linking insulin action and insulin resistance to neurodegeneration. Moreover, previous studies on cultured neurons in vitro have revealed contrary effects of insulin on τ phosphorylation, depending on the cell type investigated. In human SH-SY5Y neuroblastoma cells [99] and primary cultures of rat cortical neurons [100], insulin treatment resulted in an increase of τ phosphorylation. However, in cultured human neurons (NT2 cells), GSK-3 was inhibited, with reduced τ phosphorylation after insulin stimulation [101]. Schubert et al. tested NIRKO mice under hyperinsulinemic conditions [58, 97]. IR signalling and τ phosphorylation were completely abolished in these mice brains, indicating that cerebral IR are a direct target of peripheral administered insulin. Streptozocin (STZ) is specifically toxic for pancreatic β cells. Chronic treatment causes impairment of insulin secretion. In STZ mice, increased τ phosphorylation at multiple sites has be shown, which was reversible after peripheral insulin treatment [102]. These results show that insulin deficiency causes an increase of τ phosphorylation. Intact insulin signalling is important for promoting neuronal survival and energy metabolism, whereas impaired insulin signalling in CNS neurons results in increased GSK-3β activity, which leads to τ hyperphosphorylation. In addition, GSK-3β can be activated by hypoxic, ischaemic or metabolic injury, irrespective of growth factor stimulation [103]. Hyperphosphorylated τ fails to be transported into axons and instead accumulates and aggregates in neuronal perikarya. Besides τ hyperphosphorylation promotes further oxidative stress [104], which can cause cell death mediated by apoptosis, mitochondrial dysfunction or necrosis. The neuronal cytoskeletal lesions that correlate with dementia in AD contain hyperphosphorylated τ. Moreover, GSK-3β, activating c-Jun N-terminal-kinases (JNK) and ERK-1-2 signalling, leading to an increase in τ phosphorylation JNK and ERK-1-2, might be of importance in AD pathogenesis. In particular, Dickens et al. showed that JNK-interacting protein 1 (JIP1) caused cytoplasmic retention of JNK and inhibition of JNK-regulated gene expression [105]. In fact, the authors identified JIP1 as inhibitor of JNK signal pathway. Besides, JIP1 seems play a more direct role in targeting JNK to specific cellular regions and substrates, that is cytoplasm [105]. It is tempting to speculate that JIP1–APP interaction could provide the molecular basis for activation of signalling cascades, involving mitogen-activated and stress-activated kinases in the brain of AD patients, that may ultimately be responsible for τ hyperphosphorylation and NFT formation [102, 106]. Although the mechanisms of increased GSK3β activation in AD can be readily explained on the basis of impaired insulin-IGF-1 signalling, the increased levels of ERK [107], AKT [108] and Cdk-5 [109] detected in AD brains cannot be attributed to these abnormalities. However, as noted earlier, GSK-3β can also be activated by oxidative stress. Review of the literature revealed that in addition to IGF-1 stimulation, ERK [110], AKT [111] and Cdk-5 [110] activities also can be increased in response to oxidative stress. Hyperinsulinemia, as well as complete lack of insulin, results in increased τ phosphorylation, leading to the hypothesis that hyperphosphorylation of τ follows an imbalance of insulin-regulated τ kinases and phosphatates [100].
Brain amyloid and insulin IGF-1 signalling
The amyloid plaques is formed by amyloid β (Aβ) peptides organized in fibrils intermixed with non-fibrillar forms of this peptide and are surrounded by dystrophic dendrites, axons, reactive astrocytes and activated microglia. Aβ consists of small hydrophobic peptides with N- and C-terminal heterogeneity, that is Aβ1-40 and Aβ1-42 which are proteolytically released from a large Type 1 integral membrane glycoprotein, the APP, via sequential cleavage by two aspartyl proteases, the β- and γ-secretases [enzymatic complex, containing nicastrina, presenilina, preselin enhancer-2 (PEN-2), CD147] [112]. Initial β-secretase cleavage generates a soluble fragment from the NH2-terminus of APP, whereas the c-terminal fragment (β-CTF) stays membrane bound. Aβ40/42 activates Ca2+ influx in neurons, hyperphosphorylation of τ protein (via activation of GSK3β and CDK5), leading to deposition of neurofibrillary tangles, impaired axonal transport and finally, to neuronal death (Fig. 2). Full-length APP can undergo alternative processing by α-secretase, generating a soluble APPsα ectodomain and a membrane-bound carboxy-terminal fragment, APP-CTFα. Processing of APP by α-secretase is postulated to be protective in the context of AD, because the enzyme cleaves within the Aβ-sequence, thereby preventing the production of Aβ. Several studies have indicated that increased α-secretase-mediated processing of APP reduces the processing of APP by β-secretase and decrease Aβ-production [113, 114], however this finding has not been universally replicated [115]. Increasing α-secretase activity, as mentioned above, increases the production of APPsα, and has been reported to be neuroprotective and growth promoting [116], but the consequences of chronically up-regulating α-secretase-mediated cleavage of other substrates remain unknown. Besides, in a recent study, Adlerz et al. suggested that increased levels of APPsα and amyloid-precursor-like protein-1 (APLP1), in response to either IGF-1 or insulin, were mediated by activation of IGF-1 receptors [117]. APP, αCTF and βCTF are further cleaved by γ-secretase to generate p83 fragment and Aβ, respectively [118]. In a recent study, McElroy et al. suggested a possible link between IGF-1R and γ-secretase. IGF-1R, as several Type 1 membrane proteins, undergoes regulated intramembrane proteolysis. Afterwards metallo-protease-dependant ectodomain-shedding, IGF-1R carbossiterminal domain is cleaved by γ-secretase. These observations suggest that IGF-1R may be substrate for γ-secretase involved in mechanisms independent of its receptor tyrosine-kinase activity [119]. Multiple lines of biochemical evidence have shown γ-secretases activity to reside in a high molecular weight complex, consisting of at least four components: presenilin (PS, PS1, PS2), nicastrin, anterior pharynx-defective (APH-1) and PEN-2 [120]. The p83 fragment is rapidly degraded and widely believed to possess no important function, if any. γ-Secretase-mediated cleavage is unique in that the cleavage takes place within the membrane domain, although the exact site can vary. γ-cleavage can yield both Aβ1-40 and to a lesser extent Aβ1-42[118]. Aβ are toxic, and their accumulation is currently seen as a key step in the pathogenesis of AD. Closer examination of the amyloidogenic β- and γ-secretates discovered the membrane-anchored aspartyl protease β-site BACE-1, which acts as β-secretase and presenelin 1-2, transmembrane proteins involved in formation of the γ-secretase complex, as the responsible cleavage enzymes. Thus, alteration of their activity might be a possible target for AD treatment [121]. It has been shown that BACE-1 levels are increased in post-mortem brain sections from AD patients [122]. During aging changes in the cerebral expression levels of the neurotrophin receptors tyrosine kinase receptor A (TrkA) and p75 neurotrophin receptorv (p75NTR) have been described. In the human neuroblastoma cell line SHSY5Y as well as primary cultured neurons, chronic treatment with IGF-1 leads to a switch from TrkA to p75NTR expression as seen in aging brains [123]. This switch causes increased β-secretase activity indirectly by activation of neuronal sphingomyelinase, which is responsible for hydrolysis of sphingomyelin and active liberation of the second messenger ceramide [124]. Ceramide is responsible for the molecular stabilization of BACE-1, the β-secretase which is rate limiting for generation of Aβ [125]. This process leads to accumulation of Aβ, connecting IGF-1R signalling to neurotrophin action. Furthermore, Sotthibundhu et al. recently showed that treatment of wild-type embryonic mouse hippocampal neurons with Aβ1-42 as ligand for p75NTR resulted in significant cell death. In contrast, p75NTR deficient neurons are less affected by Aβ1-42 treatment [126]. These data might provide a molecular link between aging, pathogenesis of AD and neuronal insulin-IGF-1 signalling. Lots of research has been performed on the formation and accumulation of Aβ, however, in the last years the mechanisms of amyloid clearance came into focus. Aβ spontaneously self-aggregates into multiple coexisting physical forms, such as oligomers (2–6 peptides), intermediate assemblies, fibrils that coalesce into β pleated sheets to form insoluble fibres and amyloid plaques [127]. Although monomeric Aβ is not neurotoxic, the Aβ oligomers exhibits a marked toxicity [128]. The physiological role of β peptides is still in part unclear, but it is involved in neuronal activation and connection mechanisms [129]. Neuronal activation rapidly increase Aβ secretion at the synapse, during the process of neurotransmitters release. Normal levels of Aβ at this site may modulate neuronal transmission and prevent hyperactivity [129]. It was assumed that imbalance between production, aggregation and clearance of peptides is considered initiating factor in AD [130]. The molecular mechanisms involved in the secretion, aggregation and toxicity of Aβ are still in part unknown [130]. For Aβ clearance several mechanisms have been described: (1) enzymatic degradation by activated microglia or by insulin-degrading enzyme (IDE), neprilysin, endothelin-converting enzyme (ECE) and angiotensin-converting enzyme (ACE); (2) receptor-mediated transport across the BBB by binding to the low-density lipoprotein receptor-related protein (LRP), either directly or after binding to APO E and/or α2-macroglobulin (α2M), to be delivered to peripheral sites of degradation, for example liver and kidney [50]. Concerning insulin resistance it has been shown that IDE expression is stimulated by the insulin resistance-IGF-1R cascade (Fig. 3) [131]. Furthermore, it has been suggested that increasing circulating IGF-1 levels lead to reduction of Aβ burden in aging rats [132]. It has been recently reported that membrane associated G protein-coupled receptor kinase-5 (GRK5) deficiency occurs during early AD [133]. In deficient GRK5 mice (tg2576-APPsw), Aβ accumulation resulted significantly increased [133]. IGF-1 administration resulted in reduction of cerebral Aβ load in these mice, whereas Aβ was elevated in CSF suggesting an increased Aβ elimination across the BBB or the choroid plexus [134]. Furthermore, it has been shown that the blockade of the IGF-1R in the choroid plexus triggers AD-like pathology [134]. In contrast, Lanz et al. [135] analysed in vivo models using acute, subchronic and chronic IGF-1 treatment to evaluate Aβ levels in brain, CSF and plasma of rats and Tg2576 mice. However, no changes in Aβ were detected in any of these models. Furthermore, τ phosphorylation did not change significantly following chronic IGF-1 treatment in Tg2576 mice [134]. A possible explanation could be that the chronic increase of IGF-1 by peripheral treatment might down-regulate IGF-1R signalling. This hypothesis is supported by the finding that in a cohort of individuals with exceptional longevity serum IGF-1 levels were high but IGF-1R activity was low leading to reduced IGF-1R signalling [136]. However, induction of insulin resistance by high fat diet [137] or intake of sucrose-sweetened water [138] leads to an aggravation of amyloid pathology in mouse models of AD. Furthermore, peripheral injection of supra physiologically high insulin doses but not of physiological doses leads to transient cerebral τ phosphorylation [98], leading to the proposal that there is a dose-dependent effect of insulin resistance-IGF-1R signalling in the pathogenesis of AD.
Fig 2.
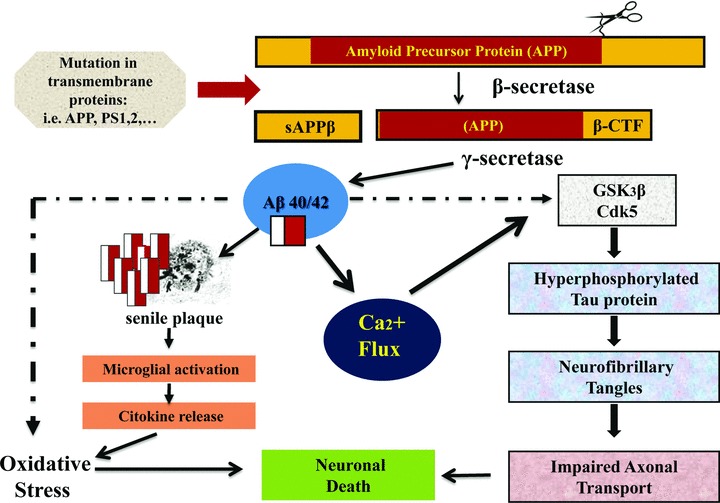
Aβ is a small peptide with N- and C-terminal heterogeneity, that is Aβ1-40 and Aβ1-42, which are released from a transmembrane glycoprotein, the amyloid precursor protein (APP) via sequential cleavage by aspartyl proteases: the β and γ secretases. Aβ/40/42 activates Ca2+ influx in neurons, hyperphosphorilation of τ protein (via activation of GSK3β and CDK5), leading to oxidative stress, deposition of neurofibrillary tangles, impaired axonal transport and, finally, to neuronal death.
Fig 3.
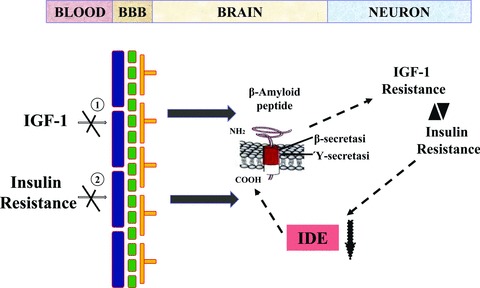
Reduced levels of (1) IGF-1 and peripheral (2) insulin resistance (IR) induced decrease uptake of IGF-1 and insulin into the brain, resulting in β amyloid accumulation. Increased levels of β amyloid antagonize insulin and IGF-1 binding to their corresponding receptors, worsening insulin/IGF-1 resistance in neurons. Neuronal IR contributes to accumulation of β amyloid by competing for insulin-degrading enzyme (IDE), a protease that degrades both insulin and β amyloid. Aβ accumulation can be linked to cellular metabolic conflict caused by τ hyperphosphorylation. Aβ accumulation can inhibit insulin actions and binding, and can also be neurotoxic.
Insulin, inflammation and AD
Inflammation has been proposed as a key pathogenetic factor for AD [139]. Elevated concentrations of interleukin (IL)-6 E2 isoprostane have been observed in CSF of patients with AD [140]. Furthermore, in vitro and animal studies suggest that inflammation interacts with processing and deposit of Aβ [141]. In the periphery, insulin modulates many aspects of the inflammatory network. Low doses of insulin exert anti-inflammatory effects [142]; however, during chronic hyperinsulinemia, insulin may exacerbate inflammatory responses and increase markers of oxidative stress [143]. In human, co-administration of insulin and lipopolysaccharide produces a synergist increase in plasma concentrations of C-reactive protein and pro-inflammatory cytokines (IL-1β, IL-6, TNF-α) [144]. Besides, recently Han and Li suggested that inflammation is able to accelerates the development Type 2 DM, through its influence on peripheral insulin sensitivity and pancreatic islet function. Consequently, cerebrovascular and central inflammation disrupts normal synaptic function, promoting AD pathological development [7]. Insulin may also modulate levels of eicosanoids such as F2-isoprostane via regulation of prostaglandin production in adypocites [145]. For example, elevated eicosanoid concentrations have been observed in hyperisulinemic Zucker rats [50]. Insulin may also contribute to inflammation in the CNS, partially through effects on Aβ. In fact, Aβ42 interacts with inflammatory agents in a cyclically reinforcing manner, such that Aβ elevations increase pro-inflammatory cytokines [146]. In vitro, soluble Aβ oligomers rapidly increase IL-1β and TNF-α levels [147]. Conversely, IL-6 and IL-1β can regulate processing of the APP from which Aβ is derived and increase production of Aβ42 [148]. The mutually reinforcing effects of Aβ, TNF-α, IL-1β and IL-6 may thus create a ‘cytokine cycle’ [146]. Insulin can regulate CNS norepinephrine [149], an endogenous anti-inflammatory neuromodulator that blocks IL-1β expression [150]. Increased Aβ plaque load in AD has been linked to neuronal loss in the locus coeruleus, the primary source of brain norepinephrine [151]. Thus, decreased norepinephrine activity in Aβ may potentate the deleterious inflammatory effects of Aβ. TNF-α may affect the transport of Aβ from brain to periphery. In fact, TNF-α may antagonize clearance action of IGF-1 mediated by carrier proteins, such as albumin and transretine [152]. Moreover, TNF-α has both neurotoxic and neuroprotective effects mediated respectively by two receptor subtypes, TNF-R1 and TNF-R2. TNF-R1 contains a death receptor domain, and has been implicated in pro-apoptotic events, whereas TNF-R2 promotes cell survival. Increased levels of TNF-R1 and decreased levels of TNF-R2 have been observed in AD brain [153], and in peripheral lymphocytes from older adults compared with younger adults [154]. Abnormal levels of soluble TNF-R1 and -R2 have been documented in adults with diabetes and impaired glucose tolerance [155], which reportedly normalize after a 3-weeks low calorie diet [156]. In the periphery, insulin reduces hepatic production of APO E and regulates its uptake by low-density lipoprotein receptor-related protein [157]. Fishel et al. showed that insulin reduced plasma APO E levels, an effect that increased with age. In contrast, insulin increased CSF APO E concentrations for older patients [158]. Increased brain APO E levels have been reported in AD in association with polymorphisms in the promoter region of the APO E gene that influence protein expression [159]. Besides, Fishel et al. observed that insulin-induced elevations of CSF APO E levels were associated with attenuated increase IL6 and TNFα levels and with higher anti-inflammatory cytokine, IL-1α concentration. This pattern suggests multiple insulin effects that modulate the role of APO E in response to inflammation in CNS [158].
