

Abstract
Mobile elements are powerful agents of genomic evolution and can be exceptionally informative markers for investigating species and population-level evolutionary history. While several studies have utilized retrotransposon-based insertional polymorphisms to resolve phylogenies, few population studies exist outside of humans. Endogenous retroviruses are LTR-retrotransposons derived from retroviruses that have become stably integrated in the host genome during past infections and transmitted vertically to subsequent generations. They offer valuable insight into host-virus co-evolution and a unique perspective on host evolutionary history because they integrate into the genome at a discrete point in time. We examined the evolutionary history of a cervid endogenous gammaretrovirus (CrERVγ) in mule deer (Odocoileus hemionus). We sequenced 14 CrERV proviruses (CrERV-in1 to -in14), and examined the prevalence and distribution of 13 proviruses in 262 deer among 15 populations from Montana, Wyoming, and Utah. CrERV absence in white-tailed deer (O. virginianus), identical 5′ and 3′ long terminal repeat (LTR) sequences, insertional polymorphism, and CrERV divergence time estimates indicated that most endogenization events occurred within the last 200000 years. Population structure inferred from CrERVs (F ST = 0.008) and microsatellites (θ = 0.01) was low, but significant, with Utah, northwestern Montana, and a Helena herd being particularly differentiated. Clustering analyses indicated regional structuring, and non-contiguous clustering could often be explained by known translocations. Cluster ensemble results indicated spatial localization of viruses, specifically in deer from northeastern and western Montana. This study demonstrates the utility of endogenous retroviruses to elucidate and provide novel insight into both ERV evolutionary history and the history of contemporary host populations.
Key words: endogenous retrovirus, gene flow, microsatellite, mule deer, population structure
Genetic variation observed among populations and species is commonly used to reconstruct ancient and contemporary histories of demography and migration. Molecular markers most commonly used to assess genetic variation include polymorphic sequence data (e.g., single nucleotide polymorphisms [SNPs], mitochondrial DNA) and simple sequence repeats (e.g., microsatellites). As genomic resources become increasingly available, structural genomic variation and polymorphic insertions of mobile elements offer promising possibilities for population and phylogenetic analyses in both model (Bamshad et al. 2003; Watkins et al. 2003; Witherspoon et al. 2006, 2013; Handsaker et al. 2011; Zichner et al. 2013) and non-model organisms (Sasaki et al. 2006; Xing et al. 2007; Li et al. 2009; Meyer et al. 2012).
Retrotransposons are discrete DNA sequences capable of moving within genomes through RNA intermediates and are the most prevalent class of mobile elements, accounting for just more than 40% of the human genome (Lander et al. 2001). Following genome integration, these elements are insertionally polymorphic and may increase in frequency over time, ultimately becoming fixed in a population or species. Insertional polymorphisms generated by retrotransposons have been regarded as being ‘nearly ideal’ genetic markers (Ray 2007). Retrotransposons have been used extensively to resolve the ancestral relationships between various primate groups (Xing et al. 2007; Li et al. 2009; McLain et al. 2012; Meyer et al. 2012), most notably the human–chimpanzee–gorilla trichotomy (Salem et al. 2003). They have also been applied to more reliably determine phylogenies of non-primate groups such as whales (Nikaido et al. 2007; Chen et al. 2011), turtles (Sasaki et al. 2006), and bats (Kawai et al. 2002), and have helped to discern ancient radiations of African cichlid fishes that were obscured by mutational saturation in sequence data (Takahashi et al. 1998). While retrotransposons have proved to be especially useful for phylogenetic reconstruction, their application to population genetics has been limited. This is due to the fact that they are typically fixed within, but polymorphic among species, a property that is useful for resolving phylogenies around the timing of activity for the particular insertion. Outside of human studies (Watkins et al. 2003; Witherspoon et al. 2006), few examples of retrotransposon-based insertional polymorphisms within species exist (but see Tarlinton et al. 2006; Liggins et al. 2008; Ávila-Arcos et al. 2013).
Endogenous retroviruses (ERVs) are retrotransponsons derived from retroviruses that have become stably integrated in the host genome through colonization of the host germline DNA during an infection. Once integrated, the proviruses are frequently transcriptionally silenced by the host and, after generations of vertical inheritance, can accumulate mutations due to host replication errors or through recombination that renders them unable to produce viral proteins (Taruscio and Mantovani 2004). The acquired provirus may ultimately be fixed in the host over time, becoming a fossilized record of past viral infections. These genomic “fossils” are abundant and ubiquitously present in all vertebrate species; for example, in humans, ERVs comprise at least 8% of the genome (Lander et al. 2001) and in mice approximately 10% of the genome (Waterston et al. 2002).
Endogenous retroviruses have provided valuable insight into the evolutionary history of modern viruses, host-viral ecology and co-evolution, and have been utilized for resolving uncertainties in host phylogenies (Shih et al. 1991; Johnson and Coffin 1999; Gifford et al. 2008; Keckesova et al. 2009; Gilbert and Feschotte 2010; Feschotte and Gilbert 2012). They can provide a discrete time stamp to investigate the history of viral epidemics and host demography. For example, the presence of endogenous lentiviruses in lemurs (Gifford et al. 2008) and related lagomorph species (van der Loo et al. 2008; Keckesova et al. 2009) indicate lentiviruses have been present in various mammalian orders for at least 12 million years. ERVs have also been used as host demographic markers to examine the history of sheep domestication (Chessa et al. 2009), revealing two independent migrations of domestic sheep and providing evidence for the origin of less common sheep breeds. However, because colonization events of most vertebrate hosts by ERVs are ancient events, the use of ERVs has been limited to establishing speciation events and viral ancestry, but they have not been used to investigate population history.
Mule deer (Odocoileus hemionus) exhibit low levels of population divergence (Cullingham et al. 2011; Powell et al. 2013), except over large geographic scales that follow subspecies designations along the US western coastline (Latch et al. 2009; Pease et al. 2009). This may be attributed to large population sizes, long generation times, and high mobility, supported by estimates of long distance movement from radiotelemetry studies (Anderson and Wallmo 1984; Mackie et al. 2003). Mule deer have been successful in a wide diversity of habitats through the species’ history, persisting in large populations throughout their vast range, which currently extends across western North America, from northern Mexico to Canada (Latch et al. 2009). In Montana (MT), overharvesting drastically reduced populations by the early 1900s, particularly in the eastern part of the state, prompting translocation efforts from western to eastern populations in an attempt to restore deer throughout their historic range (Picton and Lonner 2008). Across the state, population densities have been found to vary spatially based on presence of high-quality reproductive habitat and established populations tend to have annual fluctuations that vary independently among localities due to differences in population–habitat relationships, environmental variation and management (Mackie et al. 1998). Because deer populations tend to have local fluctuations, understanding gene flow across the landscape is necessary for the management of the species―in defining how animals have historically dispersed, developing management units for harvest and in establishing effective control strategies for infectious disease.
A recent meta-transcriptomic study of microbial biota in lymph node tissue reported on a novel cervid endogenous gammaretrovirus (CrERVγ) in mule deer (Wittekindt et al. 2010). Elleder et al. (2012) further discovered that the CrERV was polymorphic in integration profiles among deer, demonstrating that mule deer have undergone repeated germline invasion by the gammaretrovirus and that integration has occurred continually since the divergence of mule deer from its sister species, white-tailed deer (O. virginianus).
In this study, we demonstrate the unique value of CrERV data for examining both viral evolutionary history and the population history of its wildlife host. Because CrERVs are insertionally polymorphic in mule deer populations, they may reveal features of population structure that could complement traditional genetic approaches. Animals that share a CrERV integration site are related by ancestry back to the individual experiencing the original germline colonization event. Unlike microsatellites or SNP data, ERVs have a phylogenetic history that can be used to obtain estimates of the time of integration. We investigate the evolutionary history of several newly identified CrERVs in mule deer and examine how they explain the population history of their host. Our study is based on 13 CrERVs that were identified in a random screen of viral integration sites and which were evaluated for prevalence in mule deer populations from MT, Wyoming (WY), and Utah (UT). We use a variety of well-recognized and novel methods for evaluating structure connectivity and ancestral history among CrERVs and their mule deer hosts, and integrate results obtained using microsatellite data with that based on the presence or absence of CrERV loci.
Materials and Methods
Sample Collection and DNA Extraction
Retropharyngeal lymph node samples were acquired from harvested mule deer (O. hemionus) brought through hunter check stations from 2007 to 2008, with the exception of samples from Helena (HLN), which were obtained through an urban deer removal project. Approximated geographic coordinates were recorded for all samples by asking hunters to pinpoint locations using a 1:250000 gazetteer and/or report hydrological unit, township and range where the animal was obtained. Therefore, error in location estimates was expected to be within 1–5 km. Overall, we obtained samples from 13 localities within MT and one locality in northeast WY, herein referred to as “populations” (Figure 1A; n = 357; 13–34 individuals per population). Populations ranged in area from 36–14275 km2 (Supplementary Figure S1). Additional samples were acquired from localities distributed throughout UT (n = 30), to enable assessment of mule deer connectivity in a more broad scale geographic context. Samples were also obtained from white-tailed deer across MT to assess CrERV presence or absence in this species (n = 24). Tissue samples were preserved in RNA later (Ambion) and genomic DNA was extracted from lymph node tissue using a phenol/chloroform extraction method, following the manufacturer’s protocol.
