

Although it has long been assumed that insulin resistance is the leading factor in the pathogenesis of type 2 diabetes (1), evidence for the importance of the pancreatic β-cells has accumulated over the past decades. In fact, the vast majority of genes associated with type 2 diabetes have been linked to the β-cell, and impairments in β-cell mass and in insulin secretion have been reported in numerous studies in patients with type 2 diabetes. One misconception that has prevented the appreciation of the β-cell defects for a long time is the idea of a “hyperinsulinemia” in patients with type 2 diabetes. This concept has arisen from the observation that patients with type 2 diabetes often present with higher fasting insulin concentrations than nondiabetic individuals. However, if insulin concentrations are interpreted in the context of the concurrently elevated glucose levels in patients with type 2 diabetes, a relative insulin deficit rather than hyperinsulinemia becomes apparent. Furthermore, when insulin secretion is evaluated under stimulated conditions (e.g., after intravenous glucose administration), the typical defects, especially in early-phase insulin release, can be unmasked (2,3).
It has also been suggested that obesity causes type 2 diabetes through impaired insulin action. Undoubtedly, the risk of developing type 2 diabetes increases markedly with BMI. However, if obesity were really the cause of type 2 diabetes, one would expect the vast majority of obese individuals to develop hyperglycemia, whereas in reality ∼80% of obese individuals remain free of diabetes (4). These findings suggest that obesity and insulin resistance are indeed important cofactors that increase the individual risk of diabetes but that the actual cause of the disease seems to be clearly linked to the β-cells.
If one accepts this notion, the next question is whether β-cell defects are primarily functional in nature or whether a reduction in the number of insulin-secreting cells (i.e., β-cell mass) is the leading problem in type 2 diabetes. This article will summarize the arguments in favor of both sides, aiming to reach a consensus as to the importance of reduced β-cell mass and impaired β-cell function in the pathogenesis of type 2 diabetes.
Is type 2 diabetes primarily caused by a deficit in β-cell mass?
That type 2 diabetes develops largely because of a deficit in β-cell mass is supported by several lines of evidence. Autopsy studies in various populations (European, Asian, and North American) have reported significant reductions in the amount of pancreatic β-cells in patients with type 2 diabetes compared with nondiabetic individuals (5–7). The extent of this deficit ranges from ∼20% in some studies to ∼65% in others (5–7). There is also evidence for a β-cell deficit in prediabetic individuals with impaired fasting glucose (6). The reasons underlying the heterogeneous results from different studies are probably multifactorial in nature. Presumably, the individual contribution of the β-cell deficit versus that of β-cell dysfunction and insulin resistance to the overall pathogenesis of type 2 diabetes varies between different populations. While based on these studies there is no doubt that β-cell mass is reduced to a variable extent in patients with type 2 diabetes, the reasons underlying this β-cell deficit are less well established. A common view is that increased β-cell apoptosis leads to the continuous loss of β-cells (8). In support of this theory, apoptosis was found to be increased in islets from patients with type 2 diabetes compared with nondiabetic subjects based on two different studies using either immunohistochemistry or Western blot analysis (6,9). Controversy exists regarding the presumed causes of β-cell apoptosis in type 2 diabetes. Under in vitro conditions, β-cell death has been induced by various factors linked to the type 2 diabetes phenotype, such as high concentrations of glucose, free fatty acids, or human islet amyloid polypeptide (10). Also commonly assumed is that a high secretory demand in overtly hyperglycemic or obese individuals causes generation of reactive oxygen species (oxidative stress) as well as protein misfolding in the endoplasmatic reticulum (ER stress), both of which can result in the induction of apoptosis (11). Finally, inflammatory signals, such as local production of interleukin-1β within islet β-cells, have been linked to β-cell death in type 2 diabetes (12). Estimating which of these mechanisms is most important for induction of β-cell death in patients with type 2 diabetes seems difficult.
Although accelerated β-cell death would reasonably explain the overt β-cell deficit in type 2 diabetes and would also be consistent with the clinical observation of a progressive deterioration of insulin secretion in patients with type 2 diabetes over time (13), an alternative hypothesis would be insufficient islet development during the pre- and postnatal growth period (14). In support of such reasoning, we have previously noted a remarkable variation in fractional β-cell area (>30-fold) in individuals of similar age-groups throughout the pre- and postnatal growth period (15). It has also been suggested that intrauterine malnutrition as well as certain polymorphisms may predispose children to an insufficient formation of islets, which might lead to an increased risk of diabetes later in life (16).
