

Abstract
Human noroviruses constitute a significant worldwide disease burden. Each year noroviruses cause over 267 million infections, deaths in over 200,000 children under the age of five, and over 50% of U.S. food borne illness. Due to the absence of a tissue culture model or small animal model to study human norovirus, virus-like particles (VLPs) and ELISA-based biological assays have been used to answer questions about norovirus evolution and immunity as well provide a potential vaccine platform. This chapter outlines the protocols on norovirus detection in stool and norovirus VLP design, production, purification, and storage using a Venezuelan equine encephalitis virus (VEE)-based VRP expression system.
Keywords: norovirus, virus-like particle, virus replicon particle, Venezuelan equine encephalitis virus, virus-like particle purification
INTRODUCTION
Noroviruses are the leading cause of acute gastroenteritis and are responsible for 58% of all acute gastroenteritis outbreaks in the United States and Europe (2011). Clinical symptoms of norovirus gastroenteritis include vomiting and diarrhea typically over a 24–72 hour period. Although the severity of disease is usually moderate, illness can be especially severe in young children, the elderly, and the immunocompromised (Harris et al., 2008; Koopmans et al., 2000; Schorn et al., 2010). Some immunocompromised individuals may exhibit chronic symptomatic infections lasting several months to years (Schorn et al., 2010).
Noroviruses belong to the family Caliciviridae. The non-enveloped virus particles have an icosahedral symmetry enclosing an ~7.5 Kb single-stranded, positive-sense RNA genome that contains three large open reading frames (ORFs). ORF1 encodes the non-structural proteins, while ORFs 2 and 3 encode the major and minor capsid proteins, respectively (Prasad et al., 1999). Currently, noroviruses are grouped into at least five genogroups (GI-GV) with GI and GII viruses responsible for most human infections (Vinje et al., 2000). GI and GII are further subdivided into 9 and 21 different genotypes, respectively.
The prototypic norovirus strain is Norwalk virus, belonging to the GI.1 genotype. The virus was first detected by immune electron microscopy in stool samples as the cause of an outbreak of acute gastroenteritis at an elementary school in 1968 in Norwalk, Ohio (Kapikian et al., 1972). Early detection of norovirus is crucial to control the spread of disease during outbreaks. Since the discovery of the first norovirus strain in 1972, detection of norovirus has evolved from electron microscopy, to conventional RT-PCR (Jiang et al., 1996) to real-time RT-PCR assays which have become the diagnostic method in most clinical laboratories (Schultz et al., 2011; Vega et al., 2011). These molecular techniques combined with sequence analysis have resulted in a better understanding of the role of norovirus as the leading cause of epidemic gastroenteritis and as an important cause of sporadic gastroenteritis. Since the discovery of the first human norovirus strain, more than 30 norovirus genotypes have been identified, although the majority of outbreaks are caused by genotype GII.4 strains (Siebenga et al., 2009).
While tissue culture and mouse models exist for mouse norovirus (MNV), no cell culture or small animal model is available for human noroviruses. Gnotobiotic pigs (Cheetham et al., 2006; Jung et al., 2012) (Souza et al., 2007) and chimpanzees (Bok et al., 2011) have been examined as possible human norovirus animal models, but cost and feasibility make widespread use of these systems problematic. MNV can be used to study general norovirus biology and to evaluate efficacy of disinfection processes, but is not a suitable surrogate to model human norovirus immunity and evolution.
The lack of cell culture or small animal models for human norovirus has significantly delayed structural, evolutionary and immunological studies on this important group of public health relevant viruses. However the expression of the major capsid protein (ORF2) in baculovirus (Jiang et al., 1992) and Venezuelan equine encephalitis (VEE) virus (Baric et al., 2002) results in the formation of virus-like particles (VLPs) composed of 180 copies of the monomeric protein (Prasad et al., 1999). These VLPs have been key to understanding the role of genetic and immunological factors on the evolution and emergence of new strains (Donaldson et al., 2010; Lindesmith et al., 2003; Lindesmith et al., 2011). These VLPs do not contain genomic RNA and are replication deficient; however, they are nearly morphologically identical to native virions, making them appropriate surrogates for virus in biological assays to answer human norovirus-specific questions.
Norovirus VLPs have been successfully expressed using several systems including baculovirus (Jiang et al., 1992), vesicular stomatitis virus (VSV) (Ma and Li, 2011), E. coli (Tan et al., 2004), and Venezuelan equine encephalitis virus (VEE) (Baric et al., 2002) systems as well as in plant-based (Mason et al., 1996), insect based (Jiang et al., 1992), and mammalian-based (Baric et al., 2002) cell culture systems. Each system has slightly different methodologies for cloning, expression, and purification of the VLPs.
This chapter specifically addresses use of the 3526 (BSL-2) VEE expression system with mammalian cell culture (Hart et al., 2000; Pratt et al., 2003; Pushko et al., 1997; Reed et al., 2005). Protocols presented here include real-time RT-PCR detection methods which allow for simultaneous detection and differentiation of both GI and GII noroviruses in stool samples, amplification and cloning of ORF-2/ORF-3 into an expression vector, cloning of norovirus capsid gene into the VEE expression plasmid pVR21, production of VEE replicons encoding the norovirus major capsid protein, production of VLPs from replicons, purification of VLPs from BHK-21 cells, validation and quantification of VLPs, electron microscopy imaging of VLPs, and VLP storage.
CAUTION: Norovirus and VEE 3526 are Biosafety Level 2 (BSL-2) pathogens. Follow all appropriate guidelines and regulations for the use and handling of pathogenic microorganisms. See UNIT 1A.1 and other pertinent resources (APPENDIX 1B) for more information.
CAUTION: Follow all appropriate guidelines and regulations for the use and handling of human derived materials. See UNIT 1A.1 and other pertinent resources (APPENDIX 1B) for more information.
BASIC PROTOCOL 1: DUPLEX REAL-TIME RT-PCR FOR DETECTION OF HUMAN NOROVIRUS GENOGROUP I AND II
The GI/GII Norovirus Duplex real-time (TaqMan®) RT-PCR assay was designed to detect norovirus GI and GII RNA in human stool and emesis specimens (Vega et al., 2011). Norovirus RNA can be quantified by including a dilution series of a quantified norovirus RNA transcript.
Materials
-
AgPath-ID™ One-Step RT-PCR Kit (Life Technology cat. no. AM1005) containing:
2x RT-PCR Buffer
25x RT-PCR enzyme mix
Detection Enhancer
Nuclease-free water
Real-time RT-PCR oligonucleotide primers and TaqMan probes for GI and GII norovirus (Table 1, Figure 1)
RNA (See Support Protocol 1)
Table 1.
Primers and probes used in real-time RTPCR and ORF-2/ORF-3 long RT-PCR
| Primers and probes | Sequence (5′ to 3′) | Position¥ |
|---|---|---|
| Real-time RT-PCR primers and TaqMan probes | ||
| Cog 1F | CGYTGGATGCGITTYCATGA | 5276 |
| Cog 1R | CTTAGACGCCATCATCATTYAC | 5343 |
| Cog 2F | CARGARBCNATGTTYAGRTGGATGAG | 5003 |
| Cog 2R | TCGACGCCATCTTCATTCACA | 5080 |
| Ring 1C* | FAM/AGATYGCGITCICCTGTCCA/BHQ | 5314 |
| Ring 2** | QUA/TGGGAGGGCGATCGCAATCT/BHQ | 5048 |
| ORF-2/ORF3 primers | ||
| PanGIR1 | GGCARYCTWTCWGTATTRAAA | 7119 |
| PanGIIR1 | GTCCAGGAGTCCAAAA | 7428 |
| G1SKR | CCAACCCARCCATTRTACA | 5638 |
| G2SKR | CCRCCNGCATRHCCRTTRTACAT | 5373 |
| Ring2PCR | TGGGAGGGCGATCGCAATCT | 5048 |
| Ring1APCR | TGGACAGGRGATCGCRATCT | 5306 |
Nucleotide positions for real-time RT-PCR and ORF-2/ORF-3 long RT-PCR were taken from reference NoV strains in genogroups GI (Norovirus Hu/GI.1/Norwalk/1968/USA, Genbank accession no. AF093797) and GII (Norovirus Hu/GII.4/New Orleans/2010/USA GenBank accession no. JN595867)
GI TaqMan® probe is 5′-labeled with 6-carboxyfluorescein (FAM) and 3′-labeled with Black Hole quencher; Black Hole Quencher 1 is preferred.
GII TaqMan® probe is 5′-labeled with Quasar 670 or Cy5 and 3′-labeled with Black Hole quencher; Black Hole Quencher 3 is preferred
Y= C,T I= Inosine R=A,G B=C,G,T N=A,C,G,T W=A,T
Figure 1. Primers and probes used in real-time RTPCR and ORF-2/ORF-3 long RT-PCR.
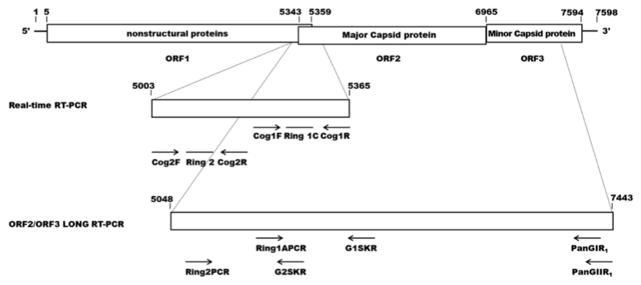
Schematic representation of Norovirus genome and target regions for real-time RT-PCR and ORF-2/ORF-3 long RT-PCR primers and probes.
Sterile 1.5 ml microcentrifuge tubes, nuclease-free
MicroAmp® Optical 96-Well Reaction Plate (Life Technology cat. no. N801-0560)
MicroAmp® Optical Adhesive Film (Life Technology cat. no. 4311971)
Applied Biosystems 7500 Real-Time PCR System (Life Technology)
-
Prepare master mix in a labeled clean 1.5 ml microcentrifuge tube by adding the following reagents for a total of 22 μl per reaction (multiply each volume by the number of samples plus one or two extra reactions for pipetting errors):
12.50 μl 2x RT-PCR buffer
1.83 μl Nuclease-free water
1.67 μl Detection enhancer
1.0 μl each of 10 μM forward and reverse oligonucleotide primers for GI and GII norovirus (Table 1, Figure 1)
-
1.0 μl 25x RT-PCR enzyme
For example, to prepare a master mix for 10 reactions, add twelve times suggested volume of each reagent, mix, and dispense 22 μl aliquots of the mix into each reaction tube.
-
Dispense 22 μl of master mix into each real-time well and add 3 μl of RNA or nuclease-free water (negative control) as template.
Each experiment should include a positive control (RNA template from a norovirus positive sample) of each genogroup, and a negative control (water). Seal the plate with Adhesive film and centrifuge the plate 1 min at 500 × g, 4°C to remove air bubbles or drops that may be present in the wells.
-
Perform real-time RT-PCR in Applied Biosystems 7500 Real-Time PCR System using the following parameters:
1 cycle: 10 min 45°C 1 cycle: 10 min 95°C 45 cycles: 15 sec 95°C 1 min 60°C Follow the manufacturer’s instructions to set up the Applied Biosystems 7500 Real-Time PCR system. The Run Mode should be set as Standard 7500, Data collection should be set at Stage 3, Step 2 (1 min, 60°C) and a final reaction volume of 25 μl should be selected. Data analysis and interpretation is provided at the Commentary section. At the end of the run, logarithmic curves representing the magnitude of the signal generated (ΔRn) versus cycle number, are displayed and should be analyzed for each target. Although baseline and threshold values can be automatically analyzed by the software, it is recommended to manually set these parameters. For the baseline, the Start cycle and End cycle fields should be set, ensuring that the amplification curve growth begins after the End Cycle value. Adjust the threshold setting bar until the threshold is within the exponential phase of the amplification curve. Analyze the data. If a sample tests positive, a Ct-value (the cycle at which the fluorescence crossed the threshold for a specific probe) will be displayed.
SUPPORT PROTOCOL 1: NOROVIRUS RNA EXTRACTION FROM STOOL SAMPLES (AUTOMATIC METHOD)
Many different protocols can be used successfully for the extraction of viral nucleic acids from stool samples and most of them rely on lysis of the virus using guanidinium thiocyanate. Most RNA extraction methods today use paramagnetic beads to bind the nucleic acids, whereas proteins and other contaminants are removed by several wash steps. The nucleic acids are then eluted off the beads using water or an elution buffer containing low concentrations of Tris-HCl buffer and EDTA.
Materials
Phosphate buffered saline (PBS) 10 mM pH7.0–7.4 (see recipe)
-
MagMAX™ −96 Viral RNA Isolation Kit (Life Technology, cat. no. 1835) including:
Lysis/Binding Solution Concentrate
Wash Solution 1 Concentrate
Wash Solution 2 Concentrate
Elution Buffer
RNA Binding Beads
Carrier RNA
Lysis/Binding Enhancer
100% ethanol, ACS grade or better
100% isopropanol, ACS grade or better
1.5 ml microcentrifuge tube
Transfer pipette or sterile sticks
Vortex
Table top high speed microcentrifuge
Sterile reservoirs
KingFisher tip comb (Thermo scientific, cat. no. 97002070)
KingFisher plate 200 μl (Thermo scientific, cat. no. 97002084)
KingFisher Magnetic Particle Processors (Thermo scientific, cat. no. 5400000)
Dispense 500 μl of PBS into 1.5 ml microcentrifuge tubes.
Add a pea-size amount of stool sample (~0.1 g) with a disposable transfer pipette or sterile stick [(~ 10–20% (w/v)]. When stool sample is liquid, use 500 μl of the specimen, it is not necessary to dilute in PBS.
-
Vortex each sample thoroughly for 1 minute. Centrifuge 5 min at 5,000 × g in a microcentrifuge to pellet the solids. The clarified supernatant can either be used directly for viral nucleic acid extraction or stored at −80°C.
When solids or lipids are present carefully remove the supernatant, as solids may interfere with the extraction. An additional centrifugation step may also help to clarify the stool. Follow the manufacturer’s directions for preparing RNA isolation reagents.
-
In a KingFisher plate add the following reagents in order:
Row A: 130 μl of Lysis/Binding Solution and 20 μl of Bead Mix Rows B and C: 150 μl of Wash Solution I Rows D and E: 150 μl of Wash Solution II Row F: 100 μl of Elution Buffer Each plate allows for up to 12 samples. Two plates can be run simultaneously. Do not set up more than 2 plates simultaneously because the buffer could evaporate before the run. Transfer the plate to a BSL-2 hood and add 50 μl of clarified 10% (v/w) stool suspension in row A.
-
Perform RNA extraction in KingFisher Magnetic Particle Processor.
After the samples have been disrupted by the lysis/binding buffer, nucleic acids are released whereas nucleases are inactivated. Paramagnetic beads with a nucleic acid binding surface bind nucleic acids. The beads/nucleic acids are captured on magnets, and proteins and other contaminants are removed by successive washes. Nucleic acids are eluted in a small volume of elution buffer, whereas paramagnetic beads remain captured on magnets. Recover RNA and store at −20°C.