Figure 1.

Map of populations sampled and clustering solutions. Population (1-13, HLN) locations and clusters based on CrERV data are shown on a map of Montana (MT) and northern Wyoming (WY) (A), with colors corresponding to the CrERV clustering solution presented in part B. Utah (UT) is not shown on the map. Agglomerative hierarchical clustering dendograms are based on a Pearson’s uncorrelated distance matrix, calculated using CrERV insertion frequencies (B) and microsatellite allele frequencies (C) within localities.
Microsatellite Genotyping and Selection
Genotyping of 16 microsatellite (μsat) loci (Bishop et al. 1994; DeWoody et al. 1995; Wilson et al. 1997; Jones et al. 2000) was conducted following protocols previously reported in a study investigating mule deer population structure in MT (Powell et al. 2013). We tested for departures from Hardy–Weinberg (H-W) equilibrium at individual loci (Supplementary Table S1) using a Markov chain exact test (Guo and Thompson 1992) and linkage disequilibrium between loci using the log-likelihood ratio statistic as implemented in GENEPOP on the web (Raymond and Rousset 1995; Rousset 2008). Significance was tested based on 1000 iterations and after applying a Bonferroni correction to account for multiple comparisons (α = 0.05). The 15 predefined populations were tested independently to account for the potential effects of population structure on the results.
Hardy–Weinberg probability tests by population revealed similar results to that which were previously reported (Pease et al. 2009). The O and Q loci indicated significant heterozygote deficiency (Supplementary Table S1) in 7 out of 15 populations each. Given this and previous suspicion of null alleles, we dropped these loci from further analyses. Also, as in Powell et al. (2013) the P locus was out of H-W equilibrium in one population (population 12). Significant linkage disequilibrium was observed between BM4107 and Rt24 (population 9), Rt30 and Rt7 (population 2), and P and G (population 2). As these observations were not a widespread phenomenon, we assumed they were an artifact of population subdivision or sampling and did not remove the loci from subsequent analyses.
CrERV Sequencing and Screening
Retroviral integration sites were identified as outlined in Elleder et al. (2012). Seven of the proviruses were previously described (CrERV-in1 to- in7; Elleder et al. 2012) and seven are reported and sequenced here for the first time (CrERV-in8 to -in14; primer information shown in Supplementary Table S2). The 3′ portions (approximately 3.3–4.5kb) of all CrERVs were sequenced directly from gel-purified PCR products in at least one individual deer (GenBank: KC934943-KC934956). For CrERV-in1, -in2, -in3, and -in5, multiple individuals were sequenced and molecular diversity indices reported (Supplementary Table S3).
Using a sub-sample of microsatellite genotyped mule deer (Table 1; n = 13–29/population), we screened for the presence or absence of 13 CrERV integration sites (CrERV-in1 to -in13), all of which were present in mule deer but not in white-tailed deer. Primers used in CrERV genotyping can be found in Supplementary Table S4. Of the total samples, 259 individuals were genotyped at both microsatellite and CrERV loci (presence/absence), and this dataset was used in subsequent marker-based comparative analyses.
Table 1.
Observed CrERV insertion frequency by geographic locality
| Location | n | Lineage I | Lineage II | Lineage III | Lineage IV | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| in1 | in3 | in12 | in9 | in6 | in10 | in13 | in5 | in8 | in2 | in4 | in7 | in11 | ||
| 1 | 17 | 1.00 | 0.41 | 0.12 | 0.50 | 0.00 | 0.00 | 0.00 | 0.35 | 0.00 | 0.35 | 0.53 | 0.00 | 0.12 |
| 2 | 17 | 1.00 | 0.47 | 0.18 | 0.71 | 0.00 | 0.00 | 0.00 | 0.41 | 0.00 | 0.29 | 0.47 | 0.12 | 0.06 |
| 3 | 17 | 1.00 | 0.53 | 0.18 | 0.56 | 0.00 | 0.00 | 0.00 | 0.35 | 0.00 | 0.47 | 0.35 | 0.12 | 0.06 |
| 4 | 18 | 1.00 | 0.50 | 0.06 | 0.56 | 0.00 | 0.00 | 0.00 | 0.22 | 0.00 | 0.11 | 0.56 | 0.06 | 0.00 |
| 5 | 17 | 1.00 | 0.29 | 0.00 | 0.50 | 0.29 | 0.12 | 0.06 | 0.12 | 0.06 | 0.47 | 0.18 | 0.00 | 0.18 |
| 6 | 16 | 1.00 | 0.53 | 0.00 | 0.56 | 0.13 | 0.00 | 0.00 | 0.19 | 0.00 | 0.25 | 0.25 | 0.00 | 0.00 |
| 7 | 18 | 1.00 | 0.56 | 0.00 | 0.72 | 0.11 | 0.00 | 0.00 | 0.17 | 0.00 | 0.22 | 0.39 | 0.00 | 0.06 |
| 8 | 13 | 1.00 | 0.46 | 0.00 | 0.77 | 0.08 | 0.00 | 0.00 | 0.23 | 0.00 | 0.54 | 0.50 | 0.00 | 0.15 |
| 9 | 15 | 1.00 | 0.20 | 0.07 | 0.60 | 0.07 | 0.07 | 0.00 | 0.20 | 0.00 | 0.40 | 0.47 | 0.00 | 0.00 |
| 10 | 17 | 1.00 | 0.50 | 0.06 | 0.76 | 0.06 | 0.00 | 0.00 | 0.29 | 0.00 | 0.29 | 0.65 | 0.00 | 0.12 |
| 11 | 15 | 1.00 | 0.57 | 0.07 | 0.53 | 0.13 | 0.00 | 0.00 | 0.13 | 0.00 | 0.40 | 0.53 | 0.00 | 0.00 |
| 12 | 18 | 1.00 | 0.33 | 0.00 | 0.83 | 0.00 | 0.00 | 0.00 | 0.17 | 0.00 | 0.22 | 0.44 | 0.00 | 0.06 |
| 13 | 17 | 1.00 | 0.47 | 0.00 | 0.41 | 0.06 | 0.00 | 0.00 | 0.38 | 0.00 | 0.24 | 0.41 | 0.00 | 0.00 |
| HLN | 18 | 1.00 | 0.47 | 0.00 | 0.56 | 0.44 | 0.00 | 0.00 | 0.29 | 0.00 | 0.47 | 0.82 | 0.00 | 0.00 |
| UT | 29 | 1.00 | 0.24 | 0.03 | 0.43 | 0.03 | 0.00 | 0.00 | 0.31 | 0.00 | 0.32 | 0.89 | 0.00 | 0.00 |
| Proportion | 1.000 | 0.426 | 0.050 | 0.593 | 0.092 | 0.011 | 0.004 | 0.258 | 0.004 | 0.331 | 0.514 | 0.019 | 0.050 | |
| N | 261 | 258 | 262 | 258 | 262 | 262 | 262 | 260 | 262 | 260 | 259 | 261 | 262 | |
Lineages corresponding to phylogenetic results (in Figure 2) are indicated.
Recombination Analyses
The occurrence of recombination can severely impact estimation of phylogenetic relationships (Posada and Crandall 2002) and downstream inferences of evolutionary history (Schierup and Hein 2000) as the assumption of a single tree is violated. The presence of recombination may augment estimates of divergence time due to an increased number of ancestral lineages back through time (Martin et al. 2011). Therefore, we tested for recombination in the mule deer CrERV dataset (alignment of 3650bp) by measuring the pairwise-homoplasy index, or PHI-statistic (Φw), which is robust to the influence of population history and can distinguish between recurrent mutation and recombination (Bruen et al. 2006). Significance of Φw was obtained through a permutation test under the null hypothesis of no recombination and implemented using the SPLITSTREE program (Huson and Bryant 2006). As the PHI-test detected a recombination signal in our dataset, we used a model-based framework in the Genetic Algorithm Recombination Detection (GARD) subroutine of the HYPHY package (Pond et al. 2006) to identify recombination breakpoints and identify non-recombinant regions useful for downstream phylogenetic analyses. A maximum likelihood model was fit to each segment and goodness of fit evaluated by Akaike Information Criteria for small sample sizes (AICc). Recombination breakpoints were verified by performing Kishino–Hasegawa (K-H) tests (Hasegawa and Kishino 1989; Kishino and Hasegawa 1989) for topological incongruence on trees generated from adjacent segments on either side of a putative breakpoint. Recombinant regions were removed from the data and the PHI-test was run to further verify a recombinant-free dispersed alignment suitable for subsequent analyses.
