
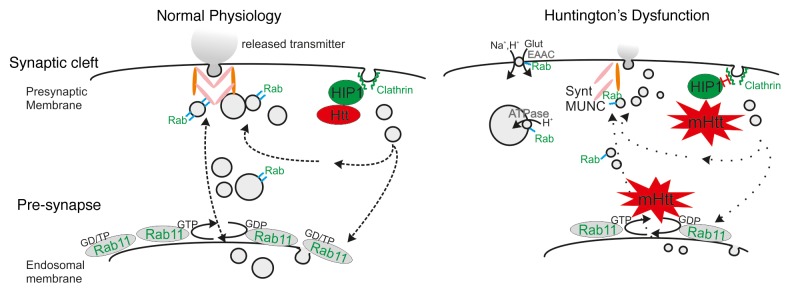
Figure 1. Model of impaired neurotransmission caused by mutant HTT. Mutant HTT leads to reduced synaptic vesicle size (right) via impaired Rab11 activity and altered interactions with HIP1, causing reduced neurotransmitter release and synaptic dysfunction. Overexpression of Rab11 may reverse these defects by several mechanisms: 1) Rab11 modulates vesicle size via enhancing proton (H+) ATPase trafficking. Rab11 has been shown to interact with the ε subunit of the vacuolar-type H+-ATPase and a possible enhanced interaction with the vesicular H+-ATPase could alleviate the mutant HTT-induced synaptic deficiency. 2) Huntington’s disease (HD) has been associated with lower cell surface expression of the glutamate/cysteine transporter EAAC141 that regulates glutamate uptake, potentially leaving the neuron with limited glutamate resources. Expression of a dominant-active Rab11 mutant in primary HD mouse neurons rectified this deficit. 3) Rab3, Rab5, and Rab11 are present at synaptic vesicles, providing a potential mechanism for direct interaction with the vesicle recycling machinery. Rab GTPases also participate in vesicle tethering, docking, and fusion events via association with v-SNARE and/or t-SNARE proteins forming trans-SNARE complexes, thereby organizing fusion competent microdomains. EAAC, glutamate/cysteine transporter; HIP, Huntingtin Interacting Protein; Glut, Glutamate; Synt, Syntaxin; Rab/Rab11, possible functions of various Rab proteins