Conclusions
DM is associated to slowly progressive end organ damage in the brain. Mild to moderate impairments of cognitive functioning has been reported both in patients with Type 1 DM and in patients with Type 2 DM. The potential impact of DM on cognitive functions in the elderly is further emphasized by several large epidemiological surveys that report an increased incidence of dementia among DM patients both AD and vascular dementia. The observation that the effects of DM on the brain are most pronounced in the elderly has been attributed to an interaction between DM and the normal aging process in the brain. Several mechanisms may be involved in accelerated cognitive decline in patients with DM. It may be possible, however, to identify process or clusters of risk factors, which explain at least part of this association, and to be targeted by preventive measures. These preventive measures may not only include improvement of glycaemic control, but could also be directed at vascular risk factors and insulin metabolism. Insulin may affect the metabolism of Aβ and τ, two proteins that represent the building blocks of amyloid plaques and neurofibrillary tangles, the neuropathological hallmarks of AD. Insulin is not a major regulator of glucose used in the brain, in contrast with its prominent action in peripheral tissue such as liver, muscle and fat. Besides, insulin and its receptor are widely distributed throughout the brain, with particular abundance in defined areas, such as the hypothalamus and the hippocampus. In addition, insulin appears to act as ‘neuromodulator’ that influences the release and reuptake of neurotransmitters, and improves learning and memory. The initial components of the insulin receptor signalling cascade in the brain are largely similar to those of the periphery, but the downstream targets of the cascade are quite different, probably involving, among others, neuronal glutamate receptors [160]. Insulin therapy plays an important role in cognitive processes and could slow dementia in AD patients; this could be explained by: (1) molecular mechanisms, insulin promotes cell membrane expression of NMDA receptors, which increases neuronal Ca2+ influx [68], that activates Ca2+-dependent enzymes, including α-dependent enzymes and strengthens neuronal synaptic association [10]. The potentiation of NMDA receptor activity insulin-induced does not occur by direct phosphorylation of the C-terminal tails of the receptor protein, but by associated targeting anchoring, or signalling protein(s). Together, these findings are consistent with a mechanism whereby insulin acting through the IR tyrosine kinase, phosphorylates one or more protein(s) involved in receptor signalling or trafficking. A recent study found that Aβ ADDL binding to particular synaptic sites and the resulting oxidative stress and synapse loss are markedly decreased by the presence of insulin [161]. (1) The protection mechanism does not involve simple competition between ADDLs and insulin, but rather is a signalling-dependent down-regulation of ADDL-binding sites [162]; (2) glucose metabolism, low concentrations of exogenous insulin may increase cerebral glucose metabolism and then modulate brain functions such as memory [163]. In fact, insulin has shown a significant effect on global brain glucose metabolism and this effect is mainly expressed in the cerebral cortex. This may be either a direct effect of insulin stimulating glucose uptake, as in peripheral tissue, or an indirect effect achieved via insulin-stimulated neuronal activation with secondary increment of glucose metabolism in cell. Either way, insulin can access the insulin receptors of the brain and have a metabolic effect, which may be maximal at basal (fasting) circulating insulin concentrations; (3) neurotransmitter modulation, low doses of insulin can reverse the amnestic effects of cholinergic blockade (Fig. 4) [164]. Disturbances in cerebral insulin signalling pathways may be involved in AD and brain aging. Aging is associated with reduction in insulin levels and the number of its receptors in the brain. In AD age-related reduction in cerebral insulin levels appears to be accompanied by disturbance of the IR signalling. Aβ is derived from the amyloid precursor protein. After secretion into the extracellular space Aβ can aggregate with other proteins, to form senile plaques. Alternatively, excessive Aβ can be cleared through LDL-receptor-related protein mediated endocytosis, or through direct extracellular proteolytic degradation. This latter process involves IDE. Insulin appears to stimulate Aβ secretion, and inhibits the extracellular degradation of Aβ by competition from IDE. A recent histopathological study of the hippocampus in patients with AD reported marked reductions in IDE expression, and IDE mRNA levels, relative to controls. Although the concepts of ‘cerebral insulin resistance’ and ‘insulin-induced amyloid pathology’ are an attractive explanation for some of the effects of DM2 on the brain, there are still many loose ends. In contrast with the increasing body of knowledge on the mechanisms of insulin resistance in peripheral tissue, we know relatively little on how DM2 and its treatment affect cerebral insulin and its receptor. It is important to point out that definitive conclusions about the value of insulinic treatment in course of AD cannot be established at this time. Further studies are required to define the effects of insulinic treatment on cognitive functions.
Fig 4.