BASIC PROTOCOL 2: AMPLIFICATION OF NOROVIRUS ORF-2 AND ORF-3 BY LONG RT-PCR
The lack of a cell culture system has significantly delayed structural and immunological studies for norovirus. This problem has been partially solved by the development of virus-like particles (VLPs). This protocol describes the amplification of the ORF-2 and ORF-3 that encode for the structural proteins of norovirus. After extraction of viral RNA from stool samples (see Support Protocol 1), ORF-2 and ORF-3 are amplified by long RT-PCR with specific oligonucleotide primers for each genogroup. The products will be used as template for Basic Protocol 3.
Materials
ORF-2/ORF-3 oligonucleotide primers for GI or GII norovirus (Table 1, Figure 1)
10 mM dNTPs mix (Life Technologies, cat. no. AM8200)
Nuclease-free water (Life Technologies, cat. no. AM9937)
RNA (see support protocol 1)
-
SuperScript™ II Reverse Transcriptase (Life Technologies, cat. no. 18080-022) including:
SuperScript™ II Reverse Transcriptase
5x First-Strand buffer
0.1 M DTT
RNase inhibitor (Life Technologies, cat. no. N808-0119)
RNase ONE™ Ribonuclease (Promega, cat. no. M4261)
DNA Clean & Concentrator™-5 (Zymo Research, cat. no. D4013)
-
Phusion® High Fidelity PCR kit (Thermo Scientific, cat. no. F-531S) including:
2x Phusion® HF buffer
Phusion® High Fidelity DNA Polymerase
400 μM of each dNTP
DMSO
Quick-Load® 1 Kb DNA ladder (BioLabs, cat. no. N0468S) or any other comparable molecular weight DNA standards)
0.7% agarose gel in 1X TAE containing GelRed™ (1:10.000)
QIAquick gel Extraction Kit (Qiagen)
1.5 ml microcentrifuge tube
0.2 ml thin-walled PCR/RT-PCR tubes
GeneAmp 9700 PCR thermal cycler (Life Technologies) or similar instrument
Ice bath
U.V. transilluminator
Dark Reader DR46B Transilluminator (Clare Chemical Research, Inc., cat. no. DR456B)
cDNA generation
-
1
Prepare oligonucleotide primer template master mix in a labeled clean 1.5 ml microcentrifuge tube by adding the following reagents for a total of 9 μl per reaction (multiply each volume by the number of samples plus one or two reactions for pipetting errors):
1.0 μl of 50 μM PanGIIR1 primer (Table 1, Figure 1)
4.0 μl of 2.5 μM dNTPs mix
4.0 μl of Nuclease-free water
For example, to make a master mix for 10 reactions, prepare a mix for 12 reactions. If GI norovirus RNA has been detected by real-time (BASIC PROTOCOL 1), use PanGIR1 primer instead of PanGIIR1. Note that the 10 mM dNTPs mix stock has been pre-diluted to 2.5 mM. -
2
Dispense 9 μl of oligonucleotide primer template master mix into 0.2 ml thin-walled PCR/RT-PCR tubes.
-
3
Add 3 μl RNA template or nuclease-free water (negative control) as template and spin tubes down.
-
4
Transfer the tubes to a GeneAmp 9700 PCR thermal cycler and denature the RNA for 5 min at 65°C.
-
5
Prepare the RT master mix in a labeled clean 1.5 ml microcentrifuge tube by adding the following reagents from SuperScript™ II Reverse Transcriptase kit for a total of 8 μl per reaction (multiply each volume by the number of samples plus one or two reactions for pipetting errors):
4.0 μl 5X First Strand Buffer
2.0 μl 0.1 M DTT
1.0 μl SuperScript® II Enzyme
1.0 μl RNase Inhibitor
-
6
Immediately and quickly move tubes from thermo cycler to ice bath.
-
7
Add 8 μl of the RT master mix into each tube and spin tubes down.
-
8
Incubate 60 min at 42°C, followed by 15 min at 70°C.
-
9
Add 1 μl of RNase ONE™ Ribonuclease (~10U).
-
10
Incubate 20 min at 37°C.
-
11
Use immediately or store at −20°C.
-
12
Add 7 volumes of DNA Binding Buffer to each volume of DNA sample.
-
13
Load the mixture into a Zymo-Spin Column and place the column into a 2 ml collection tube provided with the kit.
-
14
Centrifuge at maximum speed (≥10,000 g) for 30 seconds. Discard the flow through.
-
15
Add 200 μl of Wash Buffer included in the DNA Clean & Concentrator™-5 kit, to the column and centrifuge for 30 seconds. Discard the flow through.
-
16
Repeat the wash step.
-
17
Place the Zymo-Spin Column into a clean 1.5 ml microcentrifuge tube. Add 6–10 μl of DNA Suspension Buffer directly to the column matrix and centrifuge for 30 seconds to elute the cDNA.
ORF-2/ORF-3 PCR amplification
-
18
Prepare the PCR master mix in a labeled clean 1.5 ml microcentrifuge tube by adding the following reagents for a total of 47 μl per reaction (multiply each volume by the number of samples plus one or two reactions to adjust for pipetting errors):
21.0 μl Nuclease-free water
10.0 μl 5X HF Phusion Buffer
4.0 μl of 2.5 mM dNTPs mix
1.5 μl of DMSO
0.5 μl of Phusion® High Fidelity DNA Polymerase
Mix gently and spin down tubes.
If GI norovirus has been detected by real-time (BASIC PROTOCOL 1), replace PanGIIR1 and Ring2 PCR primers by PanGIR1 and RING1APCR primers, respectively.
-
19
Dispense 47 μl PCR master mix into 0.2 ml thin-walled PCR/RT-PCR tubes.
-
20
Add 3 μl cDNA (from step17) or nuclease-free water (negative control) as template and spin tubes down.
-
21
Transfer the tubes to a GeneAmp 9700 PCR or similar thermal cycler and perform PCR using the following parameters:
1 cycle: 30 sec 98°C 40 cycles: 10 sec 98°C 30 sec 48°C 1 min 30sec 72°C 1 cycle 10 min 72°C -
22
Run 5 μl of PCR products (along with the 1Kb DNA ladder) on a 0.7% agarose gel in 1X TAE containing GelRed™ (1:10.000) to verify the presence of the PCR product under UV light.
At this step, using a traditional transilluminator is recommended as it is more sensitive than a DarkReader to visualize PCR products. -
23
Run the remaining PCR product on 0.7% agarose gel in 1X TAE containing GelRed™ (1:10.000). Cut the bands of the appropriate size (~3Kb) using a Dark reader for visualization and gel extract the appropriate bands with QIAquick gel Extraction Kit (Qiagen) following the manufacturer’s descriptions.
A Dark reader which uses visible light is recommended to avoid DNA damage. DNA may be damaged when exposed to UV light. The use of UV-exposed DNA templates can result in reduced fidelity and incorrect DNA replication, as well as impair the biological integrity of proteins encoded by the amplified DNA.
BASIC PROTOCOL 3: TOPO TA CLONING ORF2/ORF3 INTO PLASMID VECTOR
After amplification, the ORF2/ORF3 RT-PCR product is cloned into a plasmid vector. The amplified segment could be used to express VLPs, for mutational studies, etc. The protocol described here uses the single Adenine (A) overhangs added by Taq polymerases at the 3′-end of each synthesized strand of DNA, to ligate it into any linearized vector having a thymidine (T) nucleotide overhang. The ligation reaction is then transfected into chemically competent E. coli cells. After screening for colonies with the correct insert, they should be stored with 10–15% glycerol at −70°C.
Materials
ORF2-ORF3 PCR product (see BASIC PROTOCOL 2)
-
Taq DNA polymerase, recombinant (Life Technologies, cat. no. 10342-020) including:
10x PCR Buffer
50 mM MgCl2
Taq DNA polymerase, recombinant
10 mM ATP (Life Technologies, cat.no. AM8110G)
-
TOPO TA Cloning® Kit Dual Promoter (Life Technologies, cat. no. K460040) including:
TOP10
pCR™ II-TOPO® vector
10x PCR Buffer
Salt Solution
12.5 mM dNTP Mix
0.1 μg/μl M13 Forward (−20) oligonucleotide primer
0.1 μg/μl M13 Reverse oligonucleotide primer
0.1 μg/μl Control Template
0.1 μg/μl Control PCR oligonucleotide primers
Water
-
One Shot® TOP10 Competent Cells (Life Technology, cat. no. C404003) including:
S.O.C. Medium
TOP10 cells
pUC19 Control DNA
LB agar containing 100 μg/ml ampicillin (see recipe)
LB broth containing 100 μg/ml ampicillin (see recipe)
40 μg/ml X-Gal (Promega, cat. no. PR-V3941)
DNA suspension buffer (Teknova, cat.no. T0220)
-
Taq DNA Polymerase (Roche, cat. no. 11418432001) including:
10x PCR Buffer w/MgCl2
Taq DNA polymerase
0.2 ml thin-walled PCR/RT-PCR tubes
GeneAmp 9700 PCR thermal cycler (Life Technology) or similar instrument
Water bath at 42°C
Ice bath
Rotary shaker
Autoclaved toothpicks
VWR® Digital Dry Block Heaters (VWR, cat. no. 12621-084) or similar instrument
14 ml conical tubes
Addition of 5-deoxy-Adenosine overhangs and ligation
-
1
Prepare master mix in a labeled clean 1.5 ml microcentrifuge tube by adding the following reagents for a total of 7 μl per reaction (multiply each volume by the number of samples plus one or two reactions for pipetting errors):
5.0 μl 10x PCR Buffer (Invitrogen)
1.0 μl ATP (10 μM)
1.0 μl Taq DNA Polymerase, recombinant (Invitrogen)
-
2
Dispense 7 μl of master mix into 0.2 ml thin-walled PCR/RT-PCR tubes.
-
3
Add 43 μl cDNA template (from Basic Protocol 2) or nuclease-free water (negative control) as template and spin tubes down.
-
4
Transfer the tubes to a GeneAmp 9700 PCR thermal cycler and incubate 10 min at 72°C.
-
5
For each DNA sample, prepare TOPO TA cloning® master mix (ligation reaction) in a labeled clean 0.2 ml thin-walled PCR/RT-PCR tubes by adding the following reagents for a total of 6 μl per reaction.
0.5–4.0 μl Fresh PCR template (from step 4)
1.0 μl Salt solution
1.0 μl pCR™II-TOPO® vector
q.s.f. water
-
6
Incubate tubes at least 1 hour at room temperature to ligate the PCR product with the A-overhangs into the TOPO vector.
-
7
Store at −20°C or use immediately to transform competent cells.
Transformation of chemically competent cells
-
8
Add 2 μl of ligation reaction (from step 7) into a vial of One Shot® TOP10 Competent Cells on ice.
-
9
Incubate for 5–30 minutes on ice.
-
10
Move the cells to a 42°C water bath for exactly 30 seconds, making sure the water covers the cells.
-
11
Immediately and extremely quickly place cells on ice and incubate 1–2 minutes.
-
12
Add 250 μl SOC medium at room temperature into each tube.
-
13
Incubate cells in a rotary shaker for 1 hour at 200 × g, 37°C.
-
14
Spread LB agar plates (see recipe) with 32 μl of X-gal solution approximately 30 minutes before plating transformed cells.
The addition of X-gal will cause colonies with insert to appear white, whereas a blue color indicates no insert. -
15
Spread increasing amounts of transformed cells (25 μl, 50 μl, 75 μl, and 150 μl) on 4 different LB agar plates. Let the cells absorb into the agar for 5–10 minutes.
-
16
Incubate plate upside down overnight (12–16 hrs) at 37°C.
-
17
Remove plates from incubator and identify white colonies
-
18
Aliquot 50 μl DNA suspension buffer into 1.5 ml microcentrifuge tubes (one labeled tube per colony).
-
19
Pick up to 15 individual white colonies (clearly separated from other colonies) with autoclaved toothpicks, and swirl the toothpick in the 1.5 ml microcentrifuge tube with DNA suspension buffer (one tooth pick per colony, one colony per tube). Use the same toothpick to spot the colony on a gridded LB agar with ampicillin.
-
20
Lyse the bacterial cells in a heat block for 10 min at 98°C.
Save the gridded LB agar plates at 37°C overnight (12–16 hours). -
21
Centrifuge the tubes 10 min at 18,000 × g.
-
22
Store tubes on ice or in a refrigerator.
Verify the presence of the insert
-
23
Analyze cell lysates by PCR using Taq DNA Polymerase (Roche) and the cell lysate from step 21. Prepare master mix in a labeled clean 1.5 ml microcentrifuge tube by adding the following reagents for a total of 12.5 μl per reaction (multiply each volume by the number of samples plus one or two reactions for pipetting errors):
3.53 μl Nuclease-free water
1.50 μl 10x PCR Buffer with MgCl2 (Roche)
1.40 μl of 2.5 mM dNTPs mix
3.00 μl of 5 μM Ring2PCR oligonucleotide primer (Table 1, Figure 1)
3.00 μl of 5 μM G2SKR oligonucleotide primer (Table 1, Figure 1)
1.50 μl of DMSO
-
0.08 μl of Taq DNA polymerase (Roche)
For norovirus GI products use Ring1APCR and G1SKR primes (5μM each) instead of Ring2PCR and G2SKR, respectively
-
24
Dispense 12.5 μl of master mix into 0.2 ml thin-walled PCR/RT-PCR tubes.
-
25
Add 2.5 μl cell lysate and spin the tubes down.
-
26
Place the tubes in GeneAmp 9700 PCR thermal cycler and set for the following cycling parameters:
1 cycle 2 min 94°C 40 cycles 15 sec 94°C 30 sec 40°C 45 sec 72°C 1 cycle 7 min 72°C 1 cycle 30 min 4°C -
27
Analyze the PCR product 2% agarose gel in 1X TAE containing GelRed™ (1:10.000).
Plasmid propagation
-
28
Add 5 ml LB broth to each labeled 14 ml conical tube (1 tube/colony).
-
29
Select 8 samples with visible bands from step 27.
-
30
Pick each of these 8 colonies from the gridded LB agar plate saved from step 20 and inoculate the LB broth tubes.
-
31
Loosen the caps of each tube and secure with tape. Place the tubes in a rotary shaker set at 37°C, 225 rpm, for 16–20 hours. After 16 hours of growth, continue to plasmid purification (Support Protocol 2).
The presence of the complete insert should be verified after plasmid purification. After that, 50% glycerol stock of the bacteria containing should be prepared.
SUPPORT PROTOCOL 2: PLASMID DNA PURIFICATION AND INSERT VERIFICATION
Different kits and protocols can be successfully used to concentrate and purify plasmid preparations. The protocol described here offers a pellet-free modified alkaline lysis method that bypasses bacterial culture centrifugation and resuspension steps. Constructs can be sequenced to confirm the gene and its orientation.
Materials
Bacteria containing plasmid and initially propagated in LB broth containing 100 μg/ml ampicillin (step 31, Basic protocol 3)
DNA suspension buffer (Teknova, cat.no. T0220)
-
Zyppy™ plasmid miniprep kit (Zymo Research, cat. no. D4019) including:
7x Lysis buffer
Neutralization buffer
Endo-wash buffer
Zyppy™Wash buffer
Zyppy™Elution buffer
RNase A
Zymo-Spin™ IIN columns
Collection tubes
Vortex
1.7 ml microcentrifuge
Table top high speed microcentrifuge
Purify pDNA using Zyppy plasmid miniprep kit following the manufacturer’s instructions.
Analyze the plasmid inserts by direct sequencing using genogroup specific (PanGIR1 and Ring1A for GI or PanGIIR1 and Ring2PCR for GII) and M13F and M13 R primers provided with the TOPO TA Cloning® kit.
