

Abstract
Background
Vibrio cholerae is a human intestinal pathogen and V. cholerae of the O139 serogroups are responsible for the current epidemic cholera in China. In this work, we reported the whole genome sequencing of a V. cholerae O139 strain E306 isolated from a cholera patient in the 306th Hospital of PLA, Beijing, China.
Results
We obtained the draft genome of V. cholerae O139 strain E306 with a length of 4,161,908 bps and mean G + C content of 47.7%. Phylogenetic analysis indicated that strain E306 was very close to another O139 strain, V. cholerae MO10, which was isolated during the cholera outbreak in India and Bangladesh. However, unlike MO10, strain E306 harbors the El Tor-specific RS1 element with no pre-CTX prophage (VSK), very similar to those found in some V. cholerae O1 strains. In addition, strain E306 contains a SXT/R391 family integrative conjugative element (ICE) similar to ICEVchInd4 and SXT MO10, and it carries more antibiotic resistance genes than other closest neighbors.
Conclusions
The genome sequence of the V. cholerae O139 strain E306 and its comparative analysis with other V. cholerae strains we present here will provide important information for a better understanding of the pathogenicity of V. cholerae and their molecular mechanisms to adapt different environments.
Keywords: Cholera toxin prophage, Integrative conjugative elements, Antibiotic resistance genes
Background
Vibrio cholerae is a primary causative agent of life threatening diarrheal disease, cholera. Based on the somatic O antigens, more than two hundred serogroups of V. cholerae have been identified [1], among which O1 and O139 are recognized as the two major agents for cholera epidemics. V. cholerae serogroup O1 has two biotypes and is the causative agent for the previous two cholera pandemics, in which the classical biotype was dominant in the 6th pandemic and the El Tor in the 7th [2]. In 1992, a new non-O1 strain of V. cholerae, designated as serogroup O139 was identified in an epidemic cholera in India and Bangladesh [3,4]. Since then, V. cholerae O139 has been frequently isolated in other Asian countries where the cholera epidemics have occurred. In China, V. cholerae O139 strains are the dominant contributors in cholera and have been continually isolated since it first appeared in 1993 [5].
Previous studies have identified that the major virulence of V. cholerae O1/O139 is encoded by a lysogenic bacteriophage (CTX prophage) integrated in the V. cholerae genome. Many other genetic elements, such as the toxin-linked cryptic (TLC), the RS1 element, and the pre-CTX prophage (VSK), are also known to be adjacent to the CTX prophage [6]. The CTX prophage in toxigenic V. cholerae is usually consists of two gene clusters, the core and the RS2 regions, which are functionally different [7]. The core region includes the ctxAB genes encoding cholera toxin (CT), and five other genes encoding necessary components for phage morphogenesis. The RS2 region encodes proteins involved in phage replication (RstA), integration (RstB) and regulation of site-specific recombination (RstR). Another noteworthy element in V. cholerae is the SXT/R391 family integrative conjugative element (ICE) which was first identified in a V. cholerae O139 clinical isolation in 1993 [8]. The SXT/R391 ICE in V. cholerae usually contributes to the resistance phonotype of V. cholerae, encoding resistance to several antibiotics like sulfamethoxazole and trimethoprim that had previously been used for cholera treatment.
Though great efforts have been made to understand and to control this pathogen in the past, cholera caused by V. cholerae is still occasionally outbreak in recent years [9-11]. To date, 9 complete and nearly 200 draft genomes of V. cholerae are accessible in the NCBI genome projects. However, to demonstrate the evolution and the adaption mechanism of this pathogen, detailed analysis of the genomic diversity of new clinical isolations appeared in different areas and time scales is undoubtedly needed. Here, we report the genome sequence of a V. cholerae O139 strain E306 we recently isolated from a cholera patient in Beijing, China. The genome here will shed light on the understanding of the endemicity of cholera in North China.
Methods
Strain isolation
V. cholerae O139 strain E306 was isolated from the stool sample of a cholera case in Beijing, China, on May 30, 2013. After enrichment by alkaline peptone broth, the strain was identified as O139 serogroup by combining the results of its 16S rRNA gene sequence, serum agglutination test and biochemical reaction (Vitek 2 compact, BioMerieux Corp.). This research was approved by the Research Ethics Committee of the Institute of Microbiology, Chinese Academy of Sciences, and informed consent was obtained from the patient. The strain we reported here is available in The 306th Hospital of PLA, Beijing, China.