Phylogenetic Analyses
Phylogenetic analyses that assume a relaxed molecular clock and make use of evolutionary events, such as species divergence times, to calibrate tree nodes have been suggested as a more robust method for dating ERV integrations (Martins and Villesen 2011). We used a Bayesian Markov Chain Monte Carlo (MCMC) modeling approach to jointly estimate nucleotide substitution rates, estimate relative divergence times, and reconstruct the evolutionary relationships among CrERVs using BEAST v1.6 (Drummond and Rambaut 2007). A representative sequence (i.e., either the only available or most frequent sequence) from each of the 14 mule deer CrERVs and 1 white-tailed deer CrERV (-in14) was included in the alignment constructed using the MAFFT algorithm (Katoh et al. 2002), followed by manual editing within the Geneious v5.5 interface (Drummond et al. 2011). Sequence data (totaling 3650–4143bp) consisted of contiguous regions of the 3′ portion of the pol gene, the entire env gene, and the 3′LTR. Length differences were due to deletions in the env gene in some viruses. We removed ~600bp near the 5′ end of the alignment, due to considerable sequence differences between CrERV-in12 and -in14 and the rest of the alignment, which can lead to long branch attachment and erroneous phylogenetic inference. Also, we excluded the identified recombinant regions, leaving pol, a 3′ portion of env, and the 3′LTR in the final dataset analyzed (an alignment of 1935bp).
Analyses were carried out using a Yule process speciation model (Gernhard 2008) and the transversional substitution model with 4 gamma-distributed rates (TVM + Γ4), selected as the best fit model of molecular evolution for data using the corrected Akaike Information Criteria (AICC) in jModelTest (Posada 2008). CrERV-in14 was identified in all mule deer, but only a few white-tailed deer. Based on divergence of 5′ and 3′ LTR and close homology between the two sequences, it appears to represent an older integration event in mule deer and is currently introgressing in white-tailed deer (D. Elleder, unpublished data). Therefore, as the oldest integration of the markers assessed and the only CrERV present in both O. virginianus and O. hemionus, it was specified as a monophyletic outgroup. We calibrated the white-tailed and mule deer CrERV-in14 node by specifying a uniform prior distribution with a maximum constraint equal to 1.8 million years ago (MYA), the median estimated divergence time between Odocoileus species (Pitra et al. 2004; Gilbert et al. 2006; Hedges et al. 2006). We also calibrated the internal node (most recent common ancestor to all mule deer CrERVs) based on an independent estimate of the In1 integration (0.47–1 MYA; Elleder et al. 2012). We assumed this node was older than the In1 integration, but younger than the split of Odocoileus species, by applying a conservative uniform distribution that encompassed this time period (0.47–1.8 MYA). Preliminary analyses rejected a strict molecular clock, based on the observation that the 95% highest probability density (HPD) intervals of the coefficient of variation fell well above zero. Therefore, we used a relaxed (uncorrelated, lognormal) molecular clock model (Drummond et al. 2006), applying an exponential prior distribution for evolutionary rate [exp(5e-7)] which allows for estimation of a substitution rate that is expected to fall between that of the virus and host. We repeated this analysis using the full alignment (3650bp) without excluding recombinant regions. For analyses based on both full and reduced alignments, we conducted two independent replicate runs, subsampling every 10000 steps more than 500 million steps, and discarding the first 10% steps as burnin. Additional analyses were run with an empty alignment, using prior distributions but no data, to evaluate whether the CrERV sequence data was informative in parameter estimation. The diagnostic tools in Tracer v1.4 (Rambaut and Drummond 2007) were used to inspect posterior distributions, run traces for convergence and ensure that effective sample sizes (ESS) were >200 for all parameter estimates. Consensus phylogenetic trees were constructed in TreeAnnotator v1.6 using the sampled trees from each run and visualized with the FigTree software (Rambaut 2009).
Clustering Analyses Based on CrERV Prevalence Data
To examine population-level clustering based on CrERV prevalence data, we performed an agglomerative hierarchical clustering analysis. Population frequency data takes advantage of the additional temporal information inherent in the CrERV data such that populations with similar proportions of shared CrERVs are more related. We complemented this analysis by similarly performing a clustering analysis based on microsatellite allele frequency data. Distance matrices were computed by Pearson correlation (uncentered) and clustering analyses employing average linkage was performed in Cluster 3.0 (Eisen et al. 1998). Results were visualized in TreeView (Page 1996).
We also used a cluster ensemble technique (Strehl and Ghosh 2002) to provide a framework for combining multiple clustering solutions using a matrix representation for the clustering result. This analysis was performed at the individual level, using CrERV presence–absence data. For n deer, we have an n × n co-association matrix X: Xi,j = 1 if the ith and jth deer have the same virus. Given a number of co-association matrices X(1), X(2), …, X(p), the consensus matrix can be constructed as the average of the individual co-association matrices as follows: X (c) = (X(1) + X(2) + ⋯ + X(p))/P, where P represents the number of CrERVs. The consensus matrix corresponds to a more stable representation of a partition than an individual clustering solution. As discussed above, we expect a lower prevalence among animals with CrERVs that have recently colonized the host and that animals sharing recently integrated viruses will be geographically localized. To account for this, we implement a revision of the co-association matrix for a specific virus:
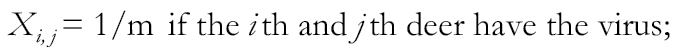 |
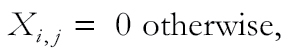 |
where m is the number of animals with the virus. The similarity between two animals is then a weighted average of co-association matrices across all viruses. To assess the significance of shared virus histories, we developed a null model under the assumption that the viruses are randomly distributed among animals. Under that null hypothesis, the probability of a pair of deer sharing the kth virus follows a binomial distribution, with 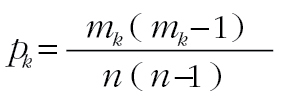 . The probability of sharing a specific subset of viruses, A, is
. The probability of sharing a specific subset of viruses, A, is 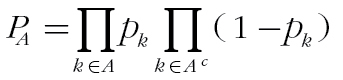 , with A
c signifying the set of all elements in the universal set that are not in A, and the corresponding similarity is
, with A
c signifying the set of all elements in the universal set that are not in A, and the corresponding similarity is 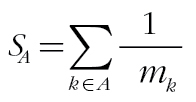 . By examining all subsets of viruses, we obtain the exact distribution of the similarity under the null model. For the observed similarity between a pair of deer, 1 minus its percentile in the null distribution gives the P value.
. By examining all subsets of viruses, we obtain the exact distribution of the similarity under the null model. For the observed similarity between a pair of deer, 1 minus its percentile in the null distribution gives the P value.
Population Structure and Differentiation
We independently assessed F-statistics for both marker types to evaluate genetic similarity among deer populations. Using microsatellite loci, we calculated an unbiased estimator of the fixation index, FST (θ; Weir and Cockerham 1984), in FSTATv2 (Goudet 1995). Significance was determined with 2000 permutations of the data, and after applying a 5% adjusted nominal level for multiple comparisons. Global θ over all populations and bootstrapping over loci was performed to determine the 95% confidence interval. Similarly, for CrERV data, global F ST was estimated in AFLP-SURV (Vekemans 2002), utilizing the Lynch and Milligan (1994) method for estimating allele frequencies.
Genetic distance between populations was also evaluated by pairwise calculations of the F ST analog, ΦST, in Arlequin v3.5 (Excoffier and Lischer 2010). We specified a Euclidean matrix of individual haplotype distances that is generalizable across marker inheritance types and, in this case, allowed for a more direct comparison of results based on binary data (CrERVs) versus co-dominant data (microsatellites). For a single-locus analysis of a microsatellite locus, with i, j, k, and l representing different alleles, the genetic distance between diploid individuals was defined as: d 2(ii,ii) = 0, d 2(ij,ij) = 0, d 2(ii,ij) = 1, d 2(ij,ik) = 1, d 2(ij,kl) = 2, d 2(ii,jk) = 3, and d 2(ii,jj) = 4 (Peakall et al. 1995). Whereas, comparable measures of genetic distance using CrERV loci were estimated as the difference in the number of shared CrERV integrations between two individual retrotypes, a method adopted from Huff et al. (1993) and Peakall et al. (1995): D = n(1 − n xy/n), where n xy represents the number of CrERVs in common between individuals x and y, and n is the total number of loci analyzed.
Hierarchical partitioning of genetic variation among populations and broad scale regions was performed through an Analysis of Molecular Variance (AMOVA; Excoffier et al. 1992) in Arlequin v3.5 (Excoffier and Lischer 2010). This analysis used Φ-statistics to estimate the proportion of genetic variability found among populations (ΦST), among populations within regions (ΦSC), and among regions (ΦCT), and utilized the haplotype distance matrices described above. non-parametric permutation procedures (10000 permutations) were performed to assess the significance of the covariance components associated with the different levels of population structure. For a priori group assignments, populations were specified as previously described and six broad-scale regions were also defined: (1) the northeast: 1-4, (2) the northwest: 5, (3) the west: 6-7 and HLN, (4) the southwest: 8-11, (5) the southeast: 12-13, and (6) Utah: UT.