After insert confirmation, store the purified plasmid at −80°C as well as 10–15% glycerol stocks of the bacteria containing the plasmid (from step 1).
BASIC PROTOCOL 4: CLONING OF NOROVIRUS CAPSID GENE INTO THE VIRUS REPLICON VECTOR (ADDING VEE LINKERS THROUGH PCR)
Expression of norovirus capsid protein is accomplished using the expression vector pVR21(Pushko et al., 1997). The pVR21 plasmid consists of the VEE genome without the capsid and envelope structural genes (Figure 2). In place of these genes, a gene encoding a protein of interest is inserted and then expressed. Viral proteins from noroviruses, coronaviruses, HIV, and dengue viruses have been successfully expressed using pVR21.
Figure 2. VEE 3526 replicon norovirus capsid expression construct.
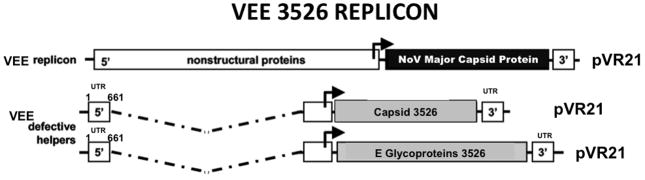
The capsid and E glycoprotein genes from Venezuelan equine encephalitis are replaced with the norovirus major capsid protein gene VP1. The VEE capsid and E glycoproteins are supplied in separate constructs. When cells are transfected with all three constructs, VEE replicons expressing norovirus VP1 are formed. These replicons can go through one round of replication, producing ample amounts of VP1 which subsequently self-assembles into VLPs.
In order to express an exogenous protein, it must first be incorporated into the DNA copy of the VEE genome in the pVR21 plasmid. To do this, the end of the desired amino acid sequence must include a linker sequence for correct in-frame alignment with the VEE promoter for subsequent expression of the protein. This nucleotide sequence can be either added onto the five-prime end of the nucleotide sequence for the gene of interest by PCR or manufactured onto the sequence if the gene is synthesized. Protocols for each of these approaches are included. This protocol inserts only ORF2 (the major capsid protein) into the replicon; adjustments will need to be made to primers to insert both ORF2 and ORF3. Basic Protocol 4 assumes the VEE linker sequences have not been added, while Alternate Protocol 4 can be followed if using a synthesized sequence already containing the linkers.
The VEE 5′ linker includes the VEE sequence from the Apa1 site through the norovirus ORF2 start site. This linker sequence can be added by using a long oligonucleotide primer with sequence AGTCTAGTCCGCCAAG followed by the first 28 nucleotides of your ORF2 sequence of interest (44 mer). The 3′ linker sequence includes the last 28 nucleotides of your gene plus an Asc1 restriction site (ggcgcgcc) followed by eight random nucleotides (44 mer). Both of these are 5′-3′ on the sense strand.
Materials
Norovirus capsid pDNA, prepared in Basic Protocol 3
Chemically Competent E. coli either lab-made or commercially purchased.
LB media (no antibiotic)—see recipe
Agar plates made with appropriate selection antibiotic (we use carbenicillin or ampicillin)
10 and 50 ml conical tubes
Qiagen QIAprep Miniprep Kit (Qiagen)
PCR oligonucleotide primers (VEE primers pVR6199 and V7564 and sequence specific norovirus capsid forward oligonucleotide primer with VEE linker and reverse oligonucleotide primer with Asc1 site)—see Table 2
Expand High Fidelity PCR Kit, (Roche)
DepC water
Sterile PCR tubes
0.8% agarose gel in 1X TAE with 0.5μg/ml EtBr
Disposable scalpel blades
Sterile 1.5 ml microcentrifuge tubes
Qiagen QIAquick Gel Extraction Kit (Qiagen)
pVR21 plasmid (not commercially available)
Molecular biology grade Enzymes (Apa1, Asc1, ligase, CIP), available from NEB
LB media (see recipe)
carbenicillin or ampicillin 1X TAE
Topo-TA cloning kit Invitrogen
Table 2.
Primers for adding VEE linkers to the Norovirus Capsid
| Primer Name | Construct | Direction | Sequence | Use* |
|---|---|---|---|---|
| G1s2V | GII.4-2006 | Forward | AGTCTAGTCCGCCAAGATGAAGATGGCGTCGAATGACGCCAACC | Adding the VEE linker sequence to the beginning of the NoV capsid gene |
| DHa2V | GII.4-2006 | Reverse | CGTTAGCAGGCGCGCCTTATAAAGCACGTCTACGCCCCGTTCCA | Adding an Asc1 site at the end of the capsid gene |
| pVR6199 | VEE | Forward | CAAAGCTGCGCAGCTTTCC | PCR amplifies the VEE piece needed for overlap PCR with NoV capsid |
| V7564 | VEE | Reverse | CATCTTGGCGGACTAGACTATGTCGTAGTCCATTCAGGTTAGCCG | PCR amplifies the VEE piece needed for overlap PCR with NoV capsid |
NoV: norovirus
42°C heat block
Glass spreading rod
Rocker
37°C incubator or warm room
Shaker that accommodates conical tubes (28°C–30°C)
Centrifuge that accommodates conical tubes
Beaker with bleach
Gel box with large comb
Gel Imager or dark reader
Thermocycler
Sequencing Primers
-
1
Add VEE linker sequences to the norovirus major capsid gene ORF2
Insert PCR and VEE arm PCR using Expand High Fidelity Kit (Figure 3, Step 1)
This adds a restriction site on the 3′ end and an overlap on the 5′ end.Insert PCR:
2 μl construct pDNA
4 μl 2 μM sense oligonucleotide primer
4 μl 2 μM antisense oligonucleotide primer
1.75 μl 10 mM dNTPs
5 μl Buffer 2
32.5 μl DepC water
0.75 μl enzyme
The sense and antisense primers for the norovirus capsid gene are sequence specific, so each capsid will have to be checked to see if oligonucleotide primer redesign is necessary. Examples are given in Table 2, G1s2V is a forward oligonucleotide primer and DHa2V is an antisense oligonucleotide primer for GII.4-2006. Note that these oligonucleotide primers will have to be checked and possibly re-designed based on sequences of other norovirus strains.
Figure 3. Cloning Strategy for inserting norovirus ORF2 into pVR21.

Step 1) Using plasmid DNA from Basic Protocol 3, an Asc1 site and a VEE linker sequence are added to ORF2 and amplified through insert PCR. Using pVR21, an overlapping region of VEE is amplified through VEE arm PCR. Step 2) Using Step 1 PCR products, a single overlap PCR product is generated that contains both the VEE arm and norovirus capsid gene. Step 3) The overlap PCR product is cloned into a TOPO TA plasmid. Step 4) Both the plasmid from Step 3 and pVR21 are cut using Apa1 and Asc1 enzymes. Step 5) The norovirus capsid gene is ligated into pVR21.
-
VEE arm PCR:
This amplifies the pVR21 piece that will overlap with the insert PCR piece.2 μl pVR21 pDNA
4 μl 2 μM pVR6199 forward oligonucleotide primer
4 μl 2 μM V7564 anti-sense oligonucleotide primer
5 μl Buffer 2
1.75 μl 10 mM dNTPs
32.5 μl DepC water
0.75 μl enzyme
-
These 50 μl reactions should be run on the following PCR program:
94°C for 3 min
94°C for 30 sec
54°C for 1 min
68°C for 3 min
Repeat steps b-d 30X
68°C for 10 min
4°C ∞
-
2
Make a 0.8% agarose gel in 1X TAE with 0.5 μg/ml ethidium bromide with a comb that will allow loading of at least 80 μl sample volume and run the samples.
-
3
Remove gel and image with a dark reader.
-
4
See Table 3 for desired band sizes for the Insert and VEE arm PCR products. Cut out the desired bands, each with a new blade, and put them in labeled microcentrifuge tubes.
-
5
Use the Qiagen QIAquick Gel Extraction kit and follow the manufacturer’s protocol to purify the digested DNA. Elute with 33 μl elution buffer.
DNA can be stored at −20°C. -
6
Overlap PCR using Expand High Fidelity PCR Kit (Figure 3, Step 2)
This joins the VEE arm and norovirus construct pieces and includes the correct restriction site to later insert this piece into the pVR21 vector.2 μl VEE arm PCR gel extract
3.5 μl norovirus capsid PCR gel extract
4 μl 2 μM 6199 forward oligonucleotide primer
4 μl 2 μM antisense norovirus oligonucleotide primer used in step 2 for insert PCR
1.75 μl 10 mM dNTPs
5 μl buffer 2
29 μl DepC water
0.75 μl enzyme
Table 3.
PCR Product and Endonuclease Digestion Band Sizes
| Plasmid or DNA* | Enzyme | Enzyme | Enzyme | Desired Band Size |
|---|---|---|---|---|
| NoV* Capsid Gene with VEE ends | Apa1 | Asc1 | N/A | ~1.8 kb |
| pVR21 | Apa1 | Asc1 | CIP | ~9.8 kb |
| Insert PCR NoV Capsid Gene | N/A | N/A | N/A | ~1.7 kb |
| VEE Arm PCR Product | N/A | N/A | N/A | ~1.2 kb |
| Overlap PCR Product | N/A | N/A | N/A | ~3 kb |
| Digested Overlap PCR Product | Apa1 | Asc1 | N/A | ~1.8 kb |
| VEE-NoV Capsid RNA | N/A | N/A | N/A | ~4 kb |
| VEE Capsid Helper RNA | N/A | N/A | N/A | ~1.5 kb |
| VEE E Glycoprotein Helper RNA | N/A | N/A | N/A | ~2 kb |
NoV: norovirus
-
This 50 μl reaction should be run on the following PCR program:
94°C for 3 min
94°C for 30 sec
55°C for 25 sec
68°C for 3 min
Repeat steps b-d 5X
94°C for 30 sec
58°C for 25 sec
68°C for 3 min
Repeat steps f-h 25X
68°C for 10 min
4°C ∞
Make a 0.8% agarose gel in 1X TAE with 0.5 μg/ml ethidium bromide with a comb that will allow loading of at least 80 μl sample volume and run the samples.
-
7
Remove gel and image with a dark reader.
-
8
See Table 3 for desired band sizes for the Insert and VEE arm PCR products. Cut out the desired bands, each with a new blade, and put them in labeled microcentrifuge tubes.
-
9
Use the Qiagen QIAquick gel extraction kit and follow the manufacturer’s protocol to purify the digested DNA. Elute with 33 μl elution buffer.
-
10
Topo TA clone overlap PCR product as directed by the manufacturer (Figure 3, Step 3).
-
11
When individual colonies are visible, for each construct label (4) 50 ml conical tubes and fill with 20 ml LB media with selection antibiotic under sterile conditions.
-
12
Under sterile conditions, carefully select an individual colony from the plate with a pipet tip and pipet up and down into the LB to inoculate the LB. Choose one colony per conical tube.
-
13
Put conical tubes on a shaker overnight at 28–30°C.
-
14
Make a library plate for your cultures (so you can grow more of your plasmid DNA if you want without having to re-transform cells). Label the bottom of an antibiotic plate with a box for each colony. Put 10 μl drop of the appropriate culture into each box, being careful not to mix them. Put at room temperature overnight, and then wrap the plate with Parafilm and store at 4°C for up to three months. Also make a glycerol stock.
A glycerol stock will keep at −80°C indefinitely, so the construct will not have to be remade from scratch again. -
15
Spin down the cultures at 800 X g for 10 minutes, and dump the media off the pelleted cells.
-
16
Use a Qiagen QIAprep Miniprep kit to purify the plasmid DNA. Elute in 50 μl elution buffer.
Since the Qiagen spin columns are not designed for cultures as large as 20 ml, prepare three spin columns for each culture that was grown. Label three microcentrifuge tubes for each conical tube culture, suspend the cell pellets in 750 μl P1 buffer, and aliquot it into three tubes of 250 μl each, which is the volume specified in the protocol. Follow the manufacturer’s protocol from this step. At the end of the purification, the tubes from the same culture can be combined (so there should be 150 μl total eluted pDNA for each bacterial culture that was grown). -
17
Digest Topo plasmid containing Overlap PCR product and pVR21 plasmid (Figure 3, Step 4).
This step cuts out the piece from the overlap PCR product that will be inserted into the pVR21 plasmid and cuts the pVR21 plasmid to allow for the ligation with the construct DNA.For construct DNA:
30 μl Topo-Overlap PCR product
4.0 μl 10X Buffer 4
0.4 μl BSA
2.0 μl Apa1 enzyme
2.0 μl Asc1 enzyme
1.6 μl DepC water
Incubate this 40 μl reaction at 25°C for 1 hour and then at 37°C for 1 hour.
-
For pVR21 DNA:
30 μl pVR21 pDNA
4.0 μl 10X Buffer 4
0.4 μl BSA
2.0 μl Apa1 enzyme
2.0 μl Asc1 enzyme
1.6 μl DepC water
Allow this 50 μl reaction to incubate at 25°C for 1 hour and then at 37°C for 1 hour.Treat digestion product with calf intestinal phosphatase (CIP).
This prevents vector self-ligation during the ligation step.40 μl digested pVR21
5 μl 10X Buffer 3
0.5 μl CIP enzyme
4.5 μl DepC water
Incubate this 50 μl reaction at 37°C for 1 hour.
Make a 0.8% agarose gel in 1X TAE with 0.5 μg/ml ethidium bromide with a comb that will allow loading of at least 80 μl sample volume and run the samples.
-
18
Remove gel and image with a dark reader.
-
19
See Table 3 for the desired band size of the digested Overlap PCR product and digested pVR21. Cut out the desired bands, each with a new blade, and put them in labeled microcentrifuge tubes.
-
20
Use the Qiagen QIAquick gel extraction kit and follow the manufacturer’s protocol to purify the digested DNA. Elute with 33 μl DepC water.
-
21
Set up a ligation reaction (Figure 3, Step 5) to insert your digested construct DNA into the digested pVR21 backbone (per reaction):
2.5 μl Asc1/Apa1/CIP digested pVR21 gel extract
3.5 μl Asc1/Apa1 digested construct Overlap PCR gel extract
1.5 μl ligase buffer
1.5 μl ligase
6.0 μl DepC water
Allow this15 μl ligation reaction to incubate overnight at 4°C.
-
22
Transform competent cells with the ligase reaction. Follow support protocol 3 starting at step 3. At step 4, instead of adding 2 μl of DNA, add all 15 μl of the ligase reaction to the competent cells. At step 10, instead of plating 20 μl and 80 μl, plate 80 μl and 150 μl of the transformed competent cells and incubate at 28–30°C overnight.
-
23
Follow steps 11–16 to choose colonies and purify pDNA.
-
24
Sequence-verify the constructs.
Design sequence specific primers about 500 bases apart. Four primers are typically used for sequencing norovirus capsid constructs in our lab.
Alternate Protocol 4: CLONING OF NOROVIRUS CAPSID GENE INTO THE VIRUS REPLICON VECTOR (SYNTHESIZED DNA ALREADY CONTAINS VEE LINKERS)
Use this protocol if DNA has been commercially synthesized and the VEE linker sequence was manufactured onto the end of the norovirus capsid sequence.
Materials
DepC water
Synthesized norovirus capsid pDNA, available from BioBasic
Chemically Competent E. coli either lab-made (Appendix 3L) or commercially purchased.