What are the consequences of a β-cell deficit for the maintenance of glucose homoeostasis? Not surprisingly, postchallenge insulin levels are reduced after a β-cell loss (17,18). There is also evidence that hyperglycemia causes additional functional impairments in insulin release that go beyond the actual β-cell deficit (19). This is most likely the result of β-cell exhaustion (i.e., depletion of insulin granules) and subsequent loss of early-phase insulin release (20). In fact, if β-cell mass is reduced by 50%, the secretory burden for the remaining β-cells increases by 100%, thereby leading to chronic β-cell stress. This is probably the reason why the functional impairment of insulin secretion (especially glucose-stimulated first-phase insulin release) in patients with type 2 diabetes often markedly exceeds the estimated deficit in β-cell mass (2,3). In turn, induction of β-cell rest by means of insulin therapy or even an overnight infusion of somatostatin has been found to largely restore the functional defect in glucose-induced insulin secretion in hyperglycemic patients with type 2 diabetes (21,22). That glucose-induced insulin secretion can be almost fully normalized even within <1 day sheds doubts on the idea of a primary functional β-cell abnormality in type 2 diabetes (23,24). Along the same line, progressive deterioration of glycemic control over time occurred despite significant improvements in β-cell function in a large randomized prospective trial (A Diabetes Outcome Progression Trial [ADOPT]) (13).
One way to address the impact of a β-cell loss is to study individuals with a β-cell deficit due to causes other than type 2 diabetes, such as chronic pancreatitis. When we examined a large group of patients who underwent partial pancreatectomy for various pancreatic diseases, we found that on average diabetes occurred when β-cell area (as quantified in the resected pancreatic tissue) was reduced by ∼65% (25). This number is consistent with the mean reduction in β-cell area reported in a recent autopsy study in patients with type 2 diabetes (6). The impact of an acute 50% reduction in β-cell mass has also been examined prospectively in individuals who donated 50% of their pancreas for transplantation (17). In this study, hemipancreatectomy led to abnormal glucose tolerance in 7 of 28 donors after 1 year, along with a significant impairment in insulin secretion (17). Four of eight patients who had been followed up for 9–18 years after the hemipancreatectomy had developed overt diabetes in the meantime (26). Notably, the risk of diabetes was greatest in obese patients (26), probably owing to the higher insulin demand in such patients. Also, disproportionate hyperproinsulinemia, which was initially believed to be a primary functional abnormality in type 2 diabetes (27), was found after hemipancreatectomy, suggesting that exaggerated secretion of proinsulin results from an increased insulin demand subsequent to the β-cell loss (28). These data from organ donors are in good agreement with studies in patients undergoing partial pancreatectomy for chronic pancreatitis or tumors showing significant impairments in insulin secretion as well as a high risk of diabetes after surgery (18).
The impact of an ∼50% reduction of β-cell mass has also been examined in various large animal models. Indeed, most of the characteristic features of type 2 diabetes, such as reduced maximum insulin secretion, reduced amplitude of pulsatile insulin secretion, reduced insulin clearance, impaired postprandial glucagon suppression, and insulin resistance, have been found after an experimental β-cell loss resembling the β-cell deficit in patients with type 2 diabetes (29,30). Studies in mice or rats suggesting preserved glucose homoeostasis after 60–90% partial pancreatectomy are difficult to interpret because of the unusually high capacity for β-cell regeneration in rodents of young age (31). Notably, studies in older animals or in adult humans have not confirmed such high potential for β-cell regeneration after partial pancreatectomy (32,33).
An important functional parameter that has been tightly linked to β-cell mass in various studies is the amplitude of pulsatile insulin secretion (34). A recent series of studies examining the interaction between pulsatile insulin secretion and hepatic insulin signaling has convincingly demonstrated that reduced pulsatile insulin secretion (which typically results from a β-cell deficit) causes impaired activation of the hepatic insulin receptor substrate (IRS)-1 and IRS-2, as well as downstream insulin-signaling molecules (35). Also, a failure to suppress glucagon levels in response to glucose administration as well as peripheral insulin resistance has been linked to abnormalities in pulsatile insulin secretion (29,36,37). Collectively, these studies lend strong support to the hypothesis that reductions in β-cell mass secondarily cause various abnormalities in β-cell function (especially pulsatile insulin secretion), α-cell function, and insulin action in patients with type 2 diabetes (38,39). The importance of β-cell mass for the maintenance of glucose homoeostasis is further emphasized by studies showing restoration of glucose control after pancreas transplantation even in insulin-resistant patients and in spite of steroid-based immunosuppressive treatment regimens (40). A working hypothesis on the consequences of reduced β-cell mass on the pathogenesis of type 2 diabetes is presented in Fig. 1.
Figure 1.
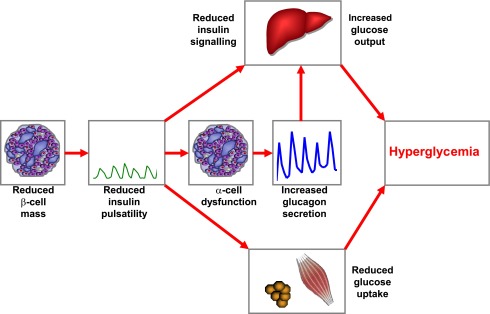
Working model for the impact of reduced β-cell mass on the pathogenesis of type 2 diabetes. In patients with type 2 diabetes, β-cell mass is reduced by ∼20–65%, leading to impaired and delayed insulin secretion and a specific reduction in the amplitude of pulsatile insulin secretion. The reduction of insulin secretion and insulin pulsatility leads to disruption of the intraislet insulin-glucagon cross-talk, causing insufficient suppression of glucagon release. Reduced pulsatile insulin secretion impairs hepatic insulin signaling and perturbs peripheral insulin action. Increased hepatic glucose release is further augmented by the exaggerated glucagon concentrations. Together, these defects cause hyperglycemia in patients with type 2 diabetes.