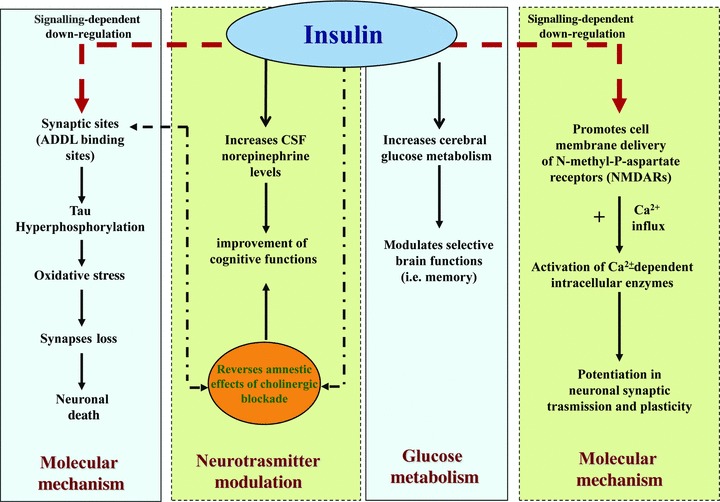
(1) Molecular mechanism insulin promotes cell membrane expression of NMDA receptors, which increases neuronal Ca2− influx-dependent enzymes and strengthens neuronal synaptic association. Besides, diffusible ligands (ADDL) binding to particular synaptic sites and the resulting oxidative stress and synapse loss are markedly decreased by the presence of insulin. This mechanism is associated with a signal-dependent down-regulation of ADDL-binding sites. (2) Glucose metabolism low concentrations of exogenous insulin may increase cerebral glucose metabolism and then modulate brain functions, such as memory. (3) Neurotransmitter modulation insulin increases CSF levels of norepinephrine and reverses the amnestics effects of cholinergic blockade.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Citron M. Alzheimer’s disease: treatment in discovery and development. Nat Neurosci. 2002;5:1055–7. doi: 10.1038/nn940. [DOI] [PubMed] [Google Scholar]
- 2.Peila R, Rodriguez BL, Lauren LJ. Type-2 diabetes, diabetes, Apo E gene and the risk for dementia and related pathogenesis: the Honolulu-Asia Aging Study. Diabetes. 2002;51:1256–62. doi: 10.2337/diabetes.51.4.1256. [DOI] [PubMed] [Google Scholar]
- 3.Ott A, Stolk RP, Hofman A, et al. Diabetes mellitus and risk of dementia: the Rotterdarm Study. Neurology. 1999;53:1907–9. doi: 10.1212/wnl.53.9.1937. [DOI] [PubMed] [Google Scholar]
- 4.Luchsinger JA, Tang MX, Shea S, et al. Hyperinsulinemia and risk of Alzheimer’s disease. Neurology. 2004;63:1187–92. doi: 10.1212/01.wnl.0000140292.04932.87. [DOI] [PubMed] [Google Scholar]
- 5.Janson J, Laedtke T, Parisi JE, et al. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes. 2004;53:474–81. doi: 10.2337/diabetes.53.2.474. [DOI] [PubMed] [Google Scholar]
- 6.Whitmer RA, Sidney S, Selby J, et al. Midlife cardiovascular risk factors and risk of dementia in late life. Neurology. 2005;64:277–81. doi: 10.1212/01.WNL.0000149519.47454.F2. [DOI] [PubMed] [Google Scholar]
- 7.Han W, Li C. Linking type 2 diabetes and Alzheimer’s disease. Proc Natl Acad Sci USA. 2010;107:6557–8. doi: 10.1073/pnas.1002555107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freude S, Schilbach K, Schubert M. The role of IGF-1 receptor and insulin receptor signaling for the pathogenesis of Alzheimer’s disease: from model organism to human disease. Curr Alzheimer Res. 2009;6:213–23. doi: 10.2174/156720509788486527. [DOI] [PubMed] [Google Scholar]
- 9.Carro E, Torres-Aleman I. The role of insulin and insulin-like growth factor I in the molecular and cellular mechanisms underlying the pathology of Alzheimer’s disease. Eur J Pharmacol. 2004;490:127–33. doi: 10.1016/j.ejphar.2004.02.050. [DOI] [PubMed] [Google Scholar]
- 10.Byrne JH. Learning and memory: basic mechanism. In: Squire LR, Bloom FE, Mc Connel SK, Roberts JL, Spitzer NC, Zigmond MJ, editors. Fundamental Neuroscience. 2. San Diego: Academic Press; 2002. pp. 299–317. [Google Scholar]
- 11.Qiu WQ, Folstein MF. Insulin, insulin-degrading enzyme and amyloid-beta peptide in Alzheimer’s disease: review and hypothesis. Neurobiol Aging. 2006;27:190–8. doi: 10.1016/j.neurobiolaging.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 12.Fisman M, Gordon B, Feleki V, et al. Metabolic changes in Alzheimer’s disease. J Am Geriatr Soc. 1988;36:298–300. doi: 10.1111/j.1532-5415.1988.tb02354.x. [DOI] [PubMed] [Google Scholar]
- 13.Plastino M, Fava A, Pirritano D, et al. Effects of insulinic therapy on cognitive impairment in patients with Alzheimer disease and diabetes mellitus type-2. J Neurol Sci. 2010;288:112–6. doi: 10.1016/j.jns.2009.09.022. [DOI] [PubMed] [Google Scholar]
- 14.Craft S, Asthana S, Newcomer JW, et al. Enhancement of Alzheimer disease with insulin and somatostatin, but not glucose. Arch Gen Psychiat. 1999;56:1135–40. doi: 10.1001/archpsyc.56.12.1135. [DOI] [PubMed] [Google Scholar]
- 15.Craft S, Asthana S, Cook DG, et al. Insulin dose-response effects on memory and plasma amyloid precursor protein in Alzheimer’s disease: interactions with Apolipoprotein-E-genotype. Psychoneuroendocrinology. 2003;28:809–22. doi: 10.1016/s0306-4530(02)00087-2. [DOI] [PubMed] [Google Scholar]
- 16.Den Heijer T, Vermeer SE, Van Dijk EJ, et al. Type 2 diabetes and atrophy of medial temporal lobe structures on brain MRI. Diabetologia. 2003;46:1604–10. doi: 10.1007/s00125-003-1235-0. [DOI] [PubMed] [Google Scholar]
- 17.Craft S, Ashana G, Schellenberg G, et al. Insulin effects on glucose metabolism, memory and plasma amyloid precursor protein in Alzheimer’s disease differ according to apolipoprotein-E-genotype. Ann NY Acad Sci. 2000;903:222–8. doi: 10.1111/j.1749-6632.2000.tb06371.x. [DOI] [PubMed] [Google Scholar]
- 18.Messier C. Diabetes, Alzheimer’s disease and apolipoprotein genotype. Exp Gerontol. 2003;38:941–6. doi: 10.1016/s0531-5565(03)00153-0. [DOI] [PubMed] [Google Scholar]
- 19.Sun LY, Al-Regaiey K, Masterkan MM, et al. Local expression of GH and IGF-1 in the hippocampus o-f GH-deficient long-lived mice. Neurobiol Aging. 2005;26:929–37. doi: 10.1016/j.neurobiolaging.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 20.Woods SC, Seeley RJ, Baskin DG, et al. Insulin and the blood brain barrier. Curr Pharm Des. 2003;9:795–800. doi: 10.2174/1381612033455323. [DOI] [PubMed] [Google Scholar]
- 21.Banks WA. The source of cerebral insulin. Eur J Pharmacol. 2004;490:5–12. doi: 10.1016/j.ejphar.2004.02.040. [DOI] [PubMed] [Google Scholar]
- 22.Schechter R, Whitmire J, Wheet GS, et al. Immunohistochemical and in situ hybridization study of an insulin-like substance in fetal neuron cell cultures. Brain Res. 1994;636:9–27. doi: 10.1016/0006-8993(94)90170-8. [DOI] [PubMed] [Google Scholar]
- 23.Devaskar SU, Giddings SJ, Rajakumar PA, et al. Insulin gene expression and insulin synthesis in mammalian neuronal cells. J Biol Chem. 1994;269:8445–54. [PubMed] [Google Scholar]
- 24.Rulifson EJ, Kim SK, Nusse R. Ablation of insulin-producing neurons in flies: growth and diabetic phenotypes. Science. 2002;296:1118–20. doi: 10.1126/science.1070058. [DOI] [PubMed] [Google Scholar]
- 25.Havrankova J, Schmechel D, Roth J, et al. Identification of insulin in rat brain. Proc Natl Acad Sci USA. 1978;75:5737–41. doi: 10.1073/pnas.75.11.5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Unger JW, Livingston JN, Moss AM. Insulin receptors in the central nervous system: localization, signaling mechanisms and functional aspects. Prog Neurobiol. 1991;36:343–62. doi: 10.1016/0301-0082(91)90015-s. [DOI] [PubMed] [Google Scholar]
- 27.Baskin DG, Porte DJ, Guest K, et al. Regional concentrations of insulin in the rat brain. Endocrinology. 1983;112:898–903. doi: 10.1210/endo-112-3-898. [DOI] [PubMed] [Google Scholar]
- 28.Johnston AM, Pirola L, Van Obberghen E. Molecular mechanisms of insulin receptor substrate protein-mediated modulation of insulin signaling. FEBS Lett. 2003;546:32–6. doi: 10.1016/s0014-5793(03)00438-1. [DOI] [PubMed] [Google Scholar]
- 29.Kahn CR, Bird KI, Jarrett DB, et al. Direct demonstration that receptor cross-linking or aggregation is important in insulin action. Proc Natl Acad Sci USA. 1978;75:4209–13. doi: 10.1073/pnas.75.9.4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun XJ, Rothenberg P, Kahn CR, et al. Structure of insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature. 1978;352:73–7. doi: 10.1038/352073a0. [DOI] [PubMed] [Google Scholar]