LB media (no antibiotic)—see recipe
Agar plates made with appropriate selection antibiotic
10 and 50 ml conical tubes
Molecular biology grade Enzymes Apa1, Asc1, CIP, ligase, available from NEB
Qiagen QIAprep Miniprep Kit
Qiagen QIAquick Gel Extraction Kit
pVR21 plasmid
LB media with appropriate selection antibiotic for the plasmid your capsid gene is in 1X TAE
42°C heat block
Glass spreading rod
Rocker
37°C incubator or warm room
Shaker that accommodates conical tubes (28°C–37°C)
Centrifuge that accommodates conical tubes
Beaker with bleach
0.8% agarose gel in 1X TAE with 0.5 μg/ml Ethidium Bromide
Gel box with large comb
Disposable scalpel blades
Sterile 1.5 ml microcentrifuge tubes
Sterile PCR tubes
Thermocycler
Dark Reader (to visualize gel bands)
Sequencing oligonucleotide primers
Spin down vials with lyophilized plasmid DNA and suspend according to manufacturer’s directions. We re-suspend our plasmid DNA in 40 μl DepC water.
Make a 20 μl 1:10 dilution of the plasmid DNA in DepC water to use as a stock (store at −20°C).
Transform competent cells with the synthesized plasmid. (See support protocol 4)
Check the plates and choose a plate with easily identifiable individual colonies to pick colonies from.
Under sterile conditions, for each construct label (3–6) 10 ml conical tubes and fill with 7 ml LB media with selection antibiotic. Also grow a culture of pVR21-expressing E. coli if there is not already purified pVR21 plasmid available.
Under sterile conditions, use a pipet tip to carefully select an individual colony from the plate and pipet up and down into the LB to inoculate the LB. Choose one colony per conical tube.
Put conical tubes on a shaker overnight (constructs in pUC57 or pVR21 can be incubated from 22–30°C).
Make a library plate for your cultures (so you can grow more of your plasmid DNA if you want without having to re-transform cells). Label the bottom of an antibiotic plate with a box for each colony. Put 10 μl drop of the appropriate culture into each box, being careful not to mix them. Put at room temperature overnight, and then wrap plate with Parafilm and store at 4°C for up to three months.
Spin down cultures at 800 × g for 10 minutes.
Dump off media into bleach, being careful not to disturb the cell pellet.
Purify the plasmid DNA using the Qiagen QIAprep Miniprep kit. Follow manufacturer directions. Elute in 33 μl elution buffer.
-
Digest both pVR21 and construct pDNA with Asc1 and Apa1 enzymes (per reaction):
40 μl pDNA
5.0 μl 10X buffer 4
0.5 μl 100X BSA
0.5 μl DepC water
2.0 μl Asc1 enzyme
-
2.0 μl Apa1 enzyme
Allow this 50 μl reaction to incubate at 25°C for 1 hour and then at 37°C for 1 hour.While the digestion is incubating, set up agarose gel.
-
Digest ONLY the pVR21 plasmid with calf intestinal phospatase (CIP) to prevent vector self-ligation later on (per reaction):
50 μl digested pVR21
6.0 μl 10X buffer 3
0.6 μl CIP enzyme
3.4 μl DepC water
Allow this 60 μl reaction to incubate at 37°C for 1 hour.
Follow Basic Protocol 2 beginning at Step 21.
Support Protocol 3: Transformation of Competent Cells
In order to produce large quantities of a particular gene, a plasmid encoding that gene can be inserted into bacterial cells through a process called transformation. Some bacteria are more able to incorporate exogenous DNA than others. Those that easily take up DNA are referred to as competent cells and are used for transformation. When the bacteria containing the gene multiply, the plasmid is copied as well, leading to the production of a huge amount of plasmid. The bacteria are then lysed and the plasmid purified away from the rest of the cell contents.
Materials
Plasmid DNA
Ice
Chemically Competent E. coli (purchased or prepared, see Appendix 3L for preparation instructions)
LB media
LB agar plates with Carbenicillin or ampicillin antibiotic
42°C heat block
glass spreading rod
rocker
37°C incubator or warm room overnight or at room temperature for 48 hours.
Thaw 50 μl (1 vial) chemically competent E. coli on ice.
Gently add 2 μl of plasmid DNA to 50 μl (1 vial) chemically competent E. coli. Gently flick tube with finger to mix (do not pipet up and down).
Incubate the cells with the plasmid DNA on ice for 30 minutes.
Incubate the cells in the 42°C heat block for 2 minutes.
Immediately incubate the cells on ice for 2 minutes.
Add 250 μl of LB broth (with no antibiotic) and incubate for 2 hours at room temperature on a rocker.
While the cells are incubating at room temperature, label two carbenicillin (or ampicillin) plates for each construct and put them at 37°C to warm them.
Under sterile conditions, add 20 μl of cells to one plate and 80 μl to the other plate and spread with a sterile glass spreading rod.
Invert plates and put them at 37°C overnight or at room temperature for two days (or until bacterial colonies are visible).
Basic Protocol 5: PRODUCTION OF STOCK NOROVIRUS RECOMBINANT VIRUS REPLICON PARTICLES
Virus replicon particles (VRPs) are constructs that are able to go through one round of replication when helper constructs are added, but on their own do not encode all of the components needed to replicate. VRPs are useful because they can express a large amount of a desired protein during a single round of infection. The VEE system utilizes pVR21, which is a plasmid encoding the VEE genome except for the capsid and E glycoproteins. In place of these genes, the gene of interest is inserted behind the 26S promoter (in this case the norovirus major capsid gene) (Figure 2). The 26S promoter facilitates production of ample amounts of exogenous protein independent of viral replication. Without the VEE capsid and E glycoproteins, no progeny virions can be produced. When separate “helper” plasmids, one encoding the capsid and the other encoding the E glycoproteins, are added along with pVR21, VEE has all the necessary components to go through one round of replication. However, because pVR21 does not encode the instructions for making the VEE capsid or E glycoproteins, it is not able to continue producing new virions unless additional helper plasmids are added. This approach ensures that norovirus capsid can be safely expressed without production of replication-competent VEE. While VEE is a BSL-3 pathogen, the 3526 VEE plasmids are BSL-2 reagents because they contain attenuating mutations. This makes the VEE replicon system a viable expression system for anyone with BSL-2 facilities. Using this system, ample amounts of norovirus VLPs are produced.
Materials
Plasmid DNA: capsid gene in pVR21, 3526 Capsid helper, 3526 Envelope glycoprotein helper
Not1 enzyme, available from NEB (comes with 10X buffer 3 and 100X BSA)
Qiagen QIAquick Gel Extraction Kit, (use PCR purification protocol)
RNAse Zap, available from Sigma-Aldrich
Ambion mMessage mMachine T7 Transcription Kit, available from Life Technologies
Filter pipet tips
Sterile PCR tubes
1X TAE
10% SDS
agarose (for gel)
2 mM EDTA, pH 8
6X loading dye, available from Promega
1 kb ladder, available from Promega
Pyrex dish (gel must fit inside during de-staining)
Ethidium bromide (EtBr)
EtBr de-staining teabags, available from Mo Bio Laboratories
Gel box with small comb
BHK-21 cells (one 80% confluent T-175 flask per VRP, plus one for a control)
BHK-21 media (see recipe)
Sterile PBS
0.05% Trypsin-EDTA
Pipets (5 ml, 10 ml, and 25 ml)
Pipetman
10 and 50 ml conical tubes
Cell culture treated flask, 75cm3
Cell culture treated flask, 175cm3
Trypan Blue
Hemocytometer
0.4 cm cuvettes, available from BioRad
Sucrose
25×89 mm Ultra-Clear Beckman Ultracentrifuge tubes, available from Beckman Coulter
Kim wipes
Parafilm
Crystal Violet Solution—see recipe
8 well chamber slides available from lab-Tech
1:1 Acetone: Methanol Solution
Pencil
Anti-norovirus mouse sera
FITC-labeled goat anti-mouse IgG antibody, available from Sigma-Aldrich
Aluminum foil (or a dark drawer)
Thermocycler, available from BioRad
Rocker
UV gel reader
Centrifuge that can accommodate conical tubes
Gene Pulser (for electroporation), available from BioRad
Digital Scale
Ultracentrifuge
Microscope (capable of imaging FITC-labeled antibodies)
Fluorescent cell imaging software
-
Digest plasmids (pVR21 construct, 3526 capsid helper, 3526 E glycoprotein helper) with Not1. Per reaction:
--50 μl pDNA
--6.5 μl buffer 3
--0.7 μl 100X BSA
--5.8 μl DepC water
--2.0 μl Not1
-
65 μl reaction, Incubate at 37°C for 1 hour.
One capsid helper and E glycoprotein helper reaction will be needed for every two VRPs made.
Clean up digested plasmid DNA using the Qiagen PCR clean-up protocol. Elute in 33 μl elution buffer.
Make RNA using the Ambion mMessage mMachine T7 Transcription Kit (see Support Protocol 4)
Check RNA production by gel analysis (see Support Protocol 5)
Electroporate BHK-21 cells with RNA (see Support Protocol 6)
Harvest VRPs (see Support Protocol 7)
Purify VRPs (see Support Protocol 8)
Safety test VRPs (see Support Protocol 9)
Titer VRPs (see Support Protocol 10).
Support Protocol 4: PRODUCTION OF RNA FOR MAKING VRPS
Norovirus and VEE are RNA viruses, so an RNA copy of the DNA must be transfected into cells in order to get expression of the desired proteins. RNA must be handled with care, as it degrades easily due to RNA-digesting enzymes called RNAses found abundantly in the environment. Degradation of even a small portion of the RNA will lead to decreased or no expression of the desired proteins. All work surfaces, gloves, equipment and reagent bottles should be wiped down with RNAse zap, which inactivates RNAses.
Materials
RNAse Zap
Not1 digested plasmid DNA (pVR21 construct, 3524 Capsid helper, 3524 Envelope glycoprotein helper)
Sterile PCR tubes
Ambion mMessage mMachine T7 Transcription Kit, available from Life Technologies
Filter pipet tips
Thermocycler
Wipe down the work surface and all tube racks and closed tubes with RNAse zap.
-
Let 10X buffer, 2X NTP/CAP, and GTP thaw at room temperature, but place 2X NTP/CAP on ice as soon as it is thawed.
10X buffer should be kept at room temperature after it thaws to prevent precipitation of the buffer. -
Mix reaction solutions in the following order at room temperature (per reaction):
--3.0 μl 10X buffer
--15 μl 2X NTP/CAP
--7.5 μl Not1 digested plasmid
--1.5 μl GTP
--3 μl T7 Enzyme Mix
30 μl reaction
Use filter tips when working with RNA. -
Put reactions in the thermocycler and program:
--2 hours at 37°C
--4°C forever
Reserve a 3 μl aliquot from each reaction to run on a gel (as described in Support Protocol 5) and freeze the RNA at −80°C until use.
Support Protocol 5: VERIFICATION OF RNA PRODUCTION BY GEL ANALYSIS
Intact RNA is necessary for VRP and VLP production. RNA is very sensitive to degradation, so running an RNA gel is a good way to ensure that RNA transcription was successful. Make sure to spray work areas, gloves, and all necessary equipment with RNAse zap when working with RNA.
Materials
1.65 liters 1X TAE with 0.1% SDS
10% SDS
1.3 grams agarose
2mM EDTA, pH 8
6X loading dye
1 kb DNA marker
Pyrex dish (gel must fit inside during destaining)
Ethidium bromide (EtBr)
0.1% SDS/2mM EDTA, pH 8.0
Gel box with small comb
Thermocycler
Rocker
EtBr teabags
UV gel reader
-
Autoclave:
--1.5L 1X TAE/0.1% SDS
--150 ml 1X TAE/0.1% SDS with 1.3 grams Ultrapure agarose
When flask with gel gets cool enough to touch, pour a gel using a small comb. Let the gel cool before removing comb.
Mix 3 μl RNA with 3 μl 0.1% SDS/2mM EDTA, pH 8.0
-
Put in thermocycler on program:
--65°C for 10 minutes
--4°C forever
This incubation step is necessary in order to remove any secondary structures present in the RNA before running on the gel. We recommend that this step be done in a thermocycler rather than a water bath to avoid contamination. Add 2 μl loading dye to each sample.
Load 10 μl 1 kb DNA marker in the first lane and then each of the samples in subsequent lanes on the gel.
Run 2/3 of the length of the gel and then rinse with water.
Put gel into a dish with approximately 250 ml water and 25 μl 10mg/ml EtBr. Make sure the water covers the gel.
Cover with aluminum foil and gently rock for at least 30 minutes and up to overnight.
-
Pour off staining solution into a container to collect EtBr waste and add an EtBr teabag to absorb the EtBr from the water. Refill dish with water and rock for 10 minutes. Repeat this 3 more times.
Ethidium bromide waste is potentially mutagenic and therefore should be allowed to sit for 24 hours with the EtBr teabag before pouring water down the drain.EtBr teabag should be put into EtBr waste. It is highly recommended to follow the disposal procedures for ethidium bromide outlined by your institution. Visualize RNA by UV light. Desired band size is indicated in Table 3 and should appear as in Figure 4. If bands are correct, proceed to the electroporation step.
Figure 4. Agarose gel analysis of RNA produced from transcription of VEE plasmids.
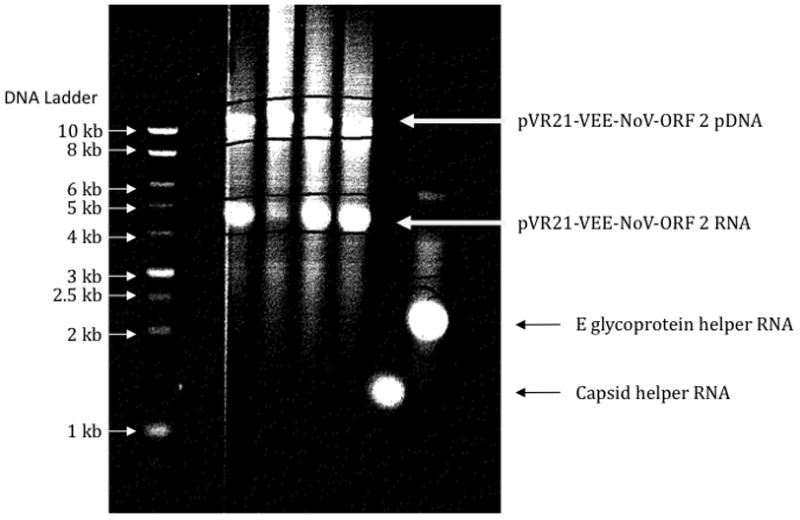
pDNA from pVR21 clones containing different norovirus ORF-2 (Lanes A–D), VEE envelope (Lane E), and VEE capsid (Lane F) genes were transcribed using a T7 in vitro transcription kit and the reaction products separated on a 0.8% agarose/0.1% SDS gel and stained with ethidium bromide. Band sizes are compared to a DNA ladder and are also described in Table 3.
Support Protocol 6: ELECTROPORATION OF BHK-21 CELLS WITH RNA
Electroporation shocks the cells and allows them to uptake exogenous RNA. Because RNA degrades quickly and easily, it is imperative that electroporation occurs immediately after adding RNA to the cuvettes.