Marker Comparison of Population Diversity and Differentiation
Within population, genetic diversity indices were calculated using both microsatellite and CrERV data. For microsatellites, genetic diversity was measured in terms of the inbreeding coefficient (F IS), observed (H O), and unbiased expected heterozygosity (H E) in GenAlEx v6.2 (Peakall and Smouse 2006), and mean allelic richness, after adjusting for unequal sample sizes through rarefaction in HP-RARE (Kalinowski 2005). For CrERV loci, we computed indices of genetic diversity following determination of allele frequencies using a Bayesian method described by Zhivotovsky (1999), implemented in AFLP-SURV v1.0 (Vekemans 2002). This approach estimates the distribution of allele frequencies based on the variability in marker presence frequency over loci, and is useful when actual allele frequencies cannot be observed directly. Estimated CrERV diversity indices include the number and proportion of polymorphic loci at the 5% level and Nei’s unbiased gene diversity (Hj, analogous to H E).
We examined the relationship between population genetic distances as calculated for CrERV versus microsatellite data. We specifically evaluated ΦST distance matrices between population pairs for both marker types and evaluated their relationship using a generalized linear model with a quasibinomial error structure and logistic link function for proportional data. Statistical analyses were conducted in R v2.14 (R Development Core Team 2011).
Results
Recombination
Retroviruses are prone to homologous recombination, which can confound estimates of phylogenetic relatedness and evolutionary rates. We first evaluated the CrERVs for recombination. The PHI test for recombination revealed statistically significant evidence for recombination in the CrERV dataset (Φw = 0.101, P < 0.001). The best-fit model in GARD suggested four recombination breakpoints (Supplementary Figure S2; positions 1093, 1380, 1952, and 2808), which included portions of the pol and env genes. We removed this region (1715bp) from the alignment and reran the PHI-test on the remaining dispersed alignment. The final 1935bp alignment showed no evidence for recombination (Φw= 0.124, P = 0.124).
Phylogenetic Relationships and Insertion Dating
Tree topologies, approximate divergence time estimates, and posterior probabilities (PP) of tree nodes based on sequence data were consistent across two independent BEAST runs. Both runs converged and all ESS values for parameter estimates were greater than 200. The resulting phylogenetic tree representing the evolutionary history of CrERVs in mule deer revealed four clades with node PP greater than 0.50 (Figure 2). However, only two clades (Lineages II and IV) were supported (PP > 0.85). Lineages III and IV together also form a highly supported clade (PP = 0.98).
Figure 2.

Evolutionary history of retroviral elements in mule deer. Median time to most recent common ancestor (tMRCA) estimated under Yule process speciation model, relaxed clock, and TVM+Γ nucleotide substitution model. Support for nodes only shown if posterior probability >0.50, and CrERV lineages are designated I–IV. CrERVs were absent in white-tailed (WT) deer with the exception of CrERV-in14. Median estimates of node age (black diamonds) and 95% highest posterior probability densities shown in insert with color used to highlight corresponding lineage. Branch widths represent relative evolutionary rate with thicker branches undergoing more rapid evolution.
The time to most recent common ancestor (tMRCA) for all CrERVs was estimated as 0.74 MYA and the divergence between CrERV-in1and all other segregating CrERVs in mule deer was estimated as 0.65MYA (Figure 2). CrERV-in2, -in4, -in7, -in11 all likely integrated into the mule deer genome <25000 years ago (ya) and CrERV-in5, -in8, -in10, -in13 <80000 ya. The estimated mean nucleotide substitution rate over all viral lineages was 3.5×10–8.
Notable similarities were observed in the phylogeny based on the full CrERV sequence alignment that did not exclude the putative recombinant region (Supplementary Figure S3). There was congruence found among phylogenies in strong support (PP = 0.96–0.99) for the affiliation of CrERV-in7 and -in11 (Lineage IV; Figure 2) which were estimated to have integrated within the last 25000 years in both trees and are likely among the youngest CrERVs as they are also found in very low prevalence in the population (Table 1). Further similarities between phylogenies is the clustering of CrERV-in2, -in4, -in7, -in8, and -in11 which all share a common ancestor ~100000 years ago. This group of five viruses also share the attribute of having intact open reading frames in the two functional genes (pol and env) encoded in the 3′ portion of the gammaretroviral genomes, which supports that they have not been present in the mule deer genome for an extended period of time. In summary, our data indicate that CrERV colonization of the mule deer genome has occurred since speciation from white-tailed deer, with most integrating within the last 200000 years, and there are significant effects of recombination among the sampled viruses that confound estimates of the integration time.
CrERV Prevalence Among Mule Deer Populations
Of the 13 CrERVs evaluated for population prevalence in O. hemionus, all but CrERV-in1 were insertionally polymorphic. We found that the integrated viruses have variable prevalence and spatial structure (Table 1, Figure 3, Supplementary Figure S4). Five (CrERV-in1, -in2, -in3, -in4, -in5, -in9) were distributed widely and found in all populations including samples from UT. CrERV-in11 was low in frequency, but had a widespread distribution in MT. CrERV-in6 and -in10 were found in western MT, although a single individual in both UT and WY also had the CrERV-in6 insertion. CrERV-in8 and -in13 were extremely rare and localized to the northwest (population 5). CrERV-in7 was found in only four deer and also had a restricted distribution in northeastern MT where populations 2, 3, and 4 meet.
Figure 3.

Spatial distribution and prevalence of CrERVs. Prevalence of CrERVs from supported viral Lineages I–IV are represented as pie charts with population proportion of presence (gray/color) or absence (white) shown. CrERVs are color coded (online version only) to correspond with inferred lineage based on the phylogenetic analysis (see Figure 2). Pie chart size is proportional to sampling intensity in each population. Distributional maps for all CrERVs can be found in Supplementary Figure S4.
Clustering Analyses Based on CrERV Prevalence Data
Agglomerative hierarchical clustering was used to cluster closely related populations based on presence/absence frequency of CrERVs in each pre-defined “population” or sampling locality (Figure 1A,B). These results suggest clustering of the northeastern populations (1–3) with population 13 in WY (blue), clustering of southwestern populations (8 and 9; yellow), and clustering of the west-central populations (6 and 7) with population 11 (green). Disjunct clustering of populations 4, 10, and 12 (orange) was also observed. Populations 5, HLN, and UT were very different and did not cluster with any other population. Hierarchical clustering based on microsatellite allele frequencies, however, did not reveal a similar level of clustering as CrERVs (Figure 1C).
Cluster ensemble analyses identified four significant clusters (P < 0.005; Figure 4). Animals in cluster 1, 2, 3, and 4 share -in12, -in11, -in7, and -in10, respectively.
Figure 4.

Cluster ensemble results. Significance (P < 0.005) of clustering solution is shown by the purple line. For each pair of animals, the p value is determined as 1 minus the percentile of their similarity in the null distribution. Significant clusters (1–4) are highlighted by the primary CrERV determining the clustering solution (in parentheses), with the cluster color (blue, green or orange) corresponding to their lineage in Figure 2. Cluster branches are color-coded by geographical region (NE = blue; NW = red; W = orange; SW = yellow, SE = green; Utah = gray), with “populations” indicated by the numbers at the end of each branch. In some cases, multiple CrERVs determine the clustering solution as indicated by the arrows for specific nodes.
Population Differentiation and Clustering Analyses
Genetic differentiation was estimated to be low among the majority of population pairs for both microsatellite and CrERV datasets, with estimates often statistically indistinguishable from zero (Table 2). Exceptions included HLN and UT, both of which were significantly differentiated from the majority of the remaining populations (Table 2) There was also evidence from both marker types that population 5 (in northwest MT) was moderately differentiated. CrERV differentiation results are in agreement with results from our clustering analyses. We also found low, but significant global differentiation among populations based on microsatellites (ΦST = 0.023, P = 0.001; θ = 0.01, P = 0.001) and CrERVs (ΦST = 0.036, P < 0.001, F ST = 0.008, P = 0.003) and AMOVA results for both marker types showed that the majority of the variation (96–98%) is found within populations (Table 3). There was relatively little differentiation between populations within the same geographical region (μsats: ΦSC = 0.008, P = 0.018; CrERVs: ΦSC = 0.008, P = 0.268), but significant differentiation was observed among regions (μsats: ΦCT = 0.014, P = 0.002; CrERVs: ΦCT = 0.025, P = 0.025).
Table 2.