Genome sequencing
The whole genome was sequenced using shotgun sequencing strategy on Illumina Genome Analyser platform. DNA Library was constructed by using the TruSeq sample preparation kit according to the manufacturer's instructions. Briefly, genomic DNA was sheared by sonication and was then end repaired. After adapters’ ligation (pair-end) with the TA cloning method, the resulting DNA fragments were size selected on a 2% agarose gel. The final DNA library was produced by PCR amplification of the selected ligation products in length of ~500 bp. DNA library (5 pM) was then loaded onto the sequencing chip; clusters were generated by using the Illumina cluster generation kit. After sequencing, image analysis and base calling were carried out by using the Illumina GA Pipeline software. Finally, a total of 6,112,322 pair-end reads were generated.
Genome assembly and annotation
The pair-end raw sequences were quality filtered by using the DynamicTrim and LengthSort Perl scripts provided in SolexaQA suite [12]. After filtering, short reads were assembled by using SOAPdenovo (http://soap.genomics.org.cn) and the gaps were closed by using SOAP GapCloser (http://soap.genomics.org.cn). Glimmer 3.02 [13] was used for prediction of open reading frames, while tRNAscan-SE [14] and RNAmmer [15] were used for tRNA and rRNA identification, respectively. The genome was further annotated with the help of the RAST program (Rapid Annotation using Subsystem Technology) [16]. The annotation results were then checked through comparisons with the databases of NCBI-NR (http://www.ncbi.nlm.nih.gov/), COG [17], and KEGG [18]. For searching the antibiotic resistance genes, the protein-coding sequences were further Blast against Antibiotic Resistance Database (ARDB) [19], using similarity thresholds as recommended in ARDB.
Comparative genomics
For comparative analysis, reference genome sequences of the closest genetic relatives of V. cholerae O139 strain E306 and representative strains belonging to important serogroups including V. cholerae O1 biovar El Tor str. N16961 (GenBank accession number AE003852 and AE003853), B33 (ACHZ00000000), V. cholerae RC9 (ACHX00000000), V. cholerae MO10 (AAKF03000000), V. cholerae MJ-1236 (CP001485 and CP001486), V. cholerae O1 classical O395 (CP000626 and CP000627), V. cholerae CIRS101 (ACVW00000000), V. cholerae IEC224 (CP003330 and CP003331), and V. cholerae O1 str. 2010EL-1786 (CP003069 and CP003070) were downloaded from the NCBI website. Whole-genome alignments and SNP identification were performed by using Progressive Mauve [20]. Concatenated SNPs in length of 23,648 bp were used to calculate the genetic distances, and a phylogenetic tree was constructed by using the neighbor-joining method in MEGA5 [21] based on these SNPs. The stability of the phylogenetic relationships was assessed by bootstrapping (1000 replicates). BWA alignment tool [22] and SAMTools [23] for SNP calling were also used for confirming the results. The genome similarities based on phylogenomic distances were analyzed using the Gegenees software [24].
Quality assurance
The genomic DNA used for sequencing was isolated from pure culture of V. cholerae O139 strain E306. The 16S rRNA gene from the draft genome sequence was further confirmed to be 16S rDNA of V. cholerae by BLSAT against the NCBI database. Sequence contamination was also assessed by RAST annotation systems.
Initial findings
Genome characteristics and phylogenetic analysis
The genome of V. cholerae O139 strain E306 was sequenced on Illumina Genome Analyzer IIx platform. A total of 6,112,322 raw reads with a mean read length of 116 bp, corresponding to 170-fold coverage of the genome were generated. After assembling, a total of 51 scaffolds with N50 length of 442,144 bp were obtained, and 9 gaps were spanned by 7 scaffolds resulting in a total length of 879,788 bp. The final assembled draft genome sequence is 4,165,057 bp with mean G + C content of 47.7%. The genome contains 3861 predicted coding DNA sequences (CDSs) and 82 RNA genes (4 rRNA genes and 78 tRNA genes). RAST annotation of the whole genome indicated the presence of 534 SEED subsystems (Figure 1A). The phylogenetic tree (Figure 1B) based on whole-genome SNPs showed that the closest ancestor for O139 strain E306 was V. cholerae MO10, which is also a member of the O139 serogroup and was isolated during the cholera outbreak in India and Bangladesh in 1992 [3,4]. The detailed comparison of the subsystems in V. cholerae O139 strain E306 and V. cholerae O139 strain MO10 is shown in Figure 1A.
Figure 1.

Gene distribution and phylogenetic analysis. A. Comparison of the distribution of genes assigned to SEED subsystems between V. cholerae O139 strain E306 and MO10. Outer circle and inner circle represent V. cholerae O139 strain E306 and MO10, respectively. Genes with less than 2% attribution are not labeled. B. Phylogenetic relationships (based on SNPs) of 11 V. cholerae strains and their genomic distance analysis. Bootstrap values less than 50% are not shown. The heat-plot of the similarity matrices is based on fragmented alignments with settings 500/500.