Materials
Cell culture treated flask 175cm3
BHK-21 cells
BHK-21 media (see recipe)
Sterile PBS, cold
5, 10, and 25 ml pipets
50 ml conical tubes
0.05% trypsin-EDTA
Trypan Blue
Hemocytometer
0.4 cm cuvettes, (BioRad)
Construct RNA and helper RNA that was made in a previous step
Filter pipet tips
Gene Pulser (For electroporation), (BioRad)
-
Prepare one T-175 flask of 80% confluent BHK-21 cells per VRP electroporation.
Make extra flasks of cells for this step since it is better to have too many cells and not use them all than to not have enough cells to make all the VRPs desired. One to two extra flasks should be sufficient. Wash cell monolayer with cold PBS. Add 2 ml trypsin to each flask and rock to distribute trypsin. Let flasks sit at room temperature until cells slide off. Do not tap to release cells.
Add 10 ml BHK-21 media to each flask and pipet a few times to wash all of the cells off the flask. Combine all of the cells into a 50 ml conical tube (or two, depending on how many flasks of cells you have).
Spin down cells for 10 minutes at 800 × g and dump off media, reserving the cell pellet.
-
Suspend in 50 ml cold, sterile PBS. Spin cells down again and dump off PBS, reserving the cell pellet.
Turn on and set the Gene Pulser while the cells are spinning down. Settings are described in step 10. Suspend washed cells in 20 ml cold, sterile PBS. Take 10 μl to count cells. Spin cells down again and count cells during this spin.
Suspend cells at 1.0 × 107 cells/ml in cold, sterile PBS.
Label one cuvette for each construct plus one for a control. Add 800 μl cells to each cuvette. Leave cuvettes at room temperature while prepping.
-
Working quickly and doing one VRP construct at a time, add 30 μl of construct RNA + 15 μl each of capsid helper RNA and E glycoprotein helper RNA to the cuvette. Tap gently to mix.
RNA is very fragile and degrades quickly and easily. It is imperative that pulses are applied to the cuvette immediately after RNA is added to avoid RNA degradation. Apply 3 pulses per cuvette on the following settings: 850V, Capacitance 25, and Resistance ∞.
Repeat steps 9 and 10 for each individual VRP construct.
Let electroporated cells sit at room temperature for 10 minutes.
Label T-75 flasks and plate cells into flasks with ~14 ml BHK-21 media. Incubate for 24 hours.
Support Protocol 7: HARVESTING VRPS
Since 3526 VEE VRPs are potentially infectious agents, it is necessary to conduct VRP harvesting steps under BSL-2 conditions in a laminar flow hood. VRPs are harvested over 2 days to ensure that as many VRPs are collected as possible. It is important to spin down the VRP-containing media after each 24-hour period to ensure that all cells are removed from the supernatant.
Materials
Pipetman
Pipets (25 ml)
BHK-21 media
10 ml and 50 ml conical tubes
Centrifuge
This protocol needs to be conducted in a laminar flow hood.
After 24 hours, collect supernatant from cells (from Support Protocol 6) in a 50 ml conical tube, and replace media with ~14 ml of fresh BHK-21 media. Return cells to incubator.
Clarify collected supernatant by spinning at 800 × g for 15 minutes at 4°C. Transfer supernatant to a new conical tube and store at 4°C.
After another 24 hours, collect ~14 ml of supernatant from the flask in a conical tube and clarify by spinning at 800 × g for 15 minutes at 4°C.
Pool clarified supernatant into one 50 ml conical tube.
Transfer 2.8 ml of the pooled clarified supernatant to a new 10 ml conical tube to use during safety testing. This sample can either be used immediately for round 1 safety testing (see support protocol 9) or frozen at −20°C for later use.
Support Protocol 8: PURIFYING VRPS
VRPs are concentrated using a sucrose cushion. After high-speed centrifugation, the VRPs are found as a pellet at the bottom of the tube. They must then be re-suspended in PBS, separated into aliquots, and frozen for future use. Instead of concentrating VRPs, it is also possible to make 2 ml aliquots of the tissue culture supernatant and freeze at −80°C. This supernatant can then be added directly to BHK-21 cells to produce VLPs (see Basic Protocol 6).
Materials
5 ml 20% sucrose in cold PBS per VRP, sterile filtered
25×89 mm Ultra-Clear Beckman Ultracentrifuge tubes (Beckman)
Sterile PBS, cold
Pipetman
Pipets (5 ml and 25 ml)
Beaker with bleach
Kim wipes
Parafilm
Filter pipet tips
Sterile 1.5 ml microcentrifuge tubes
SW28 rotor, (Beckman)
Digital Scale
Ultracentrifuge
Prepare 20% sucrose in PBS, sterile filter, and chill to 4°C. Turn on and cool Ultracentrifuge to 4°C while you are preparing the samples.
In the laminar flow hood, add collected clarified supernatant to a labeled Ultracentrifuge tube.
-
Set pipetman to slow release. Carefully underlay 5 ml of sucrose under the supernatant. If tube is not completely full, add cold, sterile PBS to just below the top of the tube.
Sucrose can also be added first and supernatant carefully overlayed on top of it. Be careful that the sucrose and supernatant layers do not mix together. Bring rotor buckets into hood. Put tubes into rotor buckets, seal, and spray with ethanol and Vesphene to decontaminate.
Take rotor buckets out of the hood to weigh. Record the weights of each rotor bucket and determine how much PBS must be added to each tube to make them all equal.
-
Take rotor buckets back to the hood and add PBS to bring them all up to the same weight.
It is VERY important to balance tubes in an Ultracentrifuge, so make sure all rotor buckets weigh the same before proceeding to the spin step. -
Centrifuge the VRPs at 131,000 × g for 3 hours at 4°C.
These settings are specific for an SW-28 rotor, and may need to be adjusted using a different rotor. In the hood, remove tube from rotor bucket and pour off the supernatant into bleach. Blot the rim of the tube with a Kim wipe. Let it sit upside down on the Kim wipe for several seconds to remove all of the supernatant. Decontaminate Kim wipes with ethanol.
Add 0.5 ml sterile PBS to the tube, wrap the tube with Parafilm, and store at 4°C overnight.
The next day, scrape the bottom of the tube with a pipet tip and pipet up and down many times to dislodge VRPs from the tube.
Transfer the VRPs to a sterile 1.5 ml microcentrifuge tube.
Add an additional 0.5 ml PBS to the pellet and repeat the scraping and washing.
Add the additional VRP wash to the 1.5 ml microcentrifuge tube (pool washes).
Remove 10 μl of the VRP prep to determine titer.
Aliquot 50–100 μl VRPs into sterile tubes and freeze at −80°C until use.
Support Protocol 9: SAFETY TESTING VRPS
Safety testing of the VEE VRPs is necessary to ensure that recombination of the virus does not produce replication-competent virus. Three separate expression plasmids encoding different parts of the VEE genome are used to make VEE VRPs to prevent recombination. Safety testing involves passaging the VRPs twice to make sure cells are not infected with wild type VEE virus. Presence of cytopathic effect (CPE) in round 2 of safety testing indicates a possible recombination event and safety testing must be repeated or VRP prep must be discarded.
Materials
Cell culture treated flask 175cm3
BHK-21 cells
BHK-21 media (see recipe)
Pipets (5 ml, 10, ml 25 ml)
Electroporation media collected during step 6 of Support Protocol 4
10 ml and 50 ml conical tubes
0.5% crystal violet solution (see recipe)
Pipetman
37°C incubator
Centrifuge (that holds conical tubes)
Microscope
-
For safety testing, prepare (1) 80% confluent T-175 flask of BHK-21 cells for each VRP, plus one to use as a control. (Split cells 1/5 the day before using).
All safety testing must be done under BSL-2 conditions in a laminar flow hood. Vero cells can also be substituted for BHK-21s during safety testing. For round 1 of safety testing, add 2.8 ml of the electroporation media to the flask.
Return flask to incubator for 1 hour, gently rocking every 15 minutes.
-
Add 25 ml of media per flask and observe for 48 hours, comparing VRP flasks to the control flask.
Cytopathic effect (CPE) should be seen during round 1 safety testing. At 48 hours, remove supernatant and clarify by centrifugation at ~3000 RPM for 15 minutes at 4°C. Transfer clarified supernatant to a new conical tube. Keep a separate tube of 5 ml of the clarified supernatant in case round 2 safety testing has to be repeated.
Either proceed to round 2 safety testing or freeze clarified supernatant.
Prepare 0.5% crystal violet in 30% methanol (see recipe) and one 60% confluent T-175 flask of BHK-21 cells for each VRP, plus one flask as a control.
Add 5 ml of round 1 testing clarified supernatant to each flask.
Return flask to incubator for 1 hour, gently rocking every 15 minutes.
At 24 hours, mark any holes in the monolayer with a black sharpie on the bottom of the flask.
Observe the flasks every 24 hours for 72 hours, comparing VRP flasks to the control flask. Watch to see if marked areas expand, new areas become bare, or cells become long and skinny (sick BHK-21s). Round bare spots that do not grow are from air bubbles, not CPE.
At 72 hours, remove tissue culture supernatant from flasks and save in case round 3 safety testing is needed (if round 2 is inconclusive).
Add 30 ml of crystal violet stain per flask. Shake around to coat all sides for 5 minutes.
Wash flasks with water until rinse is no longer blue. Let it air dry.
Evaluate flasks both macroscopically and microscopically, looking to see if marked holes got bigger or new ones formed and to see if cells around the holes look sick.
-
If the cells are both macroscopically and microscopically indistinguishable from the negative control cells, the VRPs pass safety testing for recombinant virus and can be used.
If CPE is seen or it’s inconclusive, do round 3 testing, following the same protocol for round 2 testing. If CPE is seen, discard VRP prep and remake VRPs.
Support Protocol 10: DETERMINE TITER OF VRPS
Typical yields for VEE 3526 in BHK-21 cells are between 5 × 106 and 5 × 107 VRP/ml. While it’s not vital to titer VRPs that are intended only to make VLPs, it is necessary to know the titer when immunizing animals with VRPs. Titer also gives an indication of expression efficiency in different cell types and is useful in optimizing the protocol for different conditions.
Materials
8 well chamber slides (1 per two VRPs)
BHK-21 cells
BHK-21 media
1:1 Acetone:Methanol mixture (~50 ml per slide)
50 ml conical tubes
Pencil
PBS
Anti-norovirus mouse sera or commercially available mouse monoclonal antibody (Maine Biotechnology Services)
FITC-labeled goat anti-mouse IgG antibody
Antifade (optional)
Aluminum foil (or a dark drawer)
37°C incubator
Microscope (capable of imaging FITC-labeled antibodies)
Fluorescent cell imaging software
-
Plate BHK-21 cells on 8-well chamber slides at 1 × 104 cells/well in 200 μl BHK-21 media. Let the cells adhere overnight.
Don’t try to infect cells if they are too confluent the next day. If cells are piled on top of each other, counting is inaccurate. If this occurs, plate new chamber slides, slightly reducing the number of cells per well. Serially dilute VRPs from 1 × 102 to 1 × 104 in BHK-21 media. Make at least 100 μl per dilution.
Remove media from slides and gently add 80 μl of the 102, 103, and 104 VRP dilutions and a PBS control for each VRP (2 VRPs can be titered per slide). Label slides with a pencil (which is not organic soluble).
Tap the slides to mix, cover, and incubate in a 37°C incubator for 1 hour.
-
Add 300 μl BHK-21 media per well and incubate for 18–24 hours.
Cells need to be counted before too much CPE develops, so starting infections in the afternoon and fixing the next morning works well. Prepare a 50 ml conical tube with ~50 ml 1:1 acetone:methanol mixture for each chamber slide.
Dump off the media and remove the well separators. Fix cells by putting the slides in the conical tubes with the acetone:methanol mixture and incubate at 4°C for at least one hour (and up to several days, if needed).
Prepare a conical tube with ~50 ml PBS for each chamber slide.
After fixing, rehydrate cells by putting them into the PBS conical tubes for 15 minutes at room temperature.
Wash cells with PBS 2 more times by dumping out the PBS, adding another 50 ml PBS to the same tube, and inverting the tube several times. Let it sit at room temperature for 1 minute, then dump out the PBS and refill the same tube with another 50 ml of PBS.
Take the slide out of the tube and add 100 μl 1:200 primary antibody (mouse anti-norovirus serum) to each well.
Cover slides with coverslips and incubate at room temperature for 1 hour.
Wash slides 3 times as described in steps 8–10.
-
Take the slide out of the tube and add 100 μl 1:50 secondary antibody (FITC-labeled goat anti-mouse IgG antibody) per well.
Do not re-freeze FITC-labeled antibodies, discard any remaining from the aliquot used. Put slides in a dark place (under aluminum foil or in a drawer) and allow them to incubate for 30 minutes.
Wash the slides 3 times as described in steps 8–10, except extend the soak times to 2 minutes rather than 1. Let the slides soak while setting up the imaging equipment.
When the microscope and imaging software is ready, stand a slide on end on a Kim wipe to dry it off a bit. Top with a coverslip and view (antifade can be used if desired).
Count the green cells in at least 10 fields and average the number of green cells per field. Do this for two different dilutions for each VRP. Then calculate the VRP/ml for each dilution and average those two numbers for the final value.
-
The VRP titer is calculated from the average number of green cells. The magnification of the microscope lens has to be accounted for in defining the size of the field, which is different depending on the scope.
Ours is 0.27 mm2, which is what we’ll use as an example. The volume of a well for an 8-well chamber slide is 0.79cm2. -
VRP/ml= (ave # green cells per field/area of field in mm2) × (100mm2/1cm2) × (area of well/1 well) × (dilution used/infection volume in ml).
Therefore, for example, VRP/ml= (ave # green cells/0.27mm2) × (100mm2/1cm2) × (0.79cm2/1 well) × (dil/0.8)
BASIC PROTOCOL 6: PRODUCTION OF VIRUS LIKE PARTICLES FROM REPLICONS
VRPs encoding the norovirus capsid undergo one round of replication upon infection of BHK-21 cells and produce ample amounts of VLPs. VRPs will only go through one replication cycle because they are not able to express VEE capsid or VEE E glycoproteins. During this replication cycle, the VEE replicon RNA encodes norovirus capsid mRNA, producing large amounts of norovirus capsid protein. This protein self-assembles into VLPs containing 180 copies of the capsid protein. These VLPs are nearly morphologically identical to native virions.
Materials
Cell culture treated flask 175cm3
BHK-21 cells
BHK-21 media
VRPs (made previously)
Sterile PBS
Pipetman
Pipets (5 ml, 10 ml, 25 ml)
Filter pipet tips
Prepare 1 T-175 flask to 60–80% confluency for each VLP.
Pipet media off of cells and wash cells with sterile PBS.
Add 100 μl of purified VRPs diluted in 4ml of medium or 4 ml of supernatant from an unpurified VRP prep.
Return flasks to 37°C incubator and rock gently every 15 minutes for 1 hour.
Add 20 ml BHK-21 media and return flask to incubator.
Harvest 24–30 hours later following Basic Protocol 7.
BASIC PROTOCOL 7: PURIFICATION OF NOROVIRUS VIRUS LIKE PARTICLES
There are several ways to purify VLPs depending on the expression system used and the make-up of the VLP. Norovirus VLPs made using the VEE expression system are purified using a sucrose cushion. The lysis buffer containing VLPs is layered on top of sucrose and spun at a high speed in an ultracentrifuge. The difference in densities between the lysis buffer layer and the sucrose layer allows for separation of cell components based on size and density. Norovirus VLPs are found in the sucrose layer after high speed centrifugation. These VLPs can be further purified and concentrated using a 100K MWCO centrifugation filter unit after harvesting.