Population pairwise ΦST based on binary CrERV data (13 loci), shown above the diagonal, and based on microsatellites (14 loci), shown below the diagonal
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | HLN | UT | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | – | 0.000 | 0.000 | 0.000 | 0.058 | 0.005 | 0.007 | 0.000 | 0.000 | 0.000 | 0.000 | 0.009 | 0.000 | 0.048 | 0.025 |
| 2 | 0.004 | – | 0.000 | 0.000 | 0.081 | 0.008 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.072 | 0.082 |
| 3 | 0.012 | 0.007 | – | 0.018 | 0.040 | 0.000 | 0.008 | 0.000 | 0.002 | 0.009 | 0.000 | 0.033 | 0.000 | 0.078 | 0.108 |
| 4 | 0.008 | 0.007 | 0.007 | – | 0.109 | 0.000 | 0.000 | 0.038 | 0.017 | 0.000 | 0.000 | 0.000 | 0.000 | 0.083 | 0.060 |
| 5 | 0.040 | 0.022 | 0.019 | 0.041 | – | 0.004 | 0.047 | 0.048 | 0.032 | 0.109 | 0.052 | 0.087 | 0.055 | 0.136 | 0.214 |
| 6 | 0.040 | 0.037 | 0.008 | 0.021 | 0.005 | – | 0.000 | 0.016 | 0.012 | 0.027 | 0.000 | 0.014 | 0.000 | 0.098 | 0.153 |
| 7 | 0.018 | 0.012 | 0.002 | 0.014 | 0.000 | 0.000 | – | 0.000 | 0.015 | 0.000 | 0.000 | 0.000 | 0.006 | 0.083 | 0.136 |
| 8 | 0.055 | 0.014 | 0.018 | 0.026 | 0.014 | 0.000 | 0.009 | – | 0.000 | 0.000 | 0.000 | 0.000 | 0.034 | 0.035 | 0.093 |
| 9 | 0.013 | 0.000 | 0.022 | 0.008 | 0.022 | 0.027 | 0.010 | 0.017 | – | 0.007 | 0.000 | 0.000 | 0.007 | 0.059 | 0.043 |
| 10 | 0.024 | 0.031 | 0.005 | 0.012 | 0.023 | 0.003 | 0.002 | 0.009 | 0.011 | – | 0.000 | 0.000 | 0.020 | 0.033 | 0.054 |
| 11 | 0.008 | 0.004 | 0.000 | 0.010 | 0.002 | 0.000 | 0.000 | 0.008 | 0.000 | 0.000 | – | 0.016 | 0.000 | 0.014 | 0.064 |
| 12 | 0.014 | 0.012 | 0.021 | 0.016 | 0.047 | 0.020 | 0.023 | 0.001 | 0.003 | 0.011 | 0.015 | – | 0.040 | 0.107 | 0.108 |
| 13 | 0.000 | 0.000 | 0.000 | 0.000 | 0.023 | 0.016 | 0.008 | 0.030 | 0.003 | 0.007 | 0.000 | 0.015 | – | 0.071 | 0.071 |
| HLN | 0.050 | 0.043 | 0.043 | 0.037 | 0.044 | 0.028 | 0.020 | 0.011 | 0.044 | 0.049 | 0.051 | 0.030 | 0.050 | – | 0.056 |
| UT | 0.052 | 0.038 | 0.026 | 0.051 | 0.023 | 0.035 | 0.026 | 0.051 | 0.021 | 0.036 | 0.007 | 0.039 | 0.034 | 0.085 | – |
The same individuals (n= 259) were genotyped at both marker types. Significant values (P < 0.05) determined through 10000 permutations are indicated in bold. Negative ΦST values were converted to 0.
Table 3.
Analysis of molecular variance (AMOVA) results for mule deer, grouped into 15 populations and 6 regions
| df | SS | Estimated variance | % Total | Φ-Statistics | P valuea | |
|---|---|---|---|---|---|---|
| μsats (n= 259, 14 loci) | ||||||
| Among regions | 5 | 85.363 | 0.142 | 1.44 | Φ CT = 0.014 | 0.002 |
| Among populations within regions | 9 | 98.944 | 0.082 | 0.83 | Φ SC = 0.008 | 0.018 |
| Within populations | 244 | 2353.809 | 9.647 | 97.73 | Φ ST = 0.023 | 0.001 |
| CrERVs (n= 259, 13 loci) | ||||||
| Among regions | 5 | 16.20 | 0.040 | 2.85 | Φ CT = 0.028 | 0.025 |
| Among populations within regions | 9 | 13.89 | 0.011 | 0.77 | Φ SC = 0.008 | 0.268 |
| Within populations | 244 | 332.91 | 1.364 | 96.39 | Φ ST = 0.036 | <0.001 |
The same individuals (n= 259) were genotyped at both marker types. Genetic distance matrices based on Φ-statistics and conducted separately using microsatellite and CrERV data.
a P values determined through 10000 permutations of the data.
Marker Comparison of Population Diversity and Differentiation
Expected heterozygosity was relatively similar among populations for both CrERV and microsatellite data (Supplementary Table S5; HE = 0.67–0.73, Hj = 0.15–0.22). Microsatellite allelic diversity was also similar across populations, whereas the proportion of polymorphic loci (PLP) in CrERVs varied from 46% to 69%, with the PLP highest in the northwest (population 5, 69%) and the northeast (populations 1–3, 62%). The PLP was lowest (46%) in UT, population 4, the southeast (populations 12, 13), and in population 9. We found a significant relationship between microsatellite and CrERV pair-wise population genetic distance (β = 8.30±1.46, P < 0.0001; Supplementary Figure S5).
Discussion
In this study, we provide evidence that the mule deer genome has been repeatedly colonized by gammaretroviruses over the history of the species. Endogenous retroviruses have the potential to provide a unique view of a species population history because they integrate into the germ line of an individual host at a discrete time in the species history. As they are subsequently transmitted by vertical inheritance, they also mark recipients as sharing a common ancestry. Therefore, estimates of the time of integration coupled with frequency and distribution of the ERV can provide important information on host demography and host-viral evolutionary history.
A widely used approach for dating ERV integration events involves the examination of mutational differences between the two LTRs of the virus (Mager and Freeman 1995; Johnson and Coffin 1999). This approach is based on the assumption that viral 5′ and 3′ LTR sequences are identical at the time of integration and thereafter evolve separately at an empirical evolutionary rate. Thus, the sequence divergence between the 5′ and 3′ LTR and host substitution rate can be applied to estimate integration age (Johnson and Coffin 1999). Shortcomings of this fixed rate approach include the probable differences in evolutionary rates among homologous ERV loci or LTR regions (Martins and Villesen 2011). In our dataset only CrERV-in1 (Elleder et al. 2012) and -in14 (D. Elleder, unpublished data) had differences in the 5′ and 3′ LTR. Assuming a neutral evolutionary rate of 2.3 to 5×10–9 per site per year (Johnson and Coffin 1999; Waterston et al. 2002; Pace et al. 2008), the insertion age of CrERV-in1 was previously estimated to be 0.47–1 MYA (Elleder et al. 2012). Results from our phylogenetic analysis corroborated this LTR-based estimate, with the divergence of CrERV-in1 occurring approximately 0.65 MYA (95% HPD interval = 0.47–1.4 MYA).
Multiple lines of evidence suggest the CrERVγ proviruses examined in this study are derived from evolutionarily recent invasions of the mule deer genome. First, the absence of CrERVs (CrERV-in1 to -in13) in white-tailed deer is suggestive of genome integration times following the divergence of mule and white-tailed deer, which has been estimated to be between 0.6 and 2.9 MYA (Pitra et al. 2004; Gilbert et al. 2006; Hedges et al. 2006). Furthermore, our estimates of viral divergence times (which are equivalent to maximum CrERV endogenization times) suggest that most CrERVs integrated within the last 200000 years. This is considerably younger than ERVs identified in other vertebrate species to date. For example, many human ERVs are also present in Old World monkeys and apes, implying that retroviral colonization events occurred more than 25 MYA (Shih et al. 1991; Anderssen et al. 1997; Tristem 2000), although recent data indicate that the HERV-K family is still active in humans (Jha et al. 2011). In addition, with the exception of -in1 and -in14, CrERVs have identical 5′ and 3′ LTRs and were found to be segregating in the mule deer population, supporting the premise that endogenization events in mule deer are relatively recent. Observations of insertional polymorphism within a species have been rare, though documented among domestic breeds of cats (Roca et al. 2004, 2005), sheep (Arnaud et al. 2007; Chessa et al. 2009), and pigs (Mang et al. 2001), signifying that domestication may increase colonization of exogenous retroviruses or activation of existing ERVs. In contrast, ERV insertional polymorphism within a wildlife population has only previously been reported in koala, where a circulating retrovirus has integrated into the host genome over the past two centuries and is believed to be currently invading the germ line (Ávila-Arcos et al. 2013; Tarlinton et al. 2006). Finally, Elleder et al. (2012) documented transcriptional activity of the CrERVγ genome in mule deer lymph node tissue, suggesting that some integrations may be derived from either an active endogenous provirus or a currently circulating exogenous virus. Six of the proviruses evaluated here (-in2, -in4, -in7, -in8, -in11, -in13) share the attribute of having intact open reading frames in the two functional genes (pol and env) encoded in the 3′ portion of the gammaretroviral genome. Taken together, these observations suggest many of these proviruses can be considered to be contemporary and may continue to be infiltrating the mule deer genome.
The coupling of ERV prevalence and geographical distribution with viral evolutionary history increases the information available to explore the ancestral history of the host. Insertional polymorphisms are not subject to the biases inherent in other host genetic markers (e.g., homoplasy in microsatellites, mutational site saturation or convergence in SNPs) and, therefore, are identical by descent (Salem et al. 2005), directly reflect the relationships between individuals. They differ from these commonly used population genetic markers because animals sharing a virus at the same unique position in the genome form a defined ancestral lineage; each ERV provides different information on the host’s ancestral history. Animals that share multiple ERV integration sites share ancestry over the evolutionary time frame since the virus integrated. Therefore, an ERV integration that has occurred in the recent past will mark descendants that, in the case of a wildlife population, are more likely to be localized to a discrete geographical region than individuals sharing an older ERV who have dispersed since the time of integration. In this study, we demonstrated the value of CrERV insertional polymorphism data, showing it had greater resolution than microsatellites to detect geographic clustering of related deer (Figure 1).