Is β-cell loss of function the main determinant of β-cell defects in type 2 diabetes?
The case for a prevalent role of β-cell loss of function versus β-cell loss of mass in the etiology and pathogenesis of human type 2 diabetes is a thorny issue, essentially because we have an incomplete knowledge of the exact role played by the β-cell in the natural history of this disease (41,42). In humans, only in the last decade has a reasonable consensus been reached regarding how one should measure β-cell functional mass in vivo (43). β-Cell functional mass can hardly be summarized in one single number for the simple reason that the β-cell copes with awfully complex and diverse tasks. The minimum level of description of β-cell functional mass should include measurement of both derivative, or dynamic, control (i.e., the β-cell response to the rate of glucose increase) and proportional, or static, control (i.e., the stimulus response curve relating insulin secretion rate to glucose concentration) of β-cell functional mass during both intravenous and oral glucose challenges (43) so as to also be able to quantify the incretin effect on insulin secretion (44,45).
During appropriate intravenous glucose challenges, the derivative (dynamic) control is the time-honored first-phase insulin release, whereas the stimulus response curve of the proportional (static) control embodies the traditional basal insulin secretion rate plus the second-phase insulin response (46) (Fig. 2). The incretin effect can be quantified as the amplification of insulin secretion rate (or either control of β-cell functional mass) induced by the oral versus the venous route of glucose administration (44,45). Extensive evidence supports the notion that different insulin granule pools (47) and distinct voltage-gated calcium channels (48) sustain the derivative and the proportional control of insulin secretion, whereas it is obvious that the incretin effect is served by specific β-cell receptors and signaling molecules (49). Attempts to build more sophisticated modeling of in vivo β-cell function that embodies these additional features of the insulin secretory machinery are under way (50,51).
Figure 2.

Stimulus response curve for first-phase (derivative control of β-cell function) (continuous lines) and second-phase (proportional control of β-cell function) (dotted lines) insulin release in control subjects (C) and in patients with type 2 diabetes (T2DM). All subjects underwent a number of hyperglycemic clamps at graded glucose levels to construct a stimulus response curve in each. Although both first- and second-phase insulin releases are severely impaired in the patients (P < 0.01 for both, type 2 diabetic vs. control), second phase shows a graded response to the glucose challenge, whereas first phase is virtually absent in the patients, thereby showing asymmetric functional defects. Data are redrawn from ref. 52.
Patients with type 2 diabetes display reductions in the derivative (dynamic) and proportional (static) controls of β-cell functional mass (52,53) and in the incretin effect (44). All of these impairments concur to cause β-cell failure in these patients. At this qualitative level of description, these findings may be equally compatible with a prevalent role of either a β-cell loss of function or a β-cell loss of mass in β-cell failure (41). If the latter were the only β-cell alteration, the β-cell functional profiling in human type 2 diabetes would show 1) parallel defects in both controls of β-cell functional mass, 2) no possibility of rapid reversibility of either defect, 3) no defect in the incretin effect when expressed as percent, and 4) no involvement of genes regulating β-cell function.
However, under close inspection the available data fulfill none of the above predictions, thereby lending support to the existence of β-cell loss of function independently of β-cell loss of mass in type 2 diabetes. We herein briefly review the experimental evidence falsifying the four statements above.
1. Lack of parallelism between defects of derivative (dynamic) and proportional (static) control of β-cell functional mass in patients with type 2 diabetes.
In his Banting Lecture of 1990, Daniel Porte, beautifully summarizing several decades of research on the β-cell, reported that first-phase insulin secretion (derivative or dynamic control) is disproportionately more impaired than second-phase insulin secretion (proportional or static control) in patients with overt type 2 diabetes (54). Until then, most studies were conducted with intravenous glucose challenges, in which the β-cell metrics were based on insulin concentration. Potential critiques were the (lack of) generalizability of these observations to the oral route of administration and the potential pitfalls introduced by the use of insulin concentration, which is heavily determined not only by insulin secretion rate but also by insulin catabolism, with the latter process being variably altered in states of insulin resistance such as diabetes. These potential drawbacks have been overcome by in vivo β-cell metrics resting on mathematical modeling of C-peptide (43,55,56), from which one can compute the β-cell insulin secretion rate (units: picomoles per minute) and quantify the derivative control and proportional control of β-cell functional mass. These tools have confirmed that in type 2 diabetes, there are severe impairments of both derivative (dynamic) and proportional (static) control of β-cells (53), and that these defects are evident also during an oral mixed-meal test (57).
However, during intravenous glucose challenges, the defect in the derivative (dynamic) control exceeds the impairment in the proportional (static) control of β-cell secretion (Fig. 2). Importantly, this lack of parallelism between the two defects is evident also in the prediabetes stage. While the derivative (dynamic) control displays an approximately linear decline (58), which starts already at glucose levels well within the limits of normalcy (59), the proportional (static) control is characterized by a somewhat abrupt fall in the passage from impaired glucose regulation to overt diabetes (58,60).