Materials
Ice bucket
large, flat container
Ice
Filter pipet tips
40% sucrose in PBS (4 ml per VLP)
Triton-X 100
Lysis buffer (see recipe)
Complete Mini Protease inhibitor cocktail tablets, available from Roche
10 ml and 50 ml conical tubes
Ultra-Clear Ultracentrifuge tubes, available from Beckman
Kim wipes
Ultra100K MWCO Centrifugation Filter Unit from Amicon
1.5 ml microcentrifuge tubes
SW-55 Ti rotor
Digital Scale
Ultracentrifuge
Rocker
90°C heat block
-
Prepare the 40% sucrose in PBS and the lysis buffer. Lysis buffer is 100 μl Triton-X 100 per 10 ml PBS plus 1 protease inhibitor tablet. 4 ml of lysis buffer is needed per VLP. Put both on rocker to allow the sucrose to dissolve and the Triton-X 100 to thoroughly mix. When fully dissolved/mixed, put on ice in the ice bucket. Turn on the Ultracentrifuge and set to 4°C so it can cool while samples are being prepared.
Add the protease inhibitor tablet to the lysis buffer within an hour of using the buffer. Fill a large container with ice. Remove VLP flasks from the incubator and put them on the ice.
Label 50 ml conical tubes and pipet supernatant from the flasks into the tubes.
Wash each flask thoroughly with 10 ml PBS by pipeting up and down to remove any medium left on the flask. Add this to the appropriate conical tube with the supernatant.
-
Spin supernatant down at 800 × g for 10 minutes at 4°C. Discard supernatant and keep the tube with the cell pellet.
Most of the cells are still attached to the flask, but the small number of cells in the supernatant can be recovered by pelleting via centrifugation. Suspend the cell pellet in 10 ml PBS and spin down again to remove residual medium. Discard supernatant and keep the tube with the cell pellet.
Add 4 ml of lysis buffer to each flask and rock flask back and forth to distribute lysis buffer over the cells. Take care not to make bubbles. Put flasks back on ice and incubate for 5–10 minutes.
While lysis buffer is incubating, label and then fill Ultracentrifuge tubes with 2.5 ml 40% sucrose. Two tubes will be used for each VLP if harvesting from a T-175 flask.
-
Once the lysed cells begin detaching from flask, use a pipet to carefully wash cells off of the flask, while avoiding the production of bubbles. When all cells have been removed, transfer each flask’s lysed cells (cell lysate) to the conical tube containing the appropriate cell pellet. Gently mix to suspend the cell pellet. Reserve 20 μl of the cell lysate for protein analysis.
The pelleted cells will lyse upon addition of the cell lysate to the flask, so VLPs present in these cells can be recovered. Carefully overlay ~2 ml of the cell lysate on top of the sucrose in the Ultracentrifuge tube. Make sure not to mix the sucrose and cell lysate layers. If the tubes are not full after adding all of the cell lysate, add PBS to each tube to bring the level just below the top of the tube.
Carefully transfer Ultracentrifuge tubes to rotor buckets and weigh them. Add lysis buffer as appropriate in order to bring all the tubes to the same weight.
-
Put samples in the Ultracentrifuge and set to 120,000 × g for 75 minutes at 4°C. While VLPs are being centrifuged, label tubes for VLP aliquots.
This speed is specific for SW-55Ti rotor, adjustments may be needed if using a different rotor. -
After centrifugation, remove tubes from rotor buckets, being careful not to mix the layers together. Wipe off the tube with a Kim wipe and identify the interface between the two layers.
There should be two clear layers. The top layer is the cell lysate containing soluble cellular proteins and will be discarded. The bottom layer is the sucrose cushion, which contains the VLPs. The bottom of the tube contains a white pellet consisting primarily of cellular membranes and cytoskeletal elements. Stay away from this when collecting the VLPs. Using a 1 ml pipet, carefully remove the top layer (the cell lysate layer). Reserve 20 μl of this layer for the protein gel. At the interface between the layers, stop and change pipet tips.
-
Insert the pipet tip into the sucrose cushion layer and collect the sucrose cushion in a 1.5 ml microcentrifuge tube. Be careful to avoid the pellet and any leftover lysate.
A little bit of cushion will be wasted to ensure that lysis buffer and cell pellet do not contaminate the VLP sample. Pool the VLPs that are the same and mix VLP sample gently, but thoroughly. Reserve 20 μl of the cushion (VLPs) to run on the protein gel (see Support Protocol 11) and 25 μl to quantify using a BCA protein assay (see Basic Protocol 8).
Store VLPs at 4°C until after validation of VLP production (Support Protocol 11).
Support Protocol 11: VALIDATION OF VLP PRODUCTION
Production of VLPs can be determined by running purified VLPs on an SDS-PAGE protein gel alongside cell lysate and cushion supernatant as controls. Norovirus VP1 VLPs should have a clear band at ~57kDa. Purity of the VLP prep can also be judged based on the number and intensity of background bands. While the cell lysate and cushion supernatant lanes will have many strong bands because of the many cellular proteins present, the VLP lane should contain one main band identifying the VLP and relatively few (if any), light bands that indicate undesired proteins. If the VLP prep has a lot of excess protein, it can be further purified using a 100KDa MWCO centrifugation filter unit. Production and functionality of VLPs should be verified by multiple tests including checking VLPs for protein expression (SDS-PAGE gel, BCA protein assay), purity (SDS-PAGE gel), and structural integrity (ELISA-based assays for carbohydrate and antibody binding, TEM – see Unit 2B.1).
Materials
5X sample buffer (see recipe)
Laemmli running buffer (see recipe)
30 μl 10-well Mini Protean 7.5% polyacrylamide gels, available from BioRad
Protein ladder, available from BioRad
Coomassie stain (see recipe)
10% Acetic Acid
Rocker
90°C heat block
Mix 5 μl 5X sample buffer with each of the reserved 20 μl aliquots of the cell lysate (from the flask), cushion supernatant (the lysis buffer layer), and cushion (the sucrose layer).
Heat these samples for 10 minutes at 90°C.
Load the samples on the protein gel.
Run the gel at 150 V until the bottom dye band reaches the bottom of the gel, usually 45–60 minutes.
Remove gel from plastic casing and put into a container. Cover the gel with coomassie blue stain and microwave for 30 seconds. Remove container from microwave and put it on a rocker for 30 minutes.
-
After 30 minutes drain off coomassie and cover gel with 10% acetic acid. Put back on rocker and allow it to de-stain for at least 30 minutes (until you can clearly see bands).
Put rolled up Kim wipes into the container with the acetic acid and the gel will de-stain faster as the Kim wipes absorb the dye. -
The VLP band should be visible in the cushion sample at 57kDa. Refer to Figure 5 for an example. Purity can be assessed based on the number and intensity of non-VLP protein bands.
If necessary, the VLP prep can be further purified by running the sample through an Amicon Ultra100K MWCO Centrifugation Filter Unit (Millipore). -
If EM facilities are available, norovirus VLP morphology should also be validated by transmission electron microscopy (TEM) imaging of uranyl acetate strained VLP-coated grids.
Uranyl acetate is a “negative stain” as imaging relies on residual stain around the outside of the viral particles to clearly define the structure of particles. Intact norovirus VLPs appear as round 30–40 nm particles with surface depressions (Figure 6).
Figure 5. SDS-Page analysis of norovirus VLP purification.
Norovirus VLPs were harvested from VRP-infected BHK-21 cells by detergent lysis, the lysate components separated by velocity sedimentation through in a 40% sucrose cushion and the protein components of fractions analyzed a on 7.5% acrylamide SDS-PAGE gel visualized by coomassie blue staining. After centrifugation the soluble cellular proteins remain above the sucrose cushion (Lane A) and the ~57 kDa VLP separates into the sucrose cushion (Lane B). Trace amounts of albumin contamination were removed from the VLP preparation by concentration with a 100K MWCO centrifugal filter (Lane C).
Figure 6. Electron microscopy image of norovirus VLPs.
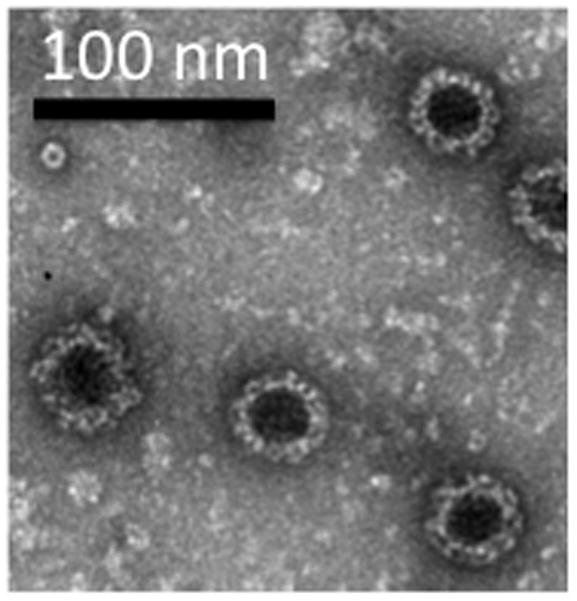
Norovirus VLPs were negative-stained with uranyl acetate and imaged by TEM. Norovirus VLPs appear as 27–40 nm round, highly-structured particles.
BASIC PROTOCOL 8: QUANTIFICATION OF VIRUS LIKE PARTICLES
Quantification of the VLP should take place after all purification steps are done (sucrose cushion and, if desired, a 100K filter). The BCA protein assay compares the protein concentration in a given sample with dilutions of known standard concentrations of BSA protein. Standards and samples are read on a plate reader and given an OD562 nm value. Standards are fit to a standard curve and the concentration of each unknown sample can be calculated using the equation for the standard curve.
Materials
25–50 μl of each VLP to be tested
-
Pierce BCA Protein Assay Kit, available from Pierce
Contains:
2 mg/ml BSA Standard
Reagent A Solution
Reagent B Solution
1.5 ml microcentrifuge tubes
96-well plate
Microplate reader
Follow manufacturer’s directions for the microplate protocol.
-
Make enough working reagent solution to run the standards and unknowns (your samples) in duplicate. A control VLP with a known concentration may be run as well.
You can choose to only run one replicate of your unknowns if quantity of VLPs is an issue. Alternatively, it is possible to reduce the amount of unknown to 10 μl, but the sensitivity of the assay is also reduced (explained in manufacturer’s instructions). Make BSA standards dilutions from 2000 μg/mL to 0 μg/mL (manufacturer’s instructions).
Pipet standards and unknowns onto the plate, add 200 μl working reagent solution to each well, and incubate at 37°C for 2 hours, per the manufacturer’s instructions.
Read plate at 562 nm.
Copy OD values for standards and unknowns into Excel.
Plot the OD values compared to each standard concentration on a XY scatter graph. (OD is x-axis, concentration is y-axis)
Copy the graph into a new sheet.
Double-click the x-axis values and change the scale to logarithmic scale. Do the same for the y-axis.
Click on “add trendline” in the chart wizard and choose “linear” for regression type.
Under options, choose to display the equation on the chart.
Substitute the unknown values for x in the equation and solve for y to get the VLP concentration in μg/ml
BASIC PROTOCOL 9: STORAGE OF VIRUS LIKE PARTICLES
Purified VLPs are suspended in 40% sucrose and are best stored at −80°C. Once a VLP aliquot is thawed, it can be refrozen or kept at 4°C. Excessive freeze/thaws can lead to degradation of the VLPs. VLPs can also be stored at 4°C after purification for a month.
Materials
Purified VLPs (from Basic Protocol 7)
1.5 ml microfuge tubes
−80°C freezer
Label microfuge tubes with appropriate VLP name and date.
-
Divide VLPs into 50 μl or 100 μl aliquots.
VLPs from Basic Protocol 7 are in 40% sucrose solution. Those that have been further concentrated and purified using Ultra100K MWCO Centrifugation Filter Unit are suspended in PBS. Store at −80°C
Once thawed, unused VLPs in an aliquot can be refrozen or stored at 4°C.
REAGENTS AND SOLUTIONS
LB agar plates
Combine the following:
24.5 g LB Media Agar (Lennox)
3.5 g NaCl
700 mL Cell culture grade water
Heat on medium-high heat with stir bar set on low
Autoclave for 1 hr
Cool down to 45°C
Add 1.4 mL of 50 mg/mL ampicillin (Fisher, cat. no. 50841075).
Pour into 100mm-diameter plates
Store plates up to 2 weeks at 4°C
LB broth with ampicillin
Combine the following:
10.5 g LB broth low sodium powder (Lennox)
2.5 g NaCl
500 mL cell culture grade water
Mix until dissolved.
Autoclave for 1 hr
Cool down to 45°C in water bath
Add 1.0 mL of 50 mg/mL ampicillin (Fisher, cat. no. 50841075).
Mix thoroughly.
Store at 4°C
Phosphate buffered saline (PBS) 10 mM pH 7.0–7.4
Combine the following:
8g NaCl
0.2g KH2PO4
0.2g KCl
2.17g Na2HPO4.7H2O
1000 mL cell culture grade water
Filter through 0.22-μm or autoclave
Store up to 6 months at 4°C
LB Media Recipe, 1 liter
Autoclave:
1 liter deionized water
10 grams Bacto tryptone, available from BD
5 grams Bacto yeast extract, available from BD
10 grams NaCl
Store at room temperature for up to 2 months.
BHK-21 Cell Media
(1) 500 mL bottle MEM
50 ml Fetal Clone II (Hyclone)
5 ml antibiotic/antimycotic
5 ml 100X Sodium Pyruvate
5 ml 100X NEAA
Store at 4°C for up to 2 months.
Coomassie Blue Stain, 1 liter
0.25 grams coomassie brilliant blue G-250, (BioRad)
100 ml acetic acid
900 ml deionized H2O
Store at room temperature for up to 1 year.
SDS-PAGE Sample Buffer, 5X
2.5 ml 0.125M Tris base, pH 6.8
2.0 ml 10% sodium dodecyl sulfate
0.2 ml 0.2% Bromphenol Blue
0.2 ml 0.2% Pyrinin Y
4.1 ml 50% Glycerol
1 ml Beta-Mercaptoethanol
Aliquot and freeze at −20°C
Laemmli Running Buffer, 10X
30.18 grams Tris base
144 grams glycine
12.98 grams SDS
Store at room temperature for up to 1 year.
Lysis Buffer
100 μl Triton-X 10 per ml PBS
4 ml lysis buffer needed per VLP
1 Protease inhibitor cocktail tablet
Prepare 4 ml lysis buffer for each VLP. In a conical tube, combine Triton-X 100, PBS, and 1 protease inhibitor cocktail tablet. Put both on rocker to allow it to thoroughly mix. When the protease inhibitor cocktail is completely dissolved, put on ice. Do not add protease inhibitor cocktail more than an hour before using lysis buffer. Do not store lysis buffer; make it fresh each time.
Crystal Violet Solution, 0.5%
2.5 grams crystal violet
5 ml 100% methanol
500 ml 30% methanol
Dissolve 2.5 grams crystal violet in 5 ml 100% methanol. Add the dissolved crystal violet solution to the 500 ml of 30% methanol. Store at room temperature for up to 1 year.