- 31.Giovannone B, Scaldaferri ML, Federici M, et al. Insulin receptor substrate (IRS) transduction system: distinct and overlapping signaling potential. Diabetes Metab Res Rev. 2000;16:434–41. doi: 10.1002/1520-7560(2000)9999:9999<::aid-dmrr159>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 32.Messier C, Teutenberg K. The role of insulin, insulin growth factor, and insulin-degrading enzyme in brain aging and Alzheimer’s disease. Neural Plast. 2005;12:311–28. doi: 10.1155/NP.2005.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Banks WA, Jaspan JB, Huang W, et al. Transport of insulin across the blood-brain barrier to insulin: saturability at euglycemic doses of insulin. Peptides. 1997;18:1423–9. doi: 10.1016/s0196-9781(97)00231-3. [DOI] [PubMed] [Google Scholar]
- 34.Clemmons DR. Role of insulin-like growth factor in maintaining normal glucose homeostasis. Horm Res. 2004;62:77–82. doi: 10.1159/000080763. [DOI] [PubMed] [Google Scholar]
- 35.Bondy CA, Cheng CM. Signaling by insulin-like growth factor 1 in brain. Eur J Pharmacol. 2004;490:25–31. doi: 10.1016/j.ejphar.2004.02.042. [DOI] [PubMed] [Google Scholar]
- 36.Tchilian EZ, Zhelezarov IE, Petkov VV, et al. 125I-insulin binding is decreased olfactory bulbs of aged rats. Neuropeptides. 1990;17:193–6. doi: 10.1016/0143-4179(90)90035-w. [DOI] [PubMed] [Google Scholar]
- 37.Adem A, Jossan SS, d’Argy R, et al. Insulin-like growth factor 1 IGF-1 receptors in the human brain: quantitative autoradiographic localization. Brain Res. 1989;503:299–303. doi: 10.1016/0006-8993(89)91678-8. [DOI] [PubMed] [Google Scholar]
- 38.Chung YH, Shin GM, Joo KM, et al. Region specific alterations in insulin-like growth factor receptor type I in the cerebral cortex and hippocampus of aged rats. Brain Res. 2002;946:307–13. doi: 10.1016/s0006-8993(02)03041-x. [DOI] [PubMed] [Google Scholar]
- 39.Frolic L, Blum-Degen D, Berstein HG, et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J Neural Transm. 1998;105:423–38. doi: 10.1007/s007020050068. [DOI] [PubMed] [Google Scholar]
- 40.Banks WA, Kastin AJ. Peptides and the blood-brain barrier: lipophilicity as a predictor of permeability. Brain Res Bull. 1985;15:287–92. doi: 10.1016/0361-9230(85)90153-4. [DOI] [PubMed] [Google Scholar]
- 41.King GL, Johnson SM. Receptor-mediated transport of insulin across endothelial cells. Science. 1987;227:1583–6. doi: 10.1126/science.3883490. [DOI] [PubMed] [Google Scholar]
- 42.Jialal I, King GL, Buchwald S, et al. Processing of insulin by bovine endothelial cells in culture. Internalization without degradation. Diabetes. 1984;33:794–800. doi: 10.2337/diab.33.8.794. [DOI] [PubMed] [Google Scholar]
- 43.Rotwein P, Burgess SK, Milbrandt JD, et al. Differential expression of insulin-like growth factor geneses in rat central nervous system. Proc Natl Acad Sci USA. 1988;85:265–9. doi: 10.1073/pnas.85.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoyer S. Brain glucose and energy metabolism abnormalities in sporadic Alzheimer’s disease. Causes and consequences: an update. Exp Gerontol. 2000;35:1363–72. doi: 10.1016/s0531-5565(00)00156-x. [DOI] [PubMed] [Google Scholar]
- 45.Blass JP, Gibson GF, Hoyer S. The role of the metabolic lesion in Alzheimer’s disease. J Alzheimer Dis. 2002;4:225–32. doi: 10.3233/jad-2002-4312. [DOI] [PubMed] [Google Scholar]
- 46.Schulingkamp RJ, Pagano TC, Hung D, et al. Insulin receptors and insulin action in the brain: review and clinical implication. Neurosci Biobehav Rev. 2000;24:855–72. doi: 10.1016/s0149-7634(00)00040-3. [DOI] [PubMed] [Google Scholar]
- 47.Apelt J, Mehlhorn G, Schliebs R. Insulin-sensitive GLUT4 glucose transporters are colocalized with GLUT3-expressing cells and demonstrate a chemically distinct neuron-specific localization in rat brain. J Neurosci Res. 1999;57:693–705. [PubMed] [Google Scholar]
- 48.Reagan LP, Gorovits N, Hoskin EK, et al. Localization and regulation of GLUTx1 glucose transporter in the hippocampus of streptozotocin diabetic rats. Proc Natl Acad Sci USA. 2001;98:2820–5. doi: 10.1073/pnas.051629798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao L, Teter B, Morihara T, et al. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implication for Alzheimer’s disease intervention. J Neurosci. 2004;24:11120–6. doi: 10.1523/JNEUROSCI.2860-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baskin DG, Stein LJ, Ikeda H, et al. Genetically obese Zucker rats have abnormally low brain insulin content. Life Sci. 1985;36:627–33. doi: 10.1016/0024-3205(85)90166-3. [DOI] [PubMed] [Google Scholar]
- 51.Moloney AM, Griffin RJ, Timmons S, et al. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signaling. Neurobiol Aging. 2010;31:224–43. doi: 10.1016/j.neurobiolaging.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 52.Messier C, White NM. Memory improvement by glucose, fructose, and two glucose analogs: a possible effect peripheral glucose transport. Behav Neural Biol. 1987;48:104–27. doi: 10.1016/s0163-1047(87)90634-0. [DOI] [PubMed] [Google Scholar]
- 53.Craft S. Insulin resistance and Alzheimer’s disease pathogenesis: potential mechanisms and implications for treatment. Curr Alzheimer Res. 2007;4:147–52. doi: 10.2174/156720507780362137. [DOI] [PubMed] [Google Scholar]
- 54.Squire LR. Mechanisms of memory. Science. 1986;232:1612–9. doi: 10.1126/science.3086978. [DOI] [PubMed] [Google Scholar]
- 55.Svensson J, Diez M, Engel J, et al. Endocrine, liver-derived IGF-1 is of importance for spatial learning and memory in old mice. J Endocrinol. 2006;189:617–27. doi: 10.1677/joe.1.06631. [DOI] [PubMed] [Google Scholar]
- 56.Dik MG, Pluijm SM, Jonker C, et al. Insulin-like growth factor 1 (IGF-1) and cognitive decline in older persons. Neurobiol Aging. 2003;24:573–81. doi: 10.1016/s0197-4580(02)00136-7. [DOI] [PubMed] [Google Scholar]
- 57.Schubert M, Gautam D, Surjo D, et al. Role for neuronal insulin resistance in neurodegenerative disease. Proc Natl Acad Sci USA. 2004;101:3100–5. doi: 10.1073/pnas.0308724101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mielke JG, Taghibiglou C, Liu L, et al. Biochemical and functional characterization of diet-induced brain insulin resistance. J Neurochem. 2005;93:1568–78. doi: 10.1111/j.1471-4159.2005.03155.x. [DOI] [PubMed] [Google Scholar]
- 59.Wan Q, Xiong ZG, Man HY, et al. Recruitment of functional GABA(A) receptors to postsynaptic domains by insulin. Nature. 1997;338:686–90. doi: 10.1038/41792. [DOI] [PubMed] [Google Scholar]
- 60.Ahmadian G, Ju W, Liu L, et al. Tyrosine phosphorylation of GluR2 is required for insulin-stimulated AMPA receptor endocytosis and LTD. EMBO J. 2004;23:1040–50. doi: 10.1038/sj.emboj.7600126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blair LA, Marshall J. IGF-1 modulates N and L calcium channels in a PI 3-kinase-dependent manner. Neuron. 1997;19:421–9. doi: 10.1016/s0896-6273(00)80950-2. [DOI] [PubMed] [Google Scholar]
- 62.Beeri Ms, Schmeidler J, Silverman JM, et al. Insulin in combination with other diabetes medication is associated with less Alzheimer neuropathology. Neurology. 2008;71:750–7. doi: 10.1212/01.wnl.0000324925.95210.6d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Craft S, Peskind E, Schellenberg G, et al. Cerebrospinal fluid and plasma insulin levels in Alzheimer’s disease differs according to apolipoprotein E genotype and gender. Neuroendocrinology. 1999;70:146–52. doi: 10.1159/000054469. [DOI] [PubMed] [Google Scholar]