2. Fast reversibility of β-cell defects in type 2 diabetes.
Looking at fast reversibility of defects in β-cell functional mass as evidence of function-related—not mass-related—impairments is predicated on the tenet that β-cells in human adults turn over very slowly. Although life span and regeneration rates of β-cells are quite arduous to measure in humans, the few current data show that the β-cell pool turns over at a very slow rate of years (61,62). Bariatric surgery performed in patients with type 2 diabetes has been reported to cause significant improvements in β-cell function in the time span of a few weeks (63) or even days (64), i.e., orders of magnitude faster than it can be accounted for by changes in mass. However, the perturbations brought about by complex and different surgical intervention always leave room for the possibility that, inadvertently, not all factors may have been controlled for appropriately in these comparisons. From this viewpoint, a recent paper by Lim et al. (65) may be of help. These authors treated patients with type 2 diabetes with a very-low-calorie diet (VLCD) and monitored changes in insulin secretion and insulin action by performing isoglycemic insulin clamps and hyperglycemic clamps, respectively. In the time frame of weeks, they detected a robust improvement in the β-cell functional mass of these patients before any change in insulin sensitivity could be documented. Similar results were reported by us in a small group of morbidly obese patients with type 2 diabetes after only 1 week of VLCD (66). Finally, in a clinical trial conducted by Weng et al. (22) patients with newly diagnosed type 2 diabetes were intensively treated for 4 weeks with insulin pump therapy, basal-bolus insulin therapy, or a number of oral hypoglycemic agents, with the goal of normalizing blood glucose levels over the entire day. At the end of the 4-week treatment period, there was a dramatic improvement in first-phase insulin release during the intravenous glucose tolerance test, which was also partially maintained after 1 year off of therapy. Therefore, different interventions, such as bariatric surgery, VLCD, or intensive diabetes treatment, can result in marked improvements in β-cell functional mass in the time frame of a few weeks.
3. Presence of an incretin defect in type 2 diabetes.
In the case of a pure β-cell loss of mass, the incretin effect would be decreased in absolute terms but normal when expressed in percent figures. However, this requires that the incretin effect be measured as insulin secretion rate—not insulin concentration. Unfortunately, in most experiments the latter metric is used rather than the former.
A few years ago, a detailed study by Muscelli et al. (44) showed that the incretin effect, computed as the ratio of total insulin secretion rate during the oral glucose challenge to total insulin secretion rate during the intravenous challenge, was decreased in type 2 diabetes. The same was also true for the incretin effect on proportional (static) control, but not on derivative (dynamic) control, of β-cell functional mass. Thus, this study provides two pieces of evidence in favor of β-cell loss of function in type 2 diabetes: 1) there is a defect in the incretin effect on insulin secretion rate when expressed as percent and 2) the defect in the incretin effect affects proportional (static) but not derivative (dynamic) control of insulin secretion, thereby highlighting one more asymmetry in β-cell functional mass defects associated with type 2 diabetes.
4. β-Cell loss-of-function gene variants are risk factors for type 2 diabetes and are associated with decreased β-cell functional mass.
Several lines of evidence, including twin studies (67), support the notion that the phenotype of β-cell functional mass is determined by genetic factors to quite a large extent. Over the last 6 years, genetic variability at >60 genetic loci has been firmly linked to type 2 diabetes risk (68). Many of these loci are believed to play a role in diabetes etiology primarily through effects on β-cell function (69), and indeed, they are associated with reduced β-cell functional mass in vivo in humans—even in patients with type 2 diabetes (70–73). However, at this level of phenotypic resolution and in the absence of an in vivo method to quantify β-cell mass, dissecting out the role(s) of β-cell loss of mass versus loss of function is only presumptive.
Studies in human islets and isolated β-cells can be helpful. Indeed, glucose-induced insulin secretion in islets taken from patients with type 2 diabetes is reduced by 50% after normalization for islet insulin content, which is a proxy for reduced β-cell number in diabetic islets (74). Most importantly, the diabetogenic variants of four loci (TCF7L2, ADRA2A, KCNJ11, and KCNQ1) were associated with reduced insulin exocytosis or altered insulin granule distribution in isolated β-cells, which implies that part, if not most, of the diabetogenic influence of these risk variants is mediated through alterations in single β-cell function (74). Thus, there is converging evidence stemming from distinct experimental settings that defects in β-cell function underlie and cause β-cell failure in type 2 diabetes. However, this does not necessarily rule out a role, even a prominent one, for β-cell loss of mass, for which extensive evidence also exists. The relative roles played by each defect in β-cell failure remain unknown.
Concluding remarks
The conundrum of whether loss of mass or loss of function underlies the β-cell defects in type 2 diabetes is not likely to be conclusively solved on the basis of the evidence we have reviewed here. Decreased cell mass and acceleration of the biological processes resulting in β-cell loss have been described in type 2 diabetes by a number of laboratories. On the other hand, several lines of evidence suggest that β-cell functional defects may exist in type 2 diabetes.
Both viewpoints tacitly assume that 1) type 2 diabetes is a rather homogeneous entity, at least when it comes to β-cell biology, and 2) overall islet secretory capacity is a linear function of the product between β-cell number and isolated β-cell function. It is possible that neither assumption holds true.