COMMENTARY
Background Information
The first known cases of norovirus occurred in 1968 at an outbreak of acute gastroenteritis in an elementary school in Norwalk, Ohio. In 1972, a virus was visualized by EM in samples collected during this outbreak. The then-unknown virus was subsequently named Norwalk virus (Kapikian et al., 1972). The negative stain image of norovirus particles typically have 27–40 nm sized round particles with rough borders. Virus like particles (VLPs) have indistinguishable morphology when compared with native viral particles. While highly infectious, with an infectious dose of as low as 18 virions, no successful human norovirus cell culture model has been developed. Within the Norovirus genus, only MNV has been cultured successfully in dendritic cells and macrophages (Wobus et al., 2004); however, similar attempts to grow human noroviruses in these and other cell types as well as using a 3D cell culture model, have failed (Duizer et al., 2004; Lay et al., 2010; Melissa M. Herbst-Kralovetz, 2013).
Research on human norovirus has relied heavily on using VLPs as a surrogate in biological assays because the viruses cannot be grown. The first norovirus VLPs were produced in 1992 using a baculovirus expression system in insect cells (Jiang et al., 1992), and this system continues to be widely used today. This effort was followed in 1996 with expression of norovirus VLPs in transgenic tobacco and potato plants (Mason et al., 1996), and since then it has been expressed in numerous other plants. In 2002, the VEE expression system in mammalian cells, which is described in this protocol (Baric et al., 2002) was developed, and more recently, in 2011 a VSV-based expression system was described (Ma and Li, 2011). All of these systems successfully express norovirus VLPs, and researchers typically choose the system that is most conveniently utilized in their own lab with the available expertise and resources. It is currently unknown whether there are biological differences among VLPs created using varied expression systems.
As there are different VLP expression systems, there are multiple purification approaches as well. In this unit, we describe a detergent lysis-sucrose cushion purification protocol, which works well to purify norovirus VLPs. The detergent lysis step is unusual and important in harvesting the VLPs from intracellular membranes. Purification by sucrose or by cesium chloride gradient centrifugation are other options. A study comparing purification of norovirus VLPs by cesium chloride followed by dialysis against a sucrose gradient followed by dialysis demonstrated that while both methods were effective in purifying VLPs, the sucrose gradient yielded more numerous, stable particles (Huhti et al., 2010). To our knowledge neither cesium chloride nor sucrose gradient purification has been experimentally compared to the detergent-lysis followed by sucrose cushion method described in this unit.
Critical Parameters and Troubleshooting
Nucleic acid isolation and molecular detection method
It is important to prevent the introduction of previously amplified products into RT-PCR reactions to avoid false positives. Therefore, it is critical to dedicate at least two separate laboratory rooms for PCR. One for pre-PCR setup and another for post RT-PCR handling. A third separate room for nucleic acid extraction is highly recommended.
Co-extracted PCR inhibitors may sometimes give false-negative results. If inhibition is suspected, the extracted RNA sample should be retested after 1:10 and 1:100 dilutions with nuclease-free water. Negative controls that turn out positive may be caused by contaminated reagents or inappropriate pipetting, or contamination of the area where the master mix was prepared, or the area where the templates were added to the reaction.
cDNA generation
Prior to cDNA synthesis, the oligonucleotide primer-template mix is incubated at 65°C to remove secondary structure of the viral RNA. It is critical to quickly move tubes from the 65°C to an ice bath to allow efficient annealing of the oligonucleotide primer to the RNA template.
ORF-2/ORF-3 PCR amplification
Different DNA polymerases can be used for PCR amplification. However, Phusion® High Fidelity DNA Polymerase is polymerase that exhibits 3′-5′ exonuclease activity for proofreading and this also guarantees that amplicons are blunt-ended. It is critical to add the Phusion® DNA Polymerase directly into the master mix preparation, since the 3′-5′ exonuclease activity can degrade primers in the absence of dNTPs. If using a thymidine overhang cloning kit, adenosines will have to be added to the amplicons using a traditional Taq polymerase. However the Phusion® DNA Polymerase should be removed first, for example by separating the PCR product on an agarose gel, because it will degrade the adenosine overhang.
If no product is obtained: 1) Verify quality and quantity of cDNA generated in previous step; 2) increase extension time or number of cycles; 3) optimize annealing temperature and/or enzyme concentration; 4) optimize denaturation temperature; 5) use freshly made high quality dNTPs. When non-specific products (High molecular weight smears or low molecular weight bands) are visible on the agarose gel: 1) Decrease enzyme, extension time and/or primers concentrations; 2) verify quantity of cDNA generated in previous step; 3) reduce number of cycles; 4) optimize Mg+2 concentration.
Cloning ORF-2/ORF-3 into plasmid vector
During standard PCR the terminal transferase activity of Taq polymerase adds a single 3′-A overhang to each strand of a PCR product. A linearized plasmid with a 5′-T overhang allows ligation and subsequent cloning of the 3′-A-tagged PCR product. The linearized plasmid includes a DNA topoisomerase I covalently bound to each 3′ phosphate to rapidly ligate the PCR product. It is critical to ensure the presence of the adenosine overhangs by removing a proof reading polymerase before adding the overhangs, as described in the protocol. Addition of salt in the TOPO cloning reaction is essential to increase the yield of the number of transformed cells.
Lower cloning efficiencies could be the result of different factors. For large inserts (>1Kb), increase the amount of insert, incubate the cloning reaction longer and gel purify the insert. The lack of enough 3′ adenosine overhangs will also decrease cloning efficiency. Increase the final extension time to ensure all 3′ ends are adenylated.
Transformation of chemically competent cells
Competent cells are sensitive to temperature and mechanical lysis caused by pipetting. Start transformation immediately after thawing the cells on ice. Mix any additions by stirring gently with a pipette tip (avoid any air bubbles). Keep the cells as cold as possible during all steps. Absence of colonies indicates that the bacteria were not competent, an incorrect antibiotic concentration was used or the plates were old. Only few colonies and all of them blue indicate no insert because of lack of 3′ A-overhangs, old PCR product, or incorrect molar ratio of vector: insert used in the ligation reaction. On the contrary, few and all white colonies indicates lack of insert due to degradation of 3′T- overhangs and re-joining of the blunt-ends.
Cloning of norovirus capsid gene into the virus replicon vector
Design of the norovirus capsid construct, primer design for VEE linker cloning, and proper bacterial transformation are the most important parameters for successful cloning. For all norovirus capsids, the sequence must be checked to make sure there are no internal Apa1 or Asc1 restriction sites apart from those used to insert the gene into pVR21. If the capsid sequence contains an internal Apa1 or Asc1 site, before ordering the synthesized DNA, the sequence should be modified in order to knock out the extra site(s) without changing the amino acid sequence. As an alternative, Swa1 can be used as a restriction site instead of Apa1 as long as there are no internal Swa1 sites. In this instance, the digestion protocol will have to be modified accordingly. The large size of pVR21 severely limits the number of restriction endonucleases that can be used for cloning your gene of interest into the vector; careful planning is required for successful cloning. Cloning the VEE linker sequences onto the ends of the capsid gene requires additional PCR steps, and design of effective norovirus capsid forward and reverse primers is critical. The intensity of the DNA bands from insert and overlap PCR reactions should indicate abundant DNA yields. If this is not the case, it may be necessary to run a positive control that has worked efficiently in the past to help narrow down the problem. Check to make sure that the norovirus capsid primers have the correct sequence, there is not potential primer dimer formation, and that the primer annealing temperature is likely to work with the PCR settings in the protocol. Changing the annealing temperatures a few degrees up or down to align with the primer properties may solve this problem. If there are big differences between the annealing temperatures of VEE and norovirus capsid primers, it may be necessary to create two PCR protocols, one for each primer set. Careful handling of competent cells during transformation is also important. Competent cells are very fragile, and can easily die if they are not handled gently. Make sure to always thaw competent cells on ice. When adding the pDNA, do not pipet up and down. Instead, flick the tube gently to mix in the DNA. During the heat shock step, follow the time the cells are at 42°C exactly to avoid killing cells, and return cells to the ice immediately. Another consideration is checking DNA concentrations at each miniprep and gel extraction step. This ensures that subsequent steps in the protocol are being performed with enough DNA to work correctly. Sometimes culture yields or gel extraction yields are uncharacteristically low and new cultures need to be grown up in order to recover enough DNA to successfully move on in the protocol.
Production of stock norovirus recombinant virus replicon particles
The most important parameters for production of VRPs are RNA production, timing of harvesting the VRPs, and the health of the BHK-21 cells used. The most common failure of VRP formation is RNA degradation. Running an RNA gel can ensure that RNA was produced during the transcription reaction, but it is still possible for RNA to degrade during storage or when added to the cells during the electroporation step before pulsing the cells. The best way to prevent RNA degradation is to spray everything used for the transcription reaction with RNAse zap, use filter tips for anything involving the RNA, and to pulse the cell cuvettes immediately after adding the RNA to them. Only nuclease free water should be used in the production of RNA, and it may be necessary to replace the water if degradation becomes a recurring issue. If RNA is not being produced during the transcription reaction, it may be the result of bad enzyme in the transcription kit. It is also possible that the transcription kit buffer was precipitated which could cause the reaction chemistry to be off. To mitigate the risk of buffer precipitation, flick the buffer tube until precipitation is no longer visible. Health of the BHK-21s and timing of the VRP harvest can affect VRP yield. BHK-21 cells that are over-confluent when transfected with RNA typically give a lower VRP yield than those that are used at 60–80% confluency.
Production of virus like particles from replicons
The critical parameters for VLP production are mainly dependent on earlier protocol steps including the cloning process and formation of VRPs. However, VLP yield can be affected by the health of the BHK-21 cells used for culture. VLP yields are typically lower in cells that are over-confluent or are harvested too early (before 24 hours) or too late (after 30 hours). If the titer has indicated that there are ample VRPs, but they do not produce VLPs, it may be helpful to check the cloning steps to make sure that the entire VEE linker sequence and norovirus capsid sequence are correct and in frame.
Purification of norovirus virus like particles
Major parameters to monitor during VLP purification are making sure cells are completely washed with PBS, making sure cells are thoroughly lysed and removed from the flask, and ensuring that the cushion and cushion supernatant layers are not mixed during VLP harvesting. When removing the media from the flasks, it is very important to wash the cells thoroughly with PBS. This removes residual media that contains albumin. If not washed properly, this albumin will be found as a contaminant band in the VLP prep and can throw off the protein quantification in later steps. It is also important to completely remove all cells and cell lysate from the flask by repeatedly washing the flask surface with the lysis buffer. This needs to be done gently since the lysis buffer contains detergent and will form bubbles easily. It is important to reduce the bubbles as much as possible to maximize the VLPs transferred to the Ultracentrifuge tube. Cells or clumps of membranes left on the flask may contain VLPs that would have been recovered during centrifugation. Finally, during the VLP harvesting step, it is sometimes hard to judge the division between the layers. It is important to be very careful to discard as much cell lysate from the top of the cushion interface and leave the sucrose portion for harvest. There is usually a small amount of cell lysate left on top of the sucrose cushion, so it is imperative to avoid taking any part of the leftover lysate as part of the VLP harvest. Doing so will contaminate the VLP prep with unwanted proteins, which may interfere with calculating the protein concentration of the VLPs accurately. To avoid contamination with the cell lysate, it is necessary to leave behind a small portion of the sucrose cushion. It is also important not to take any of the pellet that accumulates at the bottom of the ultracentrifuge tube.
Quantification of virus like particles
The most important parameters in quantifying the protein concentration are ensuring that there is minimal protein contamination from undesired cellular proteins in the VLP prep and validation of the BSA standard curve. The SDS-PAGE protein gel gives an indication of VLP prep purity. In order to quantify the VLP concentration accurately, the gel should contain few background bands in the VLP lane. If there is excessive contamination, the prep should be run through a 100KDa MWCO centrifugal filter then the prep should be re-run on a protein gel to validate purity. Once protein contamination is removed, the BCA assay can be done. It is also important that the standard curve is valid. Make sure the r2 value for the curve is higher than .95; otherwise, it should be repeated with a more reliable curve. A new standard curve must be run with each sample set.
Storage of virus like particles
Important considerations for storage of VLPs are how quickly they will be used, the concentration of the prep, and how they are typically handled. VLPs are very stable at −80°C but can also be stored at 4°C. At 4°C, VLPs are sensitive to fungal and bacterial contamination given that they are suspended in sucrose solution. Some measures to prevent contamination include the use of filter tips, opening the VLP tubes only in a biological hood, and to aliquot VLP samples to minimize potential reagent loss. Another problem that sometimes occurs at 4°C is precipitation of the VLPs, rendering them unusable. This typically occurs with VLP preps that have been concentrated to >1 mg/mL. Further, VLPs are less stable at 4°C when their protein concentration is low (less than 100 mg/mL). We usually aliquot VLPs into 50–100 μl portions and freeze them at −80°C to minimize the number of potential freeze/thaws. We also sometimes leave the unused, thawed VLPs at 4°C when we know they will be used within 1 month.
Anticipated Results
Molecular detection method
The results should be carefully interpreted in the context of the outcome of the positive and negative controls. Negative controls should not exhibit fluorescent curves that cross the threshold line (Ct) for the GI and GII detector. If a negative control crosses the threshold, the entire experiment is invalid and should be repeated. Positive control for GI and GII norovirus should exhibit fluorescent curves that cross the threshold line for the GI and GII detector, respectively. A test must be rejected if any of the positive controls are negative.
Cloning ORF2/ORF3 into plasmid vector and transformation of chemically competent cells
For all cloning experiments, each vector should yield greater than 95% cloning efficiency. For an insert size of 400–700 bp, you should obtain 50–200 colonies per plate depending on the volume plated. Of these, approximately 80% should be white on X-Gal plates. Note that ligation efficiency depends on insert size. As insert size increases, the efficiency will decrease.
Cloning of norovirus capsid gene into the virus replicon vector
Sizes associated with PCR products and digestions can be found in Table 3. After digestion with Apa1 and Asc1, the capsid gene with VEE linkers is ligated into the pVR21 plasmid behind the 26S promoter and transformed into competent E. coli. The resulting colonies are composed of a DNA copy of the VEE genome with the norovirus capsid protein gene in place of the VEE structural genes. Norovirus capsid sequence is generally stable and sequencing 1 or 2 colonies typically identifies a colony with the desired sequence.
Production of stock norovirus recombinant virus replicon particles
After the sequence verified pVR21-norovirus capsid construct pDNA is linearized, a transcription reaction will produce RNA composed of the nonstructural VEE genes with the norovirus major capsid protein gene. When this construct RNA is transfected into cells with VEE capsid helper RNA and VEE E glycoprotein helper RNA, it produces norovirus capsid-expressing VEE replicons that can be used to generate VLPs or to immunize animals. CPE (cytopathic effect) is typically evident in cells with efficient production of VRPs. Normal yields for norovirus-capsid expressing VRPs in BHK-21 cells with the 3526 VEE construct is 5 x 106 to 5 x 107 IU/mL.
Production of virus like particles from replicons
VRPs are added to BHK-21 cells, which subsequently produce norovirus VLPs. CPE is sometimes evident in the flasks and usually indicates successful VLP production. However, lack of CPE does not necessarily mean that VLPs were not produced. Purification and validation are the only sure ways to know that VLPs are produced.