- 64.Adamo M, Raizada MK, LeRoith D. Insulin and insulin growth factors receptors in the nervous system. Mol Neurobiol. 1989;3:71–100. doi: 10.1007/BF02935589. [DOI] [PubMed] [Google Scholar]
- 65.Squire LR. Memory and the hippocampus: a synthesis of findings with rats, monkeys, and humans. Psychol Rev. 1992;99(582):195–231. doi: 10.1037/0033-295x.99.2.195. [DOI] [PubMed] [Google Scholar]
- 66.Hoyer S, Henneberg N, Knapp S, et al. Brain glucose metabolism is controlled by amplification and desensitization of the neuronal insulin receptor. Ann NY Acad Sci. 1996;777:374–9. doi: 10.1111/j.1749-6632.1996.tb34448.x. [DOI] [PubMed] [Google Scholar]
- 67.Messier C, Destrade C. Insulin attenuates scopolamine-induced memory deficits. Psychobiology. 1994;22:16–21. [Google Scholar]
- 68.Skeberdis VA, Lan J, Zheng X, et al. Insulin promotes rapid delivery of N-methyl-D-aspartate receptors to the cell surface by exocytosis. Proc Natl Acad Sci USA. 2001;98:3561–6. doi: 10.1073/pnas.051634698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.De Felice FG, Vieira MN, Bomfim TR, et al. Proc Natl Acad Sci. Vol. 106. USA: 2009. Protection of synapses against Alzheimer’s linked toxins: insulin signaling prevents the pathogenic binding of A-beta oligomers; pp. 1971–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.De la Monte SM, Tong M, Lester-Coll N, et al. Therapeutic rescue of neurodegeneration experimental type-3 diabetes: relevance to Alzheimer’s disease. J Alzheimers Dis. 2006;10:89–109. doi: 10.3233/jad-2006-10113. [DOI] [PubMed] [Google Scholar]
- 71.Cohen E, Bieschle J, Perciavalle RM, et al. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–10. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- 72.Hirai K, Aliev G, Nunomura A, et al. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci. 2001;21:3017–23. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gouras GK, Almeida CG, Takahashi RH. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer’s disease. Neurobiol Aging. 2005;26:1235–44. doi: 10.1016/j.neurobiolaging.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 74.Caspersen C, Wang N, Yao J, et al. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005;19:2040–1. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 75.Smith MA, Perry G, Richey PL, et al. Oxidative damage in Alzheimer’s disease. Nature. 1996;382:120–1. doi: 10.1038/382120b0. [DOI] [PubMed] [Google Scholar]
- 76.Smith MA, Richey Harris PL, Sayre LM, et al. Widespread peroxynitrite-mediated damage Alzheimer’s disease. J Neurosci. 1997;17:2653–7. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mark RJ, Pang Z, Geddes JW, et al. Amyloid-beta peptide impairs glucose transport in hippocampal and cortical neurons: involvement lipid peroxidation. J Neurosci. 1997;17:1046–54. doi: 10.1523/JNEUROSCI.17-03-01046.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nunomura A, Perry G, Aliev G, et al. Oxidative damage is the earliest event in Alzheimer’s disease. J Neuropathol Exp Neurol. 2001;60:759–67. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 79.Cho DH, Nakamura T, Fang J, et al. S-nitrosylation of Drp l mediates beta-amyloid-related mithocondrial fissation and neuronal injury. Science. 2009;324:102–5. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yan SD, Chen M, Zhu H, et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382:685–91. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 81.Lue LF, Walker DG, Jacobson S, et al. Receptor for advanced glycation end products: its role in Alzheimer’s disease and other neurological diseases. Future Neurol. 2009;4:167–77. doi: 10.2217/14796708.4.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cho HJ, Son SM, Jin SM, et al. RAGE regulates BACE1 and A-β generation via NFAT1 activation in Alzheimer’s disease animal model. FASEB J. 2009;23:2639–49. doi: 10.1096/fj.08-126383. [DOI] [PubMed] [Google Scholar]
- 83.Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- 84.Ding Q, Keller JN. Splice variants of the receptor for advanced glycosylation end products (RAGE) in human brain. Neurosci Lett. 2005;373:67–72. doi: 10.1016/j.neulet.2004.09.059. [DOI] [PubMed] [Google Scholar]
- 85.Raucci A, Cugusi S, Antonelli A, et al. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the shedders a disintegrin and metalloprotease 10 (ADAM10) FASEB J. 2008;22:3716–27. doi: 10.1096/fj.08-109033. [DOI] [PubMed] [Google Scholar]
- 86.Lue LF, Walker DG, Brachova L, et al. Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer’s disease: identification of a cellular activation mechanism. Exp Neurol. 2001;171:29–45. doi: 10.1006/exnr.2001.7732. [DOI] [PubMed] [Google Scholar]
- 87.Deane R, Du Yan S, Submamaryan RK, et al. RAGE mediates amyloid-beta peptide transport across the blood–brain barrier and accumulation in brain. Nat Med. 2003;9:907–13. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 88.Emanuele E, D’angelo A, Tomaino C, et al. Circulating levels of soluble receptor for advanced glycation end products in Alzheimer disease and vascular dementia. Arch Neurol. 2005;62:1734–6. doi: 10.1001/archneur.62.11.1734. [DOI] [PubMed] [Google Scholar]
- 89.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Ann Rev Neurosci. 2001;24:1121–59. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 90.Iqbal K, Alonso Adel C, Chen S, et al. Tau pathology in Alzheimer’s disease and other tauopathies. Biochim Biophys Acta. 2005;1739:198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 91.Delacourte A, Sergeant N, Champain D, et al. Nonoverlapping but synergetic tau and APP pathologies in sporadic Alzheimer’s disease. Neurology. 2002;59:398–407. doi: 10.1212/wnl.59.3.398. [DOI] [PubMed] [Google Scholar]
- 92.Glenner GG, Wong CW. Alzheimer disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–90. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 93.Avila J. Tau kinases, phosphatases. J Cell Mol Med. 2008;12:258–9. doi: 10.1111/j.1582-4934.2007.00214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu R, Zhou XW, Tanila H, et al. Phosphorylated PP2A (tyrosine 307) is associated with Alzheimer neurofibrillary pathology. J Cell Mol Med. 2008;12:241–57. doi: 10.1111/j.1582-4934.2008.00249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cheng CM, Tseng V, Wang D, et al. Tau is hyperphosporylated in the insulin-like growth factor-I null brain. Endocrinology. 2005;146:5086–91. doi: 10.1210/en.2005-0063. [DOI] [PubMed] [Google Scholar]
- 96.Millward TA, Zolnierowicz S, Hemmings BA. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci. 1999;24:186–91. doi: 10.1016/s0968-0004(99)01375-4. [DOI] [PubMed] [Google Scholar]
- 97.Schubert M, Brazil DP, Burks DJ, et al. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosporylation. J Neurosc. 2003;23:7084–92. doi: 10.1523/JNEUROSCI.23-18-07084.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Freude S, Plum L, Schnitker J, et al. Peripheral hyperinsulinemia promotes Tau phosphorylation in vivo. Diabetes. 2005;54:3343–8. doi: 10.2337/diabetes.54.12.3343. [DOI] [PubMed] [Google Scholar]