Coupling the distribution of ancestrally related individuals with the ERV phylogeny and relative integration time can provide information about how individuals have dispersed over a period in time. The observation of a low frequency ERV within a host population could be explained by two alternatives: 1) a recent integration into the host or 2) an expansion of an older lineage due to immigration. Therefore, the limited presence and localized distributions of some CrERVs examined in this study (eg. -in7, -in8, -in10, -in13) may be due to either of these scenarios. By incorporating informative data on both viral phylogeny and structure with host spatial distribution, however, it is possible to make inferences that help distinguish between these hypotheses. For example, CrERV-in7 is inferred to be one of the youngest proviruses based on our phylogenetic analyses (Figure 2), while also rare and spreading locally in northeastern mule deer populations, providing support that this integration is likely of recent evolutionary origin. In contrast, three animals from western MT (two from population 5 in the north and one from population 9 in the south) cluster together because they share -in10, which is rare in the population but may be an older integration. The age and geographic distribution of animals harboring this virus suggest that it may have been introduced into the MT population due to migration or translocation events from populations where -in10 prevalence is higher. We acknowledge that our data was limited in sampling scope and that accurate assessment of CrERV frequency in mule deer would require broader sampling from the species’ distribution, a task that we are actively pursuing.
The detection of recombination within the coding region of CrERVs sampled in this study is particularly interesting. Recombination typically occurs between the two identical LTRs of a single ERV resulting in a solo LTR. The rate at which this occurs is reported to be higher for young ERV because the probability of recombination is highest when the LTRs are identical (Belshaw et al. 2007). Thus, as ERVs age in a genome, recombinant parents can be lost due to homologous recombination; the loss of parental sequences confounds detection of intergenic recombinant ERVs. This finding is important because although recombination events could be confined to viral segments, they can also lead to structural changes in the mule deer genome. A thorough analysis of host sites flanking recombinant viruses could reveal such genomic structural variation.
Previous studies found high levels of genetic diversity throughout the range of O. hemionus, with intraspecific divergences greater than between some genera (Cronin 1991; Latch et al. 2009). However, below the subspecies designation, very little population structure has previously been reported in deer (Scribner et al. 1991;Latch et al. 2009; Pease et al. 2009; Powell et al. 2013). Here, we similarly found high genetic diversity across populations in MT and low levels of genetic divergence between population pairs using standard population genetic indices. Global F ST from CrERVs (F ST = 0.008) and microsatellites (θ = 0.01) was low, but significant, and comparable to differentiation results previously reported in mule deer (F ST = 0.008; Cullingham et al. 2011). However, novel clustering analyses based on CrERV prevalence data revealed some degree of regional structuring. In particular, deer from UT, northwestern MT, and those residing in the city of HLN appeared to be different from other MT populations, results that were further corroborated with standard genetic differentiation indices (F ST).
Clustering ensemble analyses also indicated spatial localization of recent viruses, specifically in deer from northeastern and western MT. For example, 11 of the 13 animals in cluster 1 are from the east side of the continental divide, primarily from northeastern MT (populations 1–4). Cluster 3 included five animals from populations 2–4 in northeastern MT that shared a low frequency CrERV (-in7) which integrated within the last 30000 years. Cluster 4 includes only three individuals from western MT which share -in10, two of which are localized to the northwestern region (population 5). Cluster 2, however, is defined by more complex shared CrERV history, with all animals sharing -in11, which is closely related to -in7 but has a broader geographic distribution. In general, the 11 animals within this cluster are located in western MT with the 3 animals from regions in north central and southeastern MT, potentially reflecting translocations of animals. Only one animal from UT and none of the deer from the HLN herd or from north central WY were represented in these four strongly supported clusters.
Relatively lower connectivity between the northwest and the other regions in MT may be due to landscape features acting as physical barriers and/or differences in habitat selection preferences and behavior of deer in that region as compared to the surrounding regions. It is also possible that there is gene flow between the northwestern population and neighboring regions in northern Idaho or Canada, which is worthy of future investigation. The difference between urban deer from HLN and the other populations investigated here is intriguing. This result is unlikely due to a bias caused by sampling a highly related population because the inbreeding coefficient, F IS, is not significantly different from 0 and genetic diversity indices were comparable to other populations. In fact, CrERV heterozygosity was highest in HLN in comparison to all other populations, which would be unexpected given the sampling of this population covered the least area (Supplementary Figure S1).
Translocation events occurred repeatedly between areas of MT in the 1940s and 1950s for the purpose of restocking and augmenting depleted mule deer populations (Picton and Lonner 2008). In total, more than 1750 deer were translocated, by both the Montana Fish and Game Department and US Fish and Wildlife Service. Thus, apparent connectivity between disjunct populations, as indicated in our population clustering analyses (orange and green clusters; Figure 1A, B) may be a signature of these past translocation events, a hypothesis that is congruent with specific records of deer movement across the state. For example, approximately 1000 deer were moved from the National Bison Range (population 7) to supplement and restore other deer populations in the state, with approximately half of these deer moved to Golden Valley and Musselshell counties (between populations 4 and 11). Together, the pattern of population clustering observed here is consistent with the fact that the majority of movements occurred from west (populations 7 and 10) to east (locations near populations 4, 11, 12). Populations in the northeastern part of the state clustered with deer in northeast WY, and although not contiguous, these populations may have retained a genetic signature of remnant populations pre-existing prior to translocation events as there are no recorded translocations of mule deer into these areas. This is also corroborated by apparent high gene flow, indicated by low pairwise F ST values, between the regions.
Our research demonstrates the dynamic nature of a newly described ERV in the mule deer population. The viruses used in this study were not selected based on age or frequency in the population, in comparison to microsatellites, which are typically selected based on their variability. Despite this, our analyses demonstrate that CrERVs provide valuable information on mule deer population structure. We also found that both CrERV and microsatellite markers are capable of indicating similar population genetic distances; however, F ST values do not change at a similar rate. This is expected due to the differences in the nature of these marker types, with CrERVs having greater resolution to detect population differences as demonstrated by our clustering results. Further, because it is now possible to comprehensively sample integration site diversity of mobile elements (Iskow et al. 2010), there is the potential to employ hundreds of CrERVs to study host population biology. Coupled with information on relative CrERV age and diversity, these data could provide novel perspectives of historical and contemporary mule deer evolution and population history such as changes in migration routes and population mixing. Such data would be informative to management authorities to understand historical responses to environmental changes and to detect possible disease transmission routes among populations.
Supplementary Material
Supplementary material can be found at http://www.jhered.oxfordjournals.org/.
Funding
US Geological Survey (06HQAG0131); National Center for Advancing Translational Sciences (UL1TR000127), RAPIDD program of the Science and Technology Directorate; Czech Ministry of Education, Youth and Sports (LK11215); United States Department of Homeland Security; and Fogarty International Center at the National Institutes of Health.
Supplementary Material
Acknowledgments
We thank Abinash Padhi for technical assistance, Yee Ling Chong for help with figures, Beth Shapiro for insightful discussions, and two anonymous reviewers for helpful comments on the manuscript. We are grateful to Montana Fish, Wildlife and Parks (MFWP) regional biologists statewide and the MFWP Wildlife Lab for support in sample collection at hunter check stations and the hunters who voluntarily provided access to their harvested deer. We also would like to specifically thank Neil Anderson (MFWP), Jenny Sika (MFWP) and Leslie McFarlane (Utah Division of Wildlife Resources) for providing samples from Montana, Helena and Utah, respectively, and Mike Ebinger (Montana State University) for help with figures as well as coordinating and obtaining the samples used in this study. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government.