Purification of norovirus virus like particles
VLPs are purified by detergent lysis with subsequent purification through a sucrose cushion. After high-speed centrifugation, VLPs are found in the sucrose layer and are harvested by pipeting the sucrose from beneath the cushion cell lysate layer. The VLP harvesting portion of this protocol usually takes around 3 hours including prep, centrifugation, and sucrose cushion harvest times. When run on an SDS-PAGE protein gel, the sucrose cushion portion containing the VLPs will show a distinct band at ~57 kDa and relatively few background bands. The protein gel can be run on the same day as the VLP harvest or the reserved samples can be frozen to run another day. Gel analysis typically takes 2 hours including heating samples, running the gel, and staining/de-staining the gel. It takes about 90 minutes of lab time if the gel is left to de-stain overnight.
Quantification of virus like particles
VLPs are quantified by BCA protein assay, which takes about 60–90 minutes to set up, run, and analyze. It will take slightly longer if the extra purification step is done. Typical VLP yields without concentration are usually between 200–400 μg/ml at 4 ml or 800–1200 μg yield from one T-175 flask. If a 100KDa filter is used for further purification and concentration, VLP yields can be as high as 1–2 mg/ml.
Storage of virus like particles
VLPs are stable for years when stored at −80°C and can be freeze/thawed a few times without detectable degradation. VLPs can be stored at 4°C but they are more prone to contamination, degradation, and precipitation if left for a >1 month at this temperature. It takes 10–15 minutes to aliquot and freeze VLPs.
Acknowledgments
The authors would like to thanks Dr. Everardo Vega (CDC) for his work on the development of BASIC PROTOCOL 2 (AMPLIFICATION OF NOROVIRUS ORF-2 AND ORF-3 BY LONG RT-PCR). We also thank the UNC-CH Genome Analysis Facility.
This work was supported by grant AI056351 from the National Institutes of Health, Allergy and Infectious Diseases, and by grant U54-AI057157 from SERCEB (South Eastern Regional Center for Excellence and Biodefense).
The findings and conclusions in this report are those of the author(s) and do not necessarily represent the official position of the Centers for Disease Control and Prevention. Mention of company names or products does not constitute endorsement by the Centers for Disease Control and Prevention. This article did receive clearance through the appropriate channels at the CDC prior to submission.
SUPPLIERS APPENDIX
-
BioLabs, Inc.
240 County Road
Ipswich, MA 01938-2723, USA.
Phone: 978-927-5054
FAX: 978-921-1350
Website: www.neb.com
-
Clare Chemical research, Inc.
995 Railroad Ave, Unit A
Dolores, CO 81323, USA
Phone: 888-292-0356
970-882-7499
Fax: 970-882-7068
Website: www.clarechemical.com
-
Life Technologies
3175 Staley Road
Grand Island, NY 14072, USA.
Phone: 800-955-6288
Fax: 800-331-2286
Web site: www.lifetech.com
-
Promega
2800 Woods Hollow Road
Madison, WI 53711, USA.
Phone: 608-274-4330
800-356-9526
Fax: 608-277-2516
Website: www.promega.com
-
Qiagen
27220 Turnberry Lane
Suite 200
Valencia, CA 91355, USA
Phone: 800-426-8157
Fax: 800-718-2056
Website: www.qiagen.com
-
Roche
9115 Hague Road
Indianapolis, Indiana 46250, USA.
Phone: 317-521-2000
Website: www.roche.com
-
Teknova
2290 Bert Dr.
Hollister, CA 95023, USA
Phone: 831-637-1100
Fax: 831-637-2355
Website: www.teknova.com
-
Thermo scientific
Building 2B,
28 Schenck Parkway, Suite 400
Asheville, NC 28803, USA.
Phone: 866 984 3766
Website: www.thermoscientific.com
-
VWR
1050 Satellite Blvd.
Suwanee, GA 30024
Phone: 800-932-5000
Fax: (770) 232-9881
Website: http://www.vwr.com/
-
Zymo Research Corporation
17062 Murphy Ave.
Irvine, CA 92614, USA
Phone: 949-679-1190
888-882-9682
Fax: 949-266-9452
Website: www.zymoresearch.com
Contributor Information
Kari Debbink, Email: debbink@email.unc.edu, 3109 Michael Hooker Research Center, 135 Dauer Drive, Campus Box 7435, Chapel Hill, NC 27599-7435, Phone: 919-843-8558, Fax: 919-966-0584.
Veronica Costantini, Email: vcostantini@cdc.gov, CDC/NCIRD/DVD, 1600 Clifton Rd NE, MS G04, Atlanta, GA 30333, Phone: 404-639-4040, Fax: 404-639-3645.
Jesica Swanstrom, Email: jesica_swanstrom@med.unc.edu, 3109 Michael Hooker Research Center, 135 Dauer Drive, Campus Box 7435, Chapel Hill, NC 27599-7435, Phone: 919-843-8558, Fax: 919-966-0584.
Sudhakar Agnihothram, Email: agniss@email.unc.edu, 3109 Michael Hooker Research Center, 135 Dauer Drive, Campus Box 7435, Chapel Hill, NC 27599-7435, Phone: 919-966-3897, Fax: 919-966-0584.
Jan Vinjé, Email: jvinje@cdc.gov, CDC/NCIRD/DVD, 1600 Clifton Rd NE, MS G04., Atlanta, GA 30333, Phone: 404-639-3721, Fax: 404-639-3645.
Ralph Baric, Email: rbaric@email.unc.edu, 3104 Michael Hooker Research Center, 135 Dauer Drive, Campus Box 7435, Chapel Hill, NC 27599-7435, Phone: 919-966-3895, Fax: 919-966-0584.
Lisa Lindesmith, Email: lisal@unc.edu, 3109 Michael Hooker Research Center, 135 Dauer Drive, Campus Box 7435, Chapel Hill, NC 27599-7435, Phone: 919-966-4689, Fax: 919-966-0584.
LITERATURE CITED
- Anonymous. Updated norovirus outbreak management and disease prevention guidelines. MMWR Recomm Rep. 2011;60:1–18. [PubMed] [Google Scholar]
- Baric RS, Yount B, Lindesmith L, Harrington PR, Greene SR, Tseng FC, Davis N, Johnston RE, Klapper DG, Moe CL. Expression and self-assembly of norwalk virus capsid protein from venezuelan equine encephalitis virus replicons. J Virol. 2002;76:3023–3030. doi: 10.1128/JVI.76.6.3023-3030.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bok K, Parra GI, Mitra T, Abente E, Shaver CK, Boon D, Engle R, Yu C, Kapikian AZ, Sosnovtsev SV, Purcell RH, Green KY. Chimpanzees as an animal model for human norovirus infection and vaccine development. Proc Natl Acad Sci U S A. 2011;108:325–330. doi: 10.1073/pnas.1014577107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheetham S, Souza M, Meulia T, Grimes S, Han MG, Saif LJ. Pathogenesis of a genogroup II human norovirus in gnotobiotic pigs. J Virol. 2006;80:10372–10381. doi: 10.1128/JVI.00809-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson EF, Lindesmith LC, Lobue AD, Baric RS. Viral shape-shifting: norovirus evasion of the human immune system. Nat Rev Microbiol. 2010;8:231–241. doi: 10.1038/nrmicro2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duizer E, Schwab KJ, Neill FH, Atmar RL, Koopmans MP, Estes MK. Laboratory efforts to cultivate noroviruses. J Gen Virol. 2004;85:79–87. doi: 10.1099/vir.0.19478-0. [DOI] [PubMed] [Google Scholar]
- Harris JP, Edmunds WJ, Pebody R, Brown DW, Lopman BA. Deaths from norovirus among the elderly, England and Wales. Emerg Infect Dis. 2008;14:1546–1552. doi: 10.3201/eid1410.080188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart MK, Caswell-Stephan K, Bakken R, Tammariello R, Pratt W, Davis N, Johnston RE, Smith J, Steele K. Improved mucosal protection against Venezuelan equine encephalitis virus is induced by the molecularly defined, live-attenuated V3526 vaccine candidate. Vaccine. 2000;18:3067–3075. doi: 10.1016/s0264-410x(00)00042-6. [DOI] [PubMed] [Google Scholar]
- Huhti L, Blazevic V, Nurminen K, Koho T, Hytonen VP, Vesikari T. A comparison of methods for purification and concentration of norovirus GII-4 capsid virus-like particles. Arch Virol. 2010;155:1855–1858. doi: 10.1007/s00705-010-0768-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Matson DO, Cubitt WD, Estes MK. Genetic and antigenic diversity of human caliciviruses (HuCVs) using RT-PCR and new EIAs. Arch Virol Suppl. 1996;12:251–262. doi: 10.1007/978-3-7091-6553-9_27. [DOI] [PubMed] [Google Scholar]
- Jiang X, Wang M, Graham DY, Estes MK. Expression, self-assembly, and antigenicity of the Norwalk virus capsid protein. J Virol. 1992;66:6527–6532. doi: 10.1128/jvi.66.11.6527-6532.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung K, Wang Q, Kim Y, Scheuer K, Zhang Z, Shen Q, Chang KO, Saif LJ. The effects of simvastatin or interferon-alpha on infectivity of human norovirus using a gnotobiotic pig model for the study of antivirals. PLoS One. 2012;7:e41619. doi: 10.1371/journal.pone.0041619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapikian AZ, Wyatt RG, Dolin R, Thornhill TS, Kalica AR, Chanock RM. Visualization by immune electron microscopy of a 27-nm particle associated with acute infectious nonbacterial gastroenteritis. J Virol. 1972;10:1075–1081. doi: 10.1128/jvi.10.5.1075-1081.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopmans M, Vinje J, de Wit M, Leenen I, van der Poel W, van Duynhoven Y. Molecular epidemiology of human enteric caliciviruses in The Netherlands. J Infect Dis. 2000;181(Suppl 2):S262–269. doi: 10.1086/315573. [DOI] [PubMed] [Google Scholar]
- Lay MK, Atmar RL, Guix S, Bharadwaj U, He H, Neill FH, Sastry KJ, Yao Q, Estes MK. Norwalk virus does not replicate in human macrophages or dendritic cells derived from the peripheral blood of susceptible humans. Virology. 2010;406:1–11. doi: 10.1016/j.virol.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindesmith L, Moe C, Marionneau S, Ruvoen N, Jiang X, Lindblad L, Stewart P, LePendu J, Baric R. Human susceptibility and resistance to Norwalk virus infection. Nat Med. 2003;9:548–553. doi: 10.1038/nm860. [DOI] [PubMed] [Google Scholar]
- Lindesmith LC, Donaldson EF, Baric RS. Norovirus GII.4 strain antigenic variation. J Virol. 2011;85:231–242. doi: 10.1128/JVI.01364-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Li J. Vesicular stomatitis virus as a vector to deliver virus-like particles of human norovirus: a new vaccine candidate against an important noncultivable virus. J Virol. 2011;85:2942–2952. doi: 10.1128/JVI.02332-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason HS, Ball JM, Shi JJ, Jiang X, Estes MK, Arntzen CJ. Expression of Norwalk virus capsid protein in transgenic tobacco and potato and its oral immunogenicity in mice. Proc Natl Acad Sci U S A. 1996;93:5335–5340. doi: 10.1073/pnas.93.11.5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst-Kralovetz Melissa M, ALR, Lay Margarita K, Hjelm Brooke E, Bolick Alice N, Sarker Shameema S, Atmar Robert L, Kingsley David H, Arntzen Charles J, Estes Mary K, Nickerson Cheryl A. Lack of norovirus replication and histo-blood group antigen expression in 3-dimensional intestinal epithelial cells. Emerg Infect Dis. 2013;19 doi: 10.3201/eid1903.121029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad BV, Hardy ME, Dokland T, Bella J, Rossmann MG, Estes MK. X-ray crystallographic structure of the Norwalk virus capsid. Science. 1999;286:287–290. doi: 10.1126/science.286.5438.287. [DOI] [PubMed] [Google Scholar]
- Pratt WD, Davis NL, Johnston RE, Smith JF. Genetically engineered, live attenuated vaccines for Venezuelan equine encephalitis: testing in animal models. Vaccine. 2003;21:3854–3862. doi: 10.1016/s0264-410x(03)00328-1. [DOI] [PubMed] [Google Scholar]
- Pushko P, Parker M, Ludwig GV, Davis NL, Johnston RE, Smith JF. Replicon-helper systems from attenuated Venezuelan equine encephalitis virus: expression of heterologous genes in vitro and immunization against heterologous pathogens in vivo. Virology. 1997;239:389–401. doi: 10.1006/viro.1997.8878. [DOI] [PubMed] [Google Scholar]
- Reed DS, Lind CM, Lackemeyer MG, Sullivan LJ, Pratt WD, Parker MD. Genetically engineered, live, attenuated vaccines protect nonhuman primates against aerosol challenge with a virulent IE strain of Venezuelan equine encephalitis virus. Vaccine. 2005;23:3139–3147. doi: 10.1016/j.vaccine.2004.12.023. [DOI] [PubMed] [Google Scholar]
- Schorn R, Hohne M, Meerbach A, Bossart W, Wuthrich RP, Schreier E, Muller NJ, Fehr T. Chronic norovirus infection after kidney transplantation: molecular evidence for immune-driven viral evolution. Clin Infect Dis. 2010;51:307–314. doi: 10.1086/653939. [DOI] [PubMed] [Google Scholar]
- Schultz AC, Vega E, Dalsgaard A, Christensen LS, Norrung B, Hoorfar J, Vinje J. Development and evaluation of novel one-step TaqMan realtime RT-PCR assays for the detection and direct genotyping of genogroup I and II noroviruses. J Clin Virol. 2011;50:230–234. doi: 10.1016/j.jcv.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebenga JJ, Vennema H, Zheng DP, Vinje J, Lee BE, Pang XL, Ho EC, Lim W, Choudekar A, Broor S, Halperin T, Rasool NB, Hewitt J, Greening GE, Jin M, Duan ZJ, Lucero Y, O’Ryan M, Hoehne M, Schreier E, Ratcliff RM, White PA, Iritani N, Reuter G, Koopmans M. Norovirus illness is a global problem: emergence and spread of norovirus GII.4 variants, 2001–2007. J Infect Dis. 2009;200:802–812. doi: 10.1086/605127. [DOI] [PubMed] [Google Scholar]
- Souza M, Cheetham SM, Azevedo MS, Costantini V, Saif LJ. Cytokine and antibody responses in gnotobiotic pigs after infection with human norovirus genogroup II.4 (HS66 strain) J Virol. 2007;81:9183–9192. doi: 10.1128/JVI.00558-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Zhong W, Song D, Thornton S, Jiang X. E. coli-expressed recombinant norovirus capsid proteins maintain authentic antigenicity and receptor binding capability. J Med Virol. 2004;74:641–649. doi: 10.1002/jmv.20228. [DOI] [PubMed] [Google Scholar]
- Vega E, Barclay L, Gregoricus N, Williams K, Lee D, Vinje J. Novel surveillance network for norovirus gastroenteritis outbreaks, United States. Emerg Infect Dis. 2011;17:1389–1395. doi: 10.3201/eid1708.101837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinje J, Green J, Lewis DC, Gallimore CI, Brown DW, Koopmans MP. Genetic polymorphism across regions of the three open reading frames of “Norwalk-like viruses”. Arch Virol. 2000;145:223–241. doi: 10.1007/s007050050020. [DOI] [PubMed] [Google Scholar]
- Wobus CE, Karst SM, Thackray LB, Chang KO, Sosnovtsev SV, Belliot G, Krug A, Mackenzie JM, Green KY, Virgin HW. Replication of Norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol. 2004;2:e432. doi: 10.1371/journal.pbio.0020432. [DOI] [PMC free article] [PubMed] [Google Scholar]