The most likely scenario, indeed, is that a variable combination of the two processes, loss of mass and loss of function, is at work in type 2 diabetes. Indeed, there appears to be a tight relationship between mass of pancreatic β-cells and functional insulin secretion (75) (Fig. 3). A working model for the potential interaction of β-cell mass and β-cell function is presented in Fig. 4. If true, from the therapeutic viewpoint this offers an opportunity and poses a challenge.
Figure 3.

Relationship between pancreatic β-cell area, as determined from pancreatic tissue removed at surgery, and the C-peptide–to–glucose ratio determined in the fasting state (A) and 30 min after oral glucose ingestion in 8 individuals with normal glucose tolerance (NGT), 14 with impaired fasting glucose (IFG) or impaired glucose tolerance (IGT), and 11 with diabetes. r and P values were calculated by linear regression analysis. These analyses demonstrate the tight relationship between β-cell mass and β-cell function. Modified from ref. 75.
Figure 4.

Consensus model for the relationship between impaired β-cell function and mass in type 2 diabetes. A reduction in β-cell mass increases the secretory demand to the remaining β-cells, thereby disturbing β-cell function. This may lead to hyperglycemia and hyperlipidemia, which may again induce β-cell apoptosis, thereby aggravating the β-cell deficit. Along the same lines, the vicious circle may be initiated by a primary defect in β-cell function. The detrimental effects of hyperglycemia and β-cell exhaustion on β-cell mass and function may involve both oxidative stress and ER stress. FFA, free fatty acid.
The opportunity is that the defect in β-cell function is susceptible to improvement, even rapidly, with prompt beneficial effects on the patient, and it may even lead to remission of the disease (22,63–65). The challenge is that the processes leading to and the defect in β-cell mass itself need to be, at least partially, corrected to prevent an otherwise inexorable progression and to find a cure of this disease.
Acknowledgments
J.J.M. was supported by the Deutsche Forschungsgemeinschaft (DFG Me 2096/5-2) and the Ruhr University of Bochum (FoRUM). R.C.B. was supported by research grants of the University of Verona.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
R.C.B. was also supported by a research grant of the European Foundation for the Study of Diabetes/Novartis Programme. No other potential conflicts of interest relevant to this article were reported.
J.J.M. and R.C.B. researched and discussed data and wrote and reviewed the manuscript. R.C.B. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This publication is based on the presentations from the 4th World Congress on Controversies to Consensus in Diabetes, Obesity and Hypertension (CODHy). The Congress and the publication of this supplement were made possible in part by unrestricted educational grants from Abbott, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly, Ethicon Endo-Surgery, Janssen, Medtronic, Novo Nordisk, Sanofi, and Takeda.
References
- 1.DeFronzo RA. Pathogenesis of type 2 (non-insulin dependent) diabetes mellitus: a balanced overview. Diabetologia 1992;35:389–397 [DOI] [PubMed] [Google Scholar]
- 2.Ward WK, Bolgiano DC, McKnight B, Halter JB, Porte D., Jr Diminished B cell secretory capacity in patients with noninsulin-dependent diabetes mellitus. J Clin Invest 1984;74:1318–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pfeifer MA, Halter JB, Porte D., Jr Insulin secretion in diabetes mellitus. Am J Med 1981;70:579–588 [DOI] [PubMed] [Google Scholar]
- 4.Meigs JB, Wilson PW, Fox CS, et al. Body mass index, metabolic syndrome, and risk of type 2 diabetes or cardiovascular disease. J Clin Endocrinol Metab 2006;91:2906–2912 [DOI] [PubMed]
- 5.Klöppel G, Löhr M, Habich K, Oberholzer M, Heitz PU. Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv Synth Pathol Res 1985;4:110–125 [DOI] [PubMed] [Google Scholar]
- 6.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003;52:102–110 [DOI] [PubMed] [Google Scholar]
- 7.Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 2008;10(Suppl. 4):32–42 [DOI] [PubMed] [Google Scholar]
- 8.Maedler K. Beta cells in type 2 diabetes - a crucial contribution to pathogenesis. Diabetes Obes Metab 2008;10:408–420 [DOI] [PubMed] [Google Scholar]
- 9.Marchetti P, Del Guerra S, Marselli L, et al. Pancreatic islets from type 2 diabetic patients have functional defects and increased apoptosis that are ameliorated by metformin. J Clin Endocrinol Metab 2004;89:5535–5541 [DOI] [PubMed] [Google Scholar]
- 10.Maedler K, Oberholzer J, Bucher P, Spinas GA, Donath MY. Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes 2003;52:726–733 [DOI] [PubMed] [Google Scholar]