References
- Anderson A, Wallmo O. 1984. Odocoileus hemionus. Mammalian Species. 219:1–9 [Google Scholar]
- Anderssen S, Sjottem E, Svineng G, Johansen T. 1997. Comparative analyses of LTRs of the ERV-H family of primate-specific retrovirus-like elements isolated from marmoset, African green monkey, and man. Virology. 234:14–30 [DOI] [PubMed] [Google Scholar]
- Arnaud F, Caporale M, Varela M, Biek R, Chessa B, Alberti A, Golder M, Mura M, Zhang Y-p, Yu L, et al. 2007. A paradigm for virus-host coevolution: Sequential counter-adaptations between endogenous and exogenous retroviruses. Plos Pathogens. 3:1716–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ávila-Arcos MC, Ho SYW, Ishida Y, Nikolaidis N, Tsangaras K, Hönig K, Medina R, Rasmussen M, Fordyce SL, Calvignac-Spencer S, et al. 2013. One hundred twenty years of koala retrovirus evolution determined from museum skins. Mol Biol Evol. 30:299–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad MJ, Wooding S, Watkins WS, Ostler CT, Batzer MA, Jorde LB. 2003. Human population genetic structure and inference of group membership. Am J Hum Genet. 72:578–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belshaw R, Watson J, Katzourakis A, Howe A, Woolven-Allen J, Burt A, Tristem M. 2007. Rate of recombinational deletion among human endogenous retroviruses. J Virol. 81:9437–9442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop MD, Kappes SM, Keele JW, Stone RT, Sunden SLF, Hawkins GA, Toldo SS, Fries R, Grosz MD, Yoo JY, et al. 1994. A genetic linkage map for cattle. Genetics. 136:619–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruen TC, Philippe H, Bryant D. 2006. A simple and robust statistical test for detecting the presence of recombination. Genetics. 172:2665–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Xu SX, Zhou KY, Yang G. 2011. Whale phylogeny and rapid radiation events revealed using novel retroposed elements and their flanking sequences. BMC Evol Biol. 11:314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chessa B, Pereira F, Arnaud F, Amorim A, Goyache F, Mainland I, Kao RR, Pemberton JM, Beraldi D, Stear MJ, et al. 2009. Revealing the history of sheep domestication using retrovirus integrations. Science. 324:532–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin MA. 1991. Mitochondrial DNA phylogeny of deer (Cervidae). J Mammal. 72:553–566 [Google Scholar]
- Cullingham CI, Nakada SM, Merrill EH, Bollinger TK, Pybus MJ, Coltman DW. 2011. Multiscale population genetic analysis of mule deer (Odocoileus hemionus hemionus) in western Canada sheds new light on the spread of chronic wasting disease. Can J Zool. 89:134–147 [Google Scholar]
- DeWoody JA, Honeycutt RL, Skow LC. 1995. Microsatellite markers in white-tailed deer. J Heredity. 86:317–319 [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Ashton B, Buxton S, Cheung M, Cooper A, Heled J, Kearse M, Moir R, Stones-Havas S, Sturrock S, et al. 2011. Geneious v5.5 Available from: http://www.geneious.com
- Drummond AJ, Ho SYW, Phillips MJ, Rambaut A. 2006. Relaxed phylogenetics and dating with confidence. Plos Biol. 4:699–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 7:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. 1998. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 95:14863–14868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elleder D, Oekyung K, Abinash P, Bankert JG, Simeonov I, Schuster SC, Wittekindt NE, Motameny S, Poss M. 2012. Polymorphic integrations of an endogenous gammaretrovirus in the mule deer genome. J Virol. 86:2787–2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Lischer HEL. 2010. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 10:564–567 [DOI] [PubMed] [Google Scholar]
- Excoffier L, Smouse PE, Quattro JM. 1992. Analysis of molecular variance inferred from metric distances among DNA haplotypes—Application to human mitochondrial DNA restriction data. Genetics. 131:479–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feschotte C, Gilbert C. 2012. Endogenous viruses: Insights into viral evolution and impact on host biology. Nat Rev Genet. 13:283–296 [DOI] [PubMed] [Google Scholar]
- Gernhard T. 2008. The conditioned reconstructed process. J Theor Biol. 253:769–778 [DOI] [PubMed] [Google Scholar]
- Gifford RJ, Katzourakis A, Tristem M, Pybus OG, Winters M, Shafer RW. 2008. A transitional endogenous lentivirus from the genome of a basal primate and implications for lentivirus evolution. Proc Natl Acad Sci USA. 105:20362–20367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert C, Feschotte C. 2010. Genomic fossils calibrate the long term evolution of Hepadnaviruses. Plos Biol. 8:e1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert C, Ropiquet A, Hassanin A. 2006. Mitochondrial and nuclear phylogenies of Cervidae (Mammalia, Ruminantia): Systematics, morphology, and biogeography. Mol Phylogenet Evol. 40:101–117 [DOI] [PubMed] [Google Scholar]
- Goudet J. 1995. FSTAT (Version 1.2): A computer program to calculate F-statistics. J Heredity. 86:485–486 [Google Scholar]
- Guo SW, Thompson EA. 1992. Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics. 48:361–372 [PubMed] [Google Scholar]
- Handsaker RE, Korn JM, Nemesh J, McCarroll SA. 2011. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat Genet. 43:269- U126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Kishino H. 1989. Confidence-limits on the maximum-likelihood estimate of the Hominoid tree from mitochondrial-DNA sequences. Evolution. 43:672–677 [DOI] [PubMed] [Google Scholar]
- Hedges SB, Dudley J, Kumar S. 2006. TimeTree: A public knowledge-base of divergence times among organisms. Bioinformatics. 22:2971–2972 [DOI] [PubMed] [Google Scholar]
- Huff DR, Peakall R, Smouse PE. 1993. RAPD variation within and among natural populations of outcrossing buffalograss [Buchloe dactyloides (Nutt.) Engelm]. Theor Appl Genet. 86:927–934 [DOI] [PubMed] [Google Scholar]
- Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 23:254–267 [DOI] [PubMed] [Google Scholar]
- Iskow RC, McCabe MT, Mills RE, Torene S, Pittard WS, Neuwald AF, Van Meir EG, Vertino PM, Devine SE. 2010. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell. 141:1253-–U268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha AR, Nixon DF, Rosenberg MG, Martin JN, Deeks SG, Hudson RR, Garrison KE, Pillai SK. 2011. Human endogenous retrovirus K106 (HERV-K106) was infectious after the emergence of anatomically modern humans. Plos One. 6:e20234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Coffin JM. 1999. Constructing primate phylogenies from ancient retrovirus sequences. Proc Natl Acad Sci USA. 96:10254–10260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KC, Levine KF, Banks JD. 2000. DNA-based genetic markers in black-tailed and mule deer for forensic applications. Cal Fish Game. 86:115–126 [Google Scholar]
- Kalinowski ST. 2005. HP-RARE 1.0: A computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes. 5:187–189 [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30:3059–3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai K, Nikaido M, Harada M, Matsumura S, Lin LK, Wu Y, Hasegawa M, Okada N. 2002. Intra- and interfamily relationships of vespertilionidae inferred by various molecular markers including SINE insertion data. J Mol Evol. 55:284–301 [DOI] [PubMed] [Google Scholar]
- Keckesova Z, Ylinen LMJ, Towers GJ, Gifford RJ, Katzourakis A. 2009. Identification of a RELIK orthologue in the European hare (Lepus europaeus) reveals a minimum age of 12 million years for the lagomorph lentiviruses. Virology. 384:7–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishino H, Hasegawa M. 1989. Evaluation of the maximum-likelihood estimate of the evolutionary tree topologies from DNA-sequence data, and the branching order in Hominoidea. J Mol Evol. 29:170–179 [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. 2001. Initial sequencing and analysis of the human genome. Nature. 409:860–921 [DOI] [PubMed] [Google Scholar]
- Latch EK, Heffelfinger JR, Fike JA, Rhodes OE., Jr 2009. Species-wide phylogeography of North American mule deer (Odocoileus hemionus): Cryptic glacial refugia and postglacial recolonization. Mol Ecol. 18:1730–1745 [DOI] [PubMed] [Google Scholar]
- Li J, Han K, Xing JC, Kim HS, Rogers J, Ryder OA, Disotell T, Yue BS, Batzer MA. 2009. Phylogeny of the macaques (Cercopithecidae: Macaca) based on Alu elements. Gene. 448:242–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liggins L, Chapple DG, Daugherty CH, Ritchie PA. 2008. A SINE of restricted gene flow across the Alpine Fault: Phylogeography of the New Zealand common skink (Oligosoma nigriplantare polychroma). Mol Ecol. 17:3668–3683 [DOI] [PubMed] [Google Scholar]
- Lynch M, Milligan BG. 1994. Analysis of population genetic structure with RAPD markers. Mol Ecol. 3:91–99 [DOI] [PubMed] [Google Scholar]
- Mackie RJ, Kie JG, Pac DF, Hamlin KL. 2003. Mule deer. In: Feldhamer GA, Thompson BC, Chapman JA, editors. Wild mammals of North America. Baltimore, MD: The John Hopkins University Press; p. 889–905 [Google Scholar]
- Mackie RJ, Pac DF, Hamlin KL, Dusek GL. 1998. Ecology and management of mule deer and white-tailed deer in Montana. Montana Fish, Wildlife and Parks. Helena, MT: Federal Aid Project W-120-R [Google Scholar]
- Mager DL, Freeman JD. 1995. HERV-H endogenous retroviruses: Presence in the New World branch but amplification in the Old World primate lineage. Virology. 213:395–404 [DOI] [PubMed] [Google Scholar]