Cholera toxin prophage
Interestingly, though the V. cholerae O139 strain E306 is very close to V. cholerae O139 strain MO10, the gene organization of the cholera toxin-encoding CTX prophage is identical to those in the O1 strains of CIRS101, 2010EL-1786, and El Tor N16961. It is noteworthy that the genomic arrangements of the CTX prophage and the RS1 element in O139 strain E306, CIRS101, and 2010EL-1786 are opposite to that in V. cholerae O1 El Tor N16961 (Figure 2). Overall, compared with its closest neighbor, V. cholerae MO10, O139 strain E306 harbors the El Tor-specific RS1 element, and there is no VSK adjacent to the core region.
Figure 2.
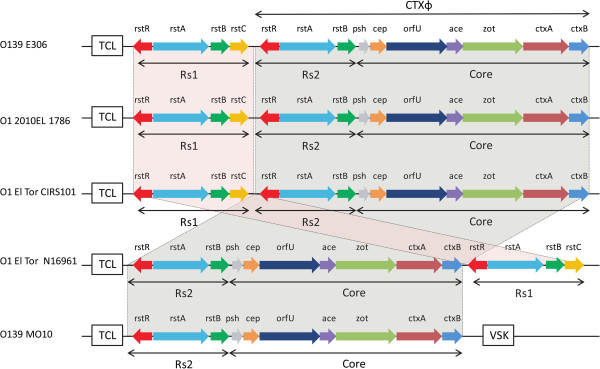
Diagrammatic indication of the structure of the CTX prophage and associated elements in V. cholerae O139 strain E306 and other 4 reference strains. The transcription direction of each gene is indicated by arrow and different genes are shaded in different colors. TLC: toxin-linked cryptic; VSK: pre-CTX prophage. The TLC and VSK elements are not drawn to scale.
Integrative conjugative elements (ICEs)
Based on the integrase gene similarity, a SXT/R391 family ICE in V. cholerae O139 strain E306 was identified inserted at the prfC locus. The general organization of this ICE was found to be highly similar to ICEVchInd4 and SXT MO10 which were previously identified in V. cholerae O139 strains. Detailed alignment indicated that ICEE306 and ICEVchInd4 only differed by 3 SNPs (3 SNPs in 3 coding regions), and ICEE306and SXTMO10 differed by 26 SNPs (17 SNPs in 14 coding regions) (Table 1); no obvious large sequence changes such as deletions and insertions were observed. These results, consistent with other study [25], suggested that these ICEs in V. cholerae are very stable over time, and because of the high degree of similarity, the dissemination of the ICE-carrying V. cholerae strains between different regions cannot be excluded.
Table 1.
SNPs identified in SXT MO10 and ICE Vch Ind4 compared with ICE E306
| Gene | UniProt GI | Product | Amino acid positions | Synonymity |
Amino acid (codon) |
||
|---|---|---|---|---|---|---|---|
| ICE E306 | SXT MO10 | ICE Vch Ind4 | |||||
| int |
21885342 |
Integrase |
326 |
syn |
I(ATA) |
I(ATT) |
. |
| |
|
|
398 |
NON |
T(ACC) |
I(ATC) |
I(ATC) |
| rumB' |
21885341 |
UV repair DNA polymerase |
104 |
syn |
T(ACT) |
. |
T(ACT) |
| s009 |
21885337 |
Unknown |
114 |
NON |
A(GCC) |
G(GGC) |
. |
| floR |
21885343 |
Florfenicol exporter |
230 |
NON |
L(CTC) |
V(GTC) |
. |
| s031 |
21885325 |
Unknown |
152 |
NON |
V(GTC) |
F(TTC) |
. |
| s035 |
21885321 |
Unknown |
398 |
NON |
W(TGG) |
G(GGG) |
. |
| s037 |
21885319 |
Unknown |
204 |
NON |
P(CCG) |
R(CGG) |
. |
| s038 |
21885318 |
Unknown |
26 |
NON |
K(AAG) |
R(AGG) |
. |
| traD |
21885274 |
Conjugative coupling factor |
430 |
NON |
D(GAC) |
A(GCC) |
. |
| |
|
|
432 |
syn |
A(GCG) |
A(GCC) |
. |
| |
|
|
456 |
syn |
V(GTG) |
V(GTC) |
. |
| s053 |
21885310 |
Unknown |
194 |
syn |
S(AGC) |
S(AGT) |
. |
| traU |
21885267 |
Sex pilus assembly |
321 |
NON |
P(CCC) |
S(TCC) |
S(TCC) |
| s060 |
21885308 |
Unknown |
194 |
NON |
P(CCG) |
T(ACG) |
. |
| |
|
Unknown |
199 |
NON |
S(TCA) |
T(ACA) |
. |
| setC |
21885287 |
Transcriptional activator |
20 |
NON |
S(TCC) |
Y(TAC) |
. |
| s083 | 21885291 | Unknown | 39 | NON | P(CCC) | R(CGC) | . |
“.” means this position has the same amino acid and codon as ICEE306.