- 11.Robertson RP, Harmon JS. Diabetes, glucose toxicity, and oxidative stress: A case of double jeopardy for the pancreatic islet beta cell. Free Radic Biol Med 2006;41:177–184 [DOI] [PubMed] [Google Scholar]
- 12.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol 2011;11:98–107 [DOI] [PubMed]
- 13.Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med 2006;355:2427–2443 [DOI] [PubMed]
- 14.Meier JJ. Linking the genetics of type 2 diabetes with low birth weight: a role for prenatal islet maldevelopment? Diabetes 2009;58:1255–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meier JJ, Butler AE, Saisho Y, et al. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 2008;57:1584–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hattersley AT, Tooke JE. The fetal insulin hypothesis: an alternative explanation of the association of low birthweight with diabetes and vascular disease. Lancet 1999;353:1789–1792 [DOI] [PubMed] [Google Scholar]
- 17.Kendall DM, Sutherland DE, Najarian JS, Goetz FC, Robertson RP. Effects of hemipancreatectomy on insulin secretion and glucose tolerance in healthy humans. N Engl J Med 1990;322:898–903 [DOI] [PubMed] [Google Scholar]
- 18.Menge BA, Schrader H, Breuer TG, et al. Metabolic consequences of a 50% partial pancreatectomy in humans. Diabetologia 2009;52:306–317 [DOI] [PubMed] [Google Scholar]
- 19.Brunzell JD, Robertson RP, Lerner RL, et al. Relationships between fasting plasma glucose levels and insulin secretion during intravenous glucose tolerance tests. J Clin Endocrinol Metab 1976;42:222–229 [DOI] [PubMed] [Google Scholar]
- 20.Toschi E, Camastra S, Sironi AM, et al. Effect of acute hyperglycemia on insulin secretion in humans. Diabetes 2002;51(Suppl. 1):S130–S133 [DOI] [PubMed] [Google Scholar]
- 21.Laedtke T, Kjems L, Pørksen N, et al. Overnight inhibition of insulin secretion restores pulsatility and proinsulin/insulin ratio in type 2 diabetes. Am J Physiol Endocrinol Metab 2000;279:E520–E528 [DOI] [PubMed] [Google Scholar]
- 22.Weng J, Li Y, Xu W, et al. Effect of intensive insulin therapy on beta-cell function and glycaemic control in patients with newly diagnosed type 2 diabetes: a multicentre randomised parallel-group trial. Lancet 2008;371:1753–1760 [DOI] [PubMed] [Google Scholar]
- 23.Kahn SE, Zraika S, Utzschneider KM, Hull RL. The beta cell lesion in type 2 diabetes: there has to be a primary functional abnormality. Diabetologia 2009;52:1003–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fehse F, Trautmann M, Holst JJ, et al. Exenatide augments first- and second-phase insulin secretion in response to intravenous glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab 2005;90:5991–5997 [DOI] [PubMed] [Google Scholar]
- 25.Meier JJ, Breuer TG, Bonadonna RC, et al. Pancreatic diabetes manifests when beta cell area declines by approximately 65% in humans. Diabetologia 2012;55:1346–1354 [DOI] [PubMed]
- 26.Robertson RP, Lanz KJ, Sutherland DE, Seaquist ER. Relationship between diabetes and obesity 9 to 18 years after hemipancreatectomy and transplantation in donors and recipients. Transplantation 2002;73:736–741 [DOI] [PubMed] [Google Scholar]
- 27.Kahn SE, Leonetti DL, Prigeon RL, Boyko EJ, Bergstrom RW, Fujimoto WY. Proinsulin as a marker for the development of NIDDM in Japanese-American men. Diabetes 1995;44:173–179 [DOI] [PubMed] [Google Scholar]
- 28.Seaquist ER, Kahn SE, Clark PM, Hales CN, Porte D, Jr, Robertson RP. Hyperproinsulinemia is associated with increased beta cell demand after hemipancreatectomy in humans. J Clin Invest 1996;97:455–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meier JJ, Kjems LL, Veldhuis JD, Lefèbvre P, Butler PC. Postprandial suppression of glucagon secretion depends on intact pulsatile insulin secretion: further evidence for the intraislet insulin hypothesis. Diabetes 2006;55:1051–1056 [DOI] [PubMed] [Google Scholar]
- 30.Goodner CJ, Koerker DJ, Weigle DS, McCulloch DK. Decreased insulin- and glucagon-pulse amplitude accompanying beta-cell deficiency induced by streptozocin in baboons. Diabetes 1989;38:925–931 [DOI] [PubMed] [Google Scholar]
- 31.Bonner-Weir S, Trent DF, Weir GC. Partial pancreatectomy in the rat and subsequent defect in glucose-induced insulin release. J Clin Invest 1983;71:1544–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rankin MM, Kushner JA. Adaptive beta-cell proliferation is severely restricted with advanced age. Diabetes 2009;58:1365–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Menge BA, Tannapfel A, Belyaev O, et al. Partial pancreatectomy in adult humans does not provoke beta-cell regeneration. Diabetes 2008;57:142–149 [DOI] [PubMed] [Google Scholar]