- Mang R, Maas J, Chen X, Goudsmit J, van der Kuyl AC. 2001. Identification of a novel type C porcine endogenous retrovirus: Evidence that copy number of endogenous retroviruses increases during host inbreeding. J Gen Virol. 82:1829–1834 [DOI] [PubMed] [Google Scholar]
- Martin DP, Lemey P, Posada D. 2011. Analysing recombination in nucleotide sequences. Mol Ecol Resour. 11:943–955 [DOI] [PubMed] [Google Scholar]
- Martins H, Villesen P. 2011. Improved integration time estimation of endogenous retroviruses with phylogenetic data. Plos One. 6:e14745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLain AT, Meyer TJ, Faulk C, Herke SW, Oldenburg JM, Bourgeois MG, Abshire CF, Roos C, Batzer MA. 2012. An alu-based phylogeny of Lemurs (Infraorder: Lemuriformes). Plos One. 7:e44035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer TJ, McLain AT, Oldenburg JM, Faulk C, Bourgeois MG, Conlin EM, Mootnick AR, de Jong PJ, Roos C, Carbone L, et al. 2012. An Alu-based phylogeny of gibbons (Hylobatidae). Mol Biol Evol. 29:3441–3450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido M, Piskurek O, Okada N. 2007. Toothed whale monophyly reassessed by SINE insertion analysis: The absence of lineage sorting effects suggests a small population of a common ancestral species. Mol Phylogenet Evol. 43:216–224 [DOI] [PubMed] [Google Scholar]
- Pace JK, II, Gilbert C, Clark MS, Feschotte C. 2008. Repeated horizontal transfer of a DNA transposon in mammals and other tetrapods. Proc Natl Acad Sci USA. 105:17023–17028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page RDM. 1996. TreeView: An application to display phylogenetic trees on personal computers. Comput Appl Biosci. 12:357–358 [DOI] [PubMed] [Google Scholar]
- Peakall R, Smouse PE. 2006. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes. 6:288–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peakall R, Smouse PE, Huff DR. 1995. Evolutionary implications of allozyme and RAPD variation in diploid populations of dioecious buffalograss Buchloe dactyloides . Mol Ecol. 4:135–147 [Google Scholar]
- Pease KM, Freedman AH, Pollinger JP, McCormack JE, Buermann W, Rodzen J, Banks J, Meredith E, Bleich VC, Schaefer RJ, et al. 2009. Landscape genetics of California mule deer (Odocoileus hemionus): The roles of ecological and historical factors in generating differentiation. Mol Ecol. 18:1848–1862 [DOI] [PubMed] [Google Scholar]
- Picton HD, Lonner TN. 2008. Montana’s Wildlife Legacy: Decimation to Restoration. Bozeman, MT: Media Works Publishing [Google Scholar]
- Pitra C, Fickel J, Meijaard E, Groves PC. 2004. Evolution and phylogeny of old world deer. Mol Phylogenet Evol. 33:880–895 [DOI] [PubMed] [Google Scholar]
- Pond SLK, Posada D, Gravenor MB, Woelk CH, Frost SDW. 2006. GARD: A genetic algorithm for recombination detection. Bioinformatics. 22:3096–3098 [DOI] [PubMed] [Google Scholar]
- Posada D. 2008. jModelTest: Phylogenetic model averaging. Mol Biol Evol. 25:1253–1256 [DOI] [PubMed] [Google Scholar]
- Posada D, Crandall KA. 2002. The effect of recombination on the accuracy of phylogeny estimation. J Mol Evol. 54:396–402 [DOI] [PubMed] [Google Scholar]
- Powell JH, Kalinowski ST, Higgs MD, Ebinger MR, Vu NV, Cross PC. 2013. Microsatellites indicate minimal barriers to mule deer Odocoileus hemionus dispersal across Montana, USA. Wildlife Biol. 19:102–110 [Google Scholar]
- R Development Core Team 2011. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Rambaut A. 2009. FigTree v1.3 [Internet]. [cited 2012. Nov 9]. Available from: http://tree.bio.ed.ac.uk/software/figtree
- Rambaut A, Drummond AJ. 2007. Tracer v1.4 Available from: http://beast.bio.ed.ac.uk/Tracer
- Ray DA. 2007. SINEs of progress: Mobile element applications to molecular ecology. Mol Ecol. 16:19–33 [DOI] [PubMed] [Google Scholar]
- Raymond M, Rousset F. 1995. GENEPOP (Version 1.2) - Population genetics software for exact tests and ecumenicism. J Heredity. 86:248–249 [Google Scholar]
- Roca AL, Nash WG, Menninger JC, Murphy WJ, O’Brien SJ. 2005. Insertional polymorphisms of endogenous feline leukemia viruses. J Virol. 79:3979–3986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca AL, Peacon-Slattery J, O’Brien SJ. 2004. Genomically intact endogenous feline leukemia viruses of recent origin. J Virol. 78:4370–4375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset F. 2008. GENEPOP ‘ 007: A complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour. 8:103–106 [DOI] [PubMed] [Google Scholar]
- Salem AH, Ray DA, Batzer MA. 2005. Identity by descent and DNA sequence variation of human SINE and LINE elements. Cytogen Genome Res. 108:63–72 [DOI] [PubMed] [Google Scholar]
- Salem AH, Ray DA, Xing J, Callinan PA, Myers JS, Hedges DJ, Garber RK, Witherspoon DJ, Jorde LB, Batzer MA. 2003. Alu elements and hominid phylogenetics. Proc Natl Acad Sci USA. 100:12787–12791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Yasukawa Y, Takahashi K, Miura S, Shedlock AM, Okada N. 2006. Extensive morphological convergence and rapid radiation in the evolutionary history of the family Geoemydidae (old world pond turtles) revealed by SINE insertion analysis. Syst Biol. 55:912–927 [DOI] [PubMed] [Google Scholar]
- Schierup MH, Hein J. 2000. Consequences of recombination on traditional phylogenetic analysis. Genetics. 156:879–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scribner KT, Smith MH, Garrott RA, Carpenter LH. 1991. Temporal, spatial, and age-specific changes in genotypic composition of mule deer. J Mammal. 72:126–137 [Google Scholar]
- Shih A, Coutavas EE, Rush MG. 1991. Evolutionary implications of primate endogenous retroviruses. Virology. 182:495–502 [DOI] [PubMed] [Google Scholar]
- Strehl A, Ghosh J. 2002. Cluster ensembles—A knowledge reuse framework for combining multiple partitions. J Mach Learn Res. 3:583–617 [Google Scholar]
- Takahashi K, Terai Y, Nishida M, Okada N. 1998. A novel family of short interspersed repetitive elements (SINEs) from cichlids: The patterns of insertion of SINEs at orthologous loci support the proposed monophyly of four major groups of cichlid fishes in Lake Tanganyika. Mol Biol Evol. 15:391–407 [DOI] [PubMed] [Google Scholar]
- Tarlinton RE, Meers J, Young PR. 2006. Retroviral invasion of the koala genome. Nature. 442:79–81 [DOI] [PubMed] [Google Scholar]
- Taruscio D, Mantovani A. 2004. Factors regulating endogenous retroviral sequences in human and mouse. Cytogen Genome Res. 105:351–362 [DOI] [PubMed] [Google Scholar]
- Tristem M. 2000. Identification and characterization of novel human endogenous retrovirus families by phylogenetic screening of the Human Genome Mapping Project database. J Virol. 74:3715–3730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Loo W, Abrantes J, Esteves PJ. 2008. Sharing of endogenous lentiviral gene fragments among leporid lineages separated for more than 12 million years. J Virol. 83:2386–2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vekemans X. 2002. AFLP-SURV version 1.0. Distributed by the author. Laboratoire de Génétique et Ecologie Végétale. Belgium: Université Libre de Bruxelles [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature. 420:520–562 [DOI] [PubMed] [Google Scholar]
- Watkins WS, Rogers AR, Ostler CT, Wooding S, Bamshad MJ, Brassington AME, Carroll ML, Nguyen SV, Walker JA, Prasad BVR, et al. 2003. Genetic variation among world populations: Inferences from 100 Alu insertion polymorphisms. Genome Res. 13:1607–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir BS, Cockerham CC. 1984. Estimating F-statistics for the analysis of population structure. Evolution. 38:1358–1370 [DOI] [PubMed] [Google Scholar]
- Wilson GA, Strobeck C, Wu L, Coffin JW. 1997. Characterization of microsatellite loci in caribou Rangifer tarandus, and their use in other artiodactyls. Mol Ecol. 6:697–699 [DOI] [PubMed] [Google Scholar]
- Witherspoon DJ, Marchani EE, Watkins WS, Ostler CT, Wooding SP, Anders BA, Fowlkes JD, Boissinot S, Furano AV, Ray DA, et al. 2006. Human population genetic structure and diversity inferred from polymorphic L1 (LINE-1) and Alu insertions. Hum Heredity. 62:30–46 [DOI] [PubMed] [Google Scholar]
- Witherspoon DJ, Zhang Y, Xing J, Watkins WS, Ha H, Batzer MA, Jorde LB. 2013. Mobile element scanning (ME-Scan) identifies thousands of novel Alu insertions in diverse human populations. Genome Res. 23:1170–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittekindt NE, Padhi A, Schuster SC, Qi J, Zhao FQ, Tomsho LP, Kasson LR, Packard M, Cross P, Poss M. 2010. Nodeomics: Pathogen detection in vertebrate lymph nodes using meta-transcriptomics. Plos One. 5:e13432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Wang H, Zhang Y, Ray DA, Tosi AJ, Disotell TR, Batzer MA. 2007. A mobile element-based evolutionary history of guenons (tribe Cercopithecini). BMC Biol. 5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhivotovsky LA. 1999. Estimating population structure in diploids with multilocus dominant DNA markers. Mol Ecol. 8:907–913 [DOI] [PubMed] [Google Scholar]
- Zichner T, Garfield DA, Rausch T, Stuetz AM, Cannavo E, Braun M, Furlong EEM, Korbel JO. 2013. Impact of genomic structural variation in Drosophila melanogaster based on population-scale sequencing. Genome Res. 23:568–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.