- 99.Lesort M, Jope RS, Johnson GV. Insulin transiently increases tau phosphorylation: involvement of glycogen synthesis kinase-3beta and Fyn tyrosine kinase. J Neurochem. 1999;72:576–84. doi: 10.1046/j.1471-4159.1999.0720576.x. [DOI] [PubMed] [Google Scholar]
- 100.Lesort M, Johnson GV. Insulin-like growth factor-1 and insulin mediate transient site-selective increases in tau phosphorylation in primary cortical neurons. Neuroscience. 2000;99:305–16. doi: 10.1016/s0306-4522(00)00200-1. [DOI] [PubMed] [Google Scholar]
- 101.Hong M, Lee VM. Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J Biol Chem. 1997;272:19547–53. doi: 10.1074/jbc.272.31.19547. [DOI] [PubMed] [Google Scholar]
- 102.Planel E, Tatebayashi Y, Miyasaka T, et al. Insulin dysfunction induces in vivo tau after hyperphosphorylation through distinct mechanisms. J Neurosci. 1997;27:13635–48. doi: 10.1523/JNEUROSCI.3949-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen GJ, Xu J, Lahousse SA, et al. Transient hypoxia causes Alzheimer-type molecular and biochemical abnormalities in cortical neurons: potential strategies for neuroprotection. J Alzheimers Dis. 2003;5:209–28. doi: 10.3233/jad-2003-5305. [DOI] [PubMed] [Google Scholar]
- 104.Mandelkow EM, Starner K, Vogel R, et al. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol Aging. 2003;24:1079–85. doi: 10.1016/j.neurobiolaging.2003.04.007. [DOI] [PubMed] [Google Scholar]
- 105.Dickens M, Rogers JS, Cavanagh J, et al. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science. 1997;277:693–6. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- 106.Whitmarsh AJ, Cavanagh J, Tournier C, et al. A mammalian scaffold complex that selectively mediates MAP kinase activation. Science. 1998;281:1671–4. doi: 10.1126/science.281.5383.1671. [DOI] [PubMed] [Google Scholar]
- 107.Zhu X, Lee HG, Raina AK, et al. The role of mitogen-activated protein kinase pathways in Alzheimer’s disease. Neurosignals. 2002;11:270–81. doi: 10.1159/000067426. [DOI] [PubMed] [Google Scholar]
- 108.Rickle A, Bogdanovic N, Volkman I, et al. AKT activity in Alzheimer’s disease and in other neurodegenerative disorders. Neuroreport. 2004;15:955–9. doi: 10.1097/00001756-200404290-00005. [DOI] [PubMed] [Google Scholar]
- 109.Patrik GN, Zukerberg L, Nikolic M, et al. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:588–9. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 110.Kelicen P, Cantuti-Castelvetri I, Pekiner C, et al. The spin trapping agent PBN stimulates H2O2-induced Erk and Src kinase activity in human neuroblastoma cells. Neuroreport. 2002;13:1057–61. doi: 10.1097/00001756-200206120-00016. [DOI] [PubMed] [Google Scholar]
- 111.Crossthwaite AJ, Hasan S, Williams RJ. Hydrogen peroxide-mediated phosphorylation of ERK 1-2, AKT, PKB and JNK in cortical neurons: dependence on Ca(2+) and P13-kinase. J Neurochem. 2002;80:24–35. doi: 10.1046/j.0022-3042.2001.00637.x. [DOI] [PubMed] [Google Scholar]
- 112.Kang J, Lemaire HG, Unterbeck A, et al. The precursor of Alzheimer’s disease amyloid E4 protein resembles a cell-surface receptor. Nature. 1987;325:733–6. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 113.Hung AY, Haass C, Nitsch RM, et al. Activation of protein kinase C inhibits cellular production of the amyloid-β-protein. J Biol Chem. 1993;268:22959–62. [PubMed] [Google Scholar]
- 114.Skovronsku DM, Moore DB, Milla Me, et al. Protein kinase C-dependent α-secretase competes with β-secretase for cleavage of amyloid-β-precursor protein in the trans-golgi network. J Biol Chem. 2000;275:2568–75. doi: 10.1074/jbc.275.4.2568. [DOI] [PubMed] [Google Scholar]
- 115.Rossner S, Beck M, Stahl T, et al. Constitutive overaction of protein kinase C in guinea pig brain increases α-secretase APP processing without decreasing β-secretase generation. Eur J Neurosci. 2000;12:3191–200. doi: 10.1046/j.1460-9568.2000.00211.x. [DOI] [PubMed] [Google Scholar]
- 116.Ring S, Weyer SW, Kilian SB, et al. The secrete β-amyloid precursor protein ectodomain APPsα is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J Neurosci. 2007;27:7817–26. doi: 10.1523/JNEUROSCI.1026-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Adlerz L, Holback S, Multhaup G, et al. IGF-1-induced processing of the amyloid precursor protein family is mediated by different signaling pathways. J Biol Chem. 2007;282:10203–9. doi: 10.1074/jbc.M611183200. [DOI] [PubMed] [Google Scholar]
- 118.Zhang Y, Thompson R, Zhang H, et al. APP processing in Alzheimer’s disease. Mol Brain. 2011;4:1–13. doi: 10.1186/1756-6606-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jacobsen KT, Adlerz L, Multhaup G, et al. Insulin-like growth factor-1 (IGF-1)-induced processing of amyloid-beta precursor protein (APP) and APP-like protein 2 is mediated by different metalloproteinases. J Biol Chem. 2010;285:10223–31. doi: 10.1074/jbc.M109.038224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kimberly WT, LaVoie MJ, Ostaszewski BL, et al. Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1 and Pen-2. Proc Natl Acad Sci USA. 2003;100:6382–7. doi: 10.1073/pnas.1037392100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vassar R, Bennett BD, Babu-Kahn S, et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–41. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 122.Holsinger RM, McLean CA, Beyreuther K, et al. Increased expression of the amyloid precursor beta-secretase in Alzheimer’s disease. Ann Neurol. 2002;51:783–6. doi: 10.1002/ana.10208. [DOI] [PubMed] [Google Scholar]
- 123.Costantini C, Scrable H, Puglielli L. An aging pathway controls the TrkA to p75NTR receptor switch and amyloid beta-peptide generation. EMBO J. 2006;25:1997–2006. doi: 10.1038/sj.emboj.7601062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Puglielli L. Aging of the brain, neurotrophic signaling, and Alzheimer’s disease: is IGF-1R the common culprit. Neurobiol Aging. 2008;29:795–811. doi: 10.1016/j.neurobiolaging.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Puglielli L, Ellis BC, Saunders AJ, et al. Ceramide stabilizes beta-size amyloid precursor protein-cleaving enzyme I and promotes amyloid beta-peptide biogenesic. J Biol Chem. 2003;278:19777–83. doi: 10.1074/jbc.M300466200. [DOI] [PubMed] [Google Scholar]
- 126.Sotthibundhu A, Sykes AM, Fox B, et al. Beta-amyloid (1-42) induces neuronal death through the p75 neurotrophin receptor. J Neurosci. 2008;28:3941–6. doi: 10.1523/JNEUROSCI.0350-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kayed RH, Thompson JL, McIntire TM, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–9. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 128.Hass C, Sedkoe DJ. Soluble protein oligomers in neuro-degeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–2. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 129.Kamenetz F, Tomita T, Hsieh H, et al. APP processing and synaptic function. Neuron. 2003;37:925–37. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 130.Tanzi RE, Moir RD, Wagner SL. Twenty years of the Alzheimer disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–55. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 131.Selkoe DJ. Translating cell biology in to therapeutic advances in Alzheimer’s disease. Nature. 1999;399:23–31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- 132.Tanzi RE, Moir RD, Wagner SL. Clearance of Alzheimer’s Abeta peptide: the many roads to perdition. Neuron. 2004;43:605–8. doi: 10.1016/j.neuron.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 133.Cheng S, Li L, He S, et al. GRK5 deficiency accelerates fl-amyloid accumulation in Tg2576 mice via impaired cholinergic activity. J Biol Chem. 2010;285:41541–8. doi: 10.1074/jbc.M110.170894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Carro E, Trejo JL, Spuch C, et al. Blockade of the insulin-like growth factor I receptor in the choroid plexus originates Alzheimer’s like neuropathology in rodents; new cues into the human disease. Neurobiol Aging. 2006;27:1618–31. doi: 10.1016/j.neurobiolaging.2005.09.039. [DOI] [PubMed] [Google Scholar]