- 34.Matveyenko AV, Veldhuis JD, Butler PC. Adaptations in pulsatile insulin secretion, hepatic insulin clearance, and beta-cell mass to age-related insulin resistance in rats. Am J Physiol Endocrinol Metab 2008;295:E832–E841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matveyenko AV, Liuwantara D, Gurlo T, et al. Pulsatile portal vein insulin delivery enhances hepatic insulin action and signaling. Diabetes 2012;61:2269–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matveyenko AV, Veldhuis JD, Butler PC. Mechanisms of impaired fasting glucose and glucose intolerance induced by an approximate 50% pancreatectomy. Diabetes 2006;55:2347–2356 [DOI] [PubMed] [Google Scholar]
- 37.Menge BA, Grüber L, Jørgensen SM, et al. Loss of inverse relationship between pulsatile insulin and glucagon secretion in patients with type 2 diabetes. Diabetes 2011;60:2160–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meier JJ, Veldhuis JD, Butler PC. Pulsatile insulin secretion dictates systemic insulin delivery by regulating hepatic insulin extraction in humans. Diabetes 2005;54:1649–1656 [DOI] [PubMed] [Google Scholar]
- 39.Meier JJ. Beta cell mass in diabetes: a realistic therapeutic target? Diabetologia 2008;51:703–713 [DOI] [PubMed] [Google Scholar]
- 40.Pox C, Ritzel R, Büsing M, et al. Combined pancreas and kidney transplantation in a lean type 2 diabetic patient. Effects on insulin secretion and sensitivity. Exp Clin Endocrinol Diabetes 2002;110:420–424 [DOI] [PubMed] [Google Scholar]
- 41.Ferrannini E. The stunned beta cell: a brief history. Cell Metab 2010;11:349–352 [DOI] [PubMed] [Google Scholar]
- 42.Gastaldelli A. Role of beta-cell dysfunction, ectopic fat accumulation and insulin resistance in the pathogenesis of type 2 diabetes mellitus. Diabetes Res Clin Pract 2011;93(Suppl. 1):S60–S65 [DOI] [PubMed] [Google Scholar]
- 43.Cobelli C, Toffolo GM, Dalla Man C, et al. Assessment of beta-cell function in humans, simultaneously with insulin sensitivity and hepatic extraction, from intravenous and oral glucose tests. Am J Physiol Endocrinol Metab 2007;293:E1–E15 [DOI] [PubMed] [Google Scholar]
- 44.Muscelli E, Mari A, Casolaro A, et al. Separate impact of obesity and glucose tolerance on the incretin effect in normal subjects and type 2 diabetic patients. Diabetes 2008;57:1340–1348 [DOI] [PubMed] [Google Scholar]
- 45.Campioni M, Toffolo G, Shuster LT, Service FJ, Rizza RA, Cobelli C. Incretin effect potentiates beta-cell responsivity to glucose as well as to its rate of change: OGTT and matched intravenous study. Am J Physiol Endocrinol Metab 2007;292:E54–E60 [DOI] [PubMed] [Google Scholar]
- 46.Bonadonna RC, Heise T, Arbet-Engels C, et al. Piragliatin (RO4389620), a novel glucokinase activator, lowers plasma glucose both in the postabsorptive state and after a glucose challenge in patients with type 2 diabetes mellitus: a mechanistic study. J Clin Endocrinol Metab 2010;95:5028–5036 [DOI] [PubMed]
- 47.Ohara-Imaizumi M, Nishiwaki C, Kikuta T, Nagai S, Nakamichi Y, Nagamatsu S. TIRF imaging of docking and fusion of single insulin granule motion in primary rat pancreatic beta-cells: different behaviour of granule motion between normal and Goto-Kakizaki diabetic rat beta-cells. Biochem J 2004;381:13–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jing X, Li DQ, Olofsson CS, et al. CaV2.3 calcium channels control second-phase insulin release. J Clin Invest 2005;115:146–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holness MJ, Hegazy S, Sugden MC. Signalling satiety and starvation to β-cell insulin secretion. Curr Diabetes Rev 2011;7:336–345 [DOI] [PubMed]
- 50.Pedersen MG, Toffolo GM, Cobelli C. Cellular modeling: insight into oral minimal models of insulin secretion. Am J Physiol Endocrinol Metab 2010;298:E597–E601 [DOI] [PubMed] [Google Scholar]
- 51.Pedersen MG, Dalla Man C, Cobelli C. Multiscale modeling of insulin secretion. IEEE Trans Biomed Eng 2011;58:3020–3023 [DOI] [PubMed] [Google Scholar]
- 52.Groop LC, Ratheiser K, Luzi L, et al. Effect of sulphonylurea on glucose-stimulated insulin secretion in healthy and non-insulin dependent diabetic subjects: a dose-response study. Acta Diabetol 1991;28:162–168 [DOI] [PubMed] [Google Scholar]
- 53.Ferrannini E, Gastaldelli A, Miyazaki Y, Matsuda M, Mari A, DeFronzo RA. beta-Cell function in subjects spanning the range from normal glucose tolerance to overt diabetes: a new analysis. J Clin Endocrinol Metab 2005;90:493–500 [DOI] [PubMed] [Google Scholar]
- 54.Porte D., Jr Banting lecture 1990. Beta-cells in type II diabetes mellitus. Diabetes 1991;40:166–180 [DOI] [PubMed] [Google Scholar]
- 55.Van Cauter E, Mestrez F, Sturis J, Polonsky KS. Estimation of insulin secretion rates from C-peptide levels. Comparison of individual and standard kinetic parameters for C-peptide clearance. Diabetes 1992;41:368–377 [DOI] [PubMed] [Google Scholar]
- 56.Mari A, Schmitz O, Gastaldelli A, Oestergaard T, Nyholm B, Ferrannini E. Meal and oral glucose tests for assessment of beta -cell function: modeling analysis in normal subjects. Am J Physiol Endocrinol Metab 2002;283:E1159–E1166 [DOI] [PubMed] [Google Scholar]
- 57.Basu A, Dalla Man C, Basu R, Toffolo G, Cobelli C, Rizza RA. Effects of type 2 diabetes on insulin secretion, insulin action, glucose effectiveness, and postprandial glucose metabolism. Diabetes Care 2009;32:866–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weiss R, Caprio S, Trombetta M, Taksali SE, Tamborlane WV, Bonadonna R. Beta-cell function across the spectrum of glucose tolerance in obese youth. Diabetes 2005;54:1735–1743 [DOI] [PubMed] [Google Scholar]
- 59.Bonadonna RC, Stumvoll M, Fritsche A, et al. Altered homeostatic adaptation of first- and second-phase beta-cell secretion in the offspring of patients with type 2 diabetes: studies with a minimal model to assess beta-cell function. Diabetes 2003;52:470–480 [DOI] [PubMed] [Google Scholar]
- 60.Cali’ AM, Bonadonna RC, Trombetta M, Weiss R, Caprio S. Metabolic abnormalities underlying the different prediabetic phenotypes in obese adolescents. J Clin Endocrinol Metab 2008;93:1767–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cnop M, Hughes SJ, Igoillo-Esteve M, et al. The long lifespan and low turnover of human islet beta cells estimated by mathematical modelling of lipofuscin accumulation. Diabetologia 2010;53:321–330 [DOI] [PubMed] [Google Scholar]
- 62.Perl S, Kushner JA, Buchholz BA, et al. Significant human beta-cell turnover is limited to the first three decades of life as determined by in vivo thymidine analog incorporation and radiocarbon dating. J Clin Endocrinol Metab 2010;.95:E234–E239 [DOI] [PMC free article] [PubMed]
- 63.Ferrannini E, Mingrone G. Impact of different bariatric surgical procedures on insulin action and beta-cell function in type 2 diabetes. Diabetes Care 2009;32:514–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guidone C, Manco M, Valera-Mora E, et al. Mechanisms of recovery from type 2 diabetes after malabsorptive bariatric surgery. Diabetes 2006;55:2025–2031 [DOI] [PubMed] [Google Scholar]
- 65.Lim EL, Hollingsworth KG, Aribisala BS, Chen MJ, Mathers JC, Taylor R. Reversal of type 2 diabetes: normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia 2011;54:2506–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Malandrucco I, Pasqualetti P, Giordani I, et al. Very-low-calorie diet: a quick therapeutic tool to improve β cell function in morbidly obese patients with type 2 diabetes. Am J Clin Nutr 2012;95:609–613 [DOI] [PubMed]
- 67.Lehtovirta M, Kaprio J, Groop L, Trombetta M, Bonadonna RC. Heritability of model-derived parameters of beta cell secretion during intravenous and oral glucose tolerance tests: a study of twins. Diabetologia 2005;48:1604–1613 [DOI] [PubMed] [Google Scholar]
- 68.Wheeler E, Barroso I. Genome-wide association studies and type 2 diabetes. Brief Funct Genomics 2011;10:52–60 [DOI] [PubMed]
- 69.Ashcroft FM, Rorsman P. Diabetes mellitus and the β cell: the last ten years. Cell 2012;148:1160–1171 [DOI] [PMC free article] [PubMed]
- 70.Bonetti S, Trombetta M, Malerba G, et al. Variants and haplotypes of TCF7L2 are associated with β-cell function in patients with newly diagnosed type 2 diabetes: the Verona Newly Diagnosed Type 2 Diabetes Study (VNDS) 1. J Clin Endocrinol Metab 2011;96:E389–E93 [DOI] [PubMed]
- 71.Bonetti S, Trombetta M, Boselli ML, et al. Variants of GCKR affect both β-cell and kidney function in patients with newly diagnosed type 2 diabetes: the Verona newly diagnosed type 2 diabetes study 2. Diabetes Care 2011;34:1205–1210 [DOI] [PMC free article] [PubMed]
- 72.Lyssenko V, Eliasson L, Kotova O, et al. Pleiotropic effects of GIP on islet function involve osteopontin. Diabetes 2011;60:2424–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Trombetta M, Bonetti S, Boselli M, et al. CACNA1E variants affect beta cell function in patients with newly diagnosed type 2 diabetes. The Verona newly diagnosed type 2 diabetes study (VNDS) 3. PLoS One 2012;7:e32755 [DOI] [PMC free article] [PubMed]
- 74.Rosengren AH, Braun M, Mahdi T, et al. Reduced insulin exocytosis in human pancreatic β-cells with gene variants linked to type 2 diabetes. Diabetes 2012;61:1726–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Meier JJ, Menge BA, Breuer TG, et al. Functional assessment of pancreatic beta-cell area in humans. Diabetes 2009;58:1595–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]