- 135.Lanz TA, Salatto CT, Semproni AR, et al. Peripheral elevation of IGF-1 fails to alter A-beta clearance in multiple in vivo models. Biochem Pharmacol. 2008;75:1093–103. doi: 10.1016/j.bcp.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 136.Suh Y, Atzomon G, Cho Mo, et al. Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc Natl Acad Sci USA. 2008;105:3438–42. doi: 10.1073/pnas.0705467105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ho L, Qin W, Pompl PN, et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2004;18:902–4. doi: 10.1096/fj.03-0978fje. [DOI] [PubMed] [Google Scholar]
- 138.Cao D, Lu H, Lewis TI, et al. Intake of sucrose-sweetened water induced insulin resistance and exacerbates memory deficits and amyloidosis in a transgenic mouse model of Alzheimer disease. J Biol Chem. 2007;282:36275–82. doi: 10.1074/jbc.M703561200. [DOI] [PubMed] [Google Scholar]
- 139.Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Montine TJ, Kaye JA, Montine KS, et al. Cerebrospinal fluid abeta-42, tau, and f2-isoprostane concentrations in patients with Alzheimer disease, other dementias, and age-matched controls. Arch Pathol Lab Med. 2001;125:510–2. doi: 10.5858/2001-125-0510-CFATAF. [DOI] [PubMed] [Google Scholar]
- 141.Sheng JG, Bora SH, Xu G, et al. Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPsw transgenic mice. Neurobiol Dis. 2003;14:133–45. doi: 10.1016/s0969-9961(03)00069-x. [DOI] [PubMed] [Google Scholar]
- 142.Dandona P. Endothelium, inflammation and diabetes. Curr Diab Rep. 2002;2:311–5. doi: 10.1007/s11892-002-0019-0. [DOI] [PubMed] [Google Scholar]
- 143.Krogh-Madsen R, Plomgaard P, Keller P, et al. Insulin stimulates interlukin-6 and tumor necrosis factor-alpha gene expression in human subcutaneous adipose tissue. Am J Physiol Endocrinol Metab. 2004;286:234–8. doi: 10.1152/ajpendo.00274.2003. [DOI] [PubMed] [Google Scholar]
- 144.Soop M, Duxubury H, Agwunobi AO, et al. Euglycemic hyperinsulinemia augments the cytokine and endocrine responses to endotoxin in humans. Am J Physiol Endocrinol Metab. 2002;282:1276–85. doi: 10.1152/ajpendo.00535.2001. [DOI] [PubMed] [Google Scholar]
- 145.Axelrod L. Insulin, prostaglandins, and the pathogenesis of hypertension. Diabetes. 1991;40:1223–7. doi: 10.2337/diab.40.10.1223. [DOI] [PubMed] [Google Scholar]
- 146.Mrak RE, Griffin WS. Interleukin-1, neuroinflammation, and Alzheimer’s disease. Neurobiol Aging. 2001;22:903–8. doi: 10.1016/s0197-4580(01)00287-1. [DOI] [PubMed] [Google Scholar]
- 147.White JA, Manelli AM, Holmberg KH, et al. Differential effects of oligomeric and fibrillar amyloid-beta 1-42 on astrocyte mediated inflammation. Neurobiol Dis. 2005;18:459–65. doi: 10.1016/j.nbd.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 148.Papassotiropoulos A, Hock C, Nitsch RM. Genetics of interleukin 6: implications for Alzheimer’s disease. Neurobiol Aging. 2001;22:863–71. doi: 10.1016/s0197-4580(01)00294-9. [DOI] [PubMed] [Google Scholar]
- 149.Figlewicz DP, Bentson K, Octrant I. The effect of insulin on norepinephrine uptake by PC12 cells. Brain Res. 1993;602:161–4. doi: 10.1016/0361-9230(93)90210-3. [DOI] [PubMed] [Google Scholar]
- 150.Heneka MT, Galea E, Gavriluyk V, et al. Noradrenergic depletion potentiates beta-amyloid-induced cortical inflammation: implications for Alzheimer’s disease. J Neurosci. 2002;22:2434–42. doi: 10.1523/JNEUROSCI.22-07-02434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bondareff W, Mountjoy CQ, Roth M, et al. Neuronal degeneration in locus ceruleus and cortical correlates of Alzheimer disease. Alzheimer Dis Assoc Disord. 1987;1:256–62. doi: 10.1097/00002093-198701040-00005. [DOI] [PubMed] [Google Scholar]
- 152.Carro E, Trejo JL, Gomez-Isla T, et al. Serum insulin-like growth factor I regulates brain amyloid-beta levels. Nat Med. 2002;8:1390–7. doi: 10.1038/nm1202-793. [DOI] [PubMed] [Google Scholar]
- 153.Zhao M, Cribbs DH, Anderson AJ, et al. The induction of the TNF alpha death domain signaling pathway in Alzheimer’s disease brain. Neurochem Res. 2003;28:307–18. doi: 10.1023/a:1022337519035. [DOI] [PubMed] [Google Scholar]
- 154.Aggarwal S, Gollapudi S, Gupta S. Increased TNF-alpha-induced apoptosis in lymphocytes from aged humans: changes in TNF-alpha receptor expression and activation of caspases. J Immunol. 1999;162:2154–61. [PubMed] [Google Scholar]
- 155.Dzienis-Straczkowski M, Straczkowski M, Szelachowska M, et al. Soluble tumor necrosis factor-alpha receptors in young obese subjects with normal and impaired glucose tolerance. Diabetes Care. 2003;26:875–80. doi: 10.2337/diacare.26.3.875. [DOI] [PubMed] [Google Scholar]
- 156.Bastard JP, Jardel C, Bruckert E, et al. Variations in plasma soluble tumor necrosis factor receptors after diet-induced weight loss in obesity. Diabetes Obes Metab. 2000;2:323–5. doi: 10.1046/j.1463-1326.2000.00090.x. [DOI] [PubMed] [Google Scholar]
- 157.Descamps O, Bilheimer D, Herz J. Insulin stimulates receptor-mediated uptake of Apo E-enriched lipoproteins and activated alpha 2-macroglobulin in adypocytes. J Biol Chem. 1993;268:974–81. [PubMed] [Google Scholar]
- 158.Fishel MA, Watson GS, Montine TJ, et al. Hyperinsulinemia provokes synchronous increases in central inflammation and β-amyloid in normal adults. Arch Neurol. 2005;62:1539–44. doi: 10.1001/archneur.62.10.noc50112. [DOI] [PubMed] [Google Scholar]
- 159.Yamada T, Kondo A, Takamatsu J, et al. Apolipoprotein E mRNA in the brains of patients with Alzheimer’s disease. J Neurol Sci. 1995;129:56–61. doi: 10.1016/0022-510x(94)00249-n. [DOI] [PubMed] [Google Scholar]
- 160.Zheng WH, Quirion R. Glutamate acting on N-methyl-D-aspartate receptors attenuates insulin-like growth factor-1 receptor tyrosine phosphorylation and its survival signaling properties in rat hippocampal neurons. J Biol Chem. 2009;284:855–61. doi: 10.1074/jbc.M807914200. [DOI] [PubMed] [Google Scholar]
- 161.Torres-Aleman I. Targeting insulin-like growth factor-1 to treat Alzheimer’s disease. Expert Opin Ther Targets. 2007;11:1535–42. doi: 10.1517/14728222.11.12.1535. [DOI] [PubMed] [Google Scholar]
- 162.De Felice FG, Lambert MP, Fernandez SJ, et al. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging. 2008;29:1334–47. doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bingham EM, Hopkins D, Smith D, et al. The role of insulin in human brain glucose metabolism: an 18fluoro-deoxyglucose positron emission tomography study. Diabetes. 2002;51:3384–90. doi: 10.2337/diabetes.51.12.3384. [DOI] [PubMed] [Google Scholar]
- 164.Blanchard JG, Duncan PM. Effect in combinations of insulin, glucose and scopolamine on radial arm maze performance. Pharmacol Biochem Behav. 1997;58:209–14. doi: 10.1016/s0091-3057(97)00064-6. [DOI] [PubMed] [Google Scholar]