Antibiotic resistance genes
We compared all the predicted protein-coding genes from 11 V. cholerae strains with known antibiotic resistance genes (BLASTp against the ARDB database [19]), yielding 50 matches to antibiotic resistance genes, mainly aminoglycoside resistance genes and tetracycline resistance genes (Table 2). A chloramphenicol resistance gene type (catb5) encoding Group B chloramphenicol acetyltransferase is present in 9 out of the 11 genomes, which is the most common resistance gene type. Interestingly, V. cholerae O139 strain E306 has 9 resistance genes, but no resistance gene was identified in O395 and only one was found in N16961. These results implied that different V. cholerae strains have different resistance profiles; the new isolation V. cholerae O139 strain E306 seems to have accumulated more antibiotic resistance in an environment with rapid growth rate of drug resistance [26].
Table 2.
Antibiotic resistance genes in the V. cholerae strains predicted by using the antibiotic resistance genes database
| Resistance type | Description | Resistance profile | B33 | CIRS101 | IEC224 | M66 2 | MJ 1236 | MO10 | O1 2010EL | N16961 | O395 | RC9 | E306 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ant2ia |
Aminoglycoside O-nucleotidylyltransferaseadenylylation. |
Tobramycin, gentamicin, dibekacin, sisomicin, kanamycin |
|
|
|
|
|
|
|
|
|
* |
|
| ant3ia |
Spectinomycin, streptomycin |
* |
|
|
|
|
|
|
|
|
|
|
|
| aph33ib |
Aminoglycoside O-phosphotransferase |
Streptomycin |
* |
* |
|
|
** |
* |
* |
|
|
* |
* |
| aph3ia |
Paromomycin, kanamycin, neomycin, ribostamycin, lividomycin, gentamincin_b |
|
|
|
|
|
|
|
|
|
|
* |
|
| aph6id |
Streptomycin |
* |
* |
|
|
* |
* |
* |
|
|
* |
* |
|
| catb5 |
Group B chloramphenicol acetyltransferase |
Chloramphenicol |
* |
* |
* |
|
* |
* |
* |
* |
|
* |
* |
| cml_e3 |
Major facilitator superfamily transporter, chloramphenicol efflux pump. |
Chloramphenicol |
|
* |
|
|
* |
* |
* |
|
|
|
* |
| dfra1 |
Group A drug-insensitive dihydrofolate reductase |
Trimethoprim |
|
|
|
|
* |
|
* |
|
|
|
|
| dfra23 |
Trimethoprim |
|
|
|
|
|
|
|
|
|
* |
|
|
| sul1 |
Sulfonamide-resistant dihydropteroate synthase |
Sulfonamide |
* |
|
|
|
|
|
|
|
|
* |
* |
| sul2 |
Sulfonamide |
* |
* |
|
|
* |
* |
* |
|
|
* |
* |
|
| teta |
Major facilitator superfamily transporter, tetracycline efflux pump |
Tetracycline |
|
|
|
|
* |
|
|
|
|
|
|
| tetc |
Tetracycline |
|
|
|
|
|
* |
|
|
|
|
|
|
| tetd |
Tetracycline |
|
|
|
|
|
|
|
|
|
* |
* |
|
| tetm | Ribosomal protection protein | Tetracycline | * |
Stars mean the number of antibiotic resistance genes found.
Future directions
Compared to the epidemic lineages of V. cholerae serogroup O1, our understanding of the genomic properties and their diversity of V. cholerae serogroup O139 is very limited. In this study, we sequenced the whole genome of a newly isolated strain of V. cholerae O139. This strain, carrying an El Tor-specific RS1 element that was found in V. cholerae O1 serogroup and more antibiotic resistance genes than other sequenced strains, highlights its high ability to adapt to new environments and poses a risk of causing new epidemic cholera. Moreover, the genome here will be of great interests for future V. cholerae comparative genomics.
Availability of supporting data
This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession AWWA00000000. The version described in this paper is version AWWA01000000.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YY and FL interpreted the sequencing data and prepared the manuscript. NL, JL and RFZ generated the sequencing data. YFH participated all discussions of data analysis and rewrite the manuscript. YFH, YY, BLZ and YC were involved in overall experimental design. All authors have read the manuscript and approved.
Contributor Information
Yong Yi, Email: yiyong1471@163.com.
Na Lu, Email: lvn@im.ac.cn.
Fei Liu, Email: liuf@im.ac.cn.
Jing Li, Email: lijing@im.ac.cn.
Ruifen Zhang, Email: zhangr@im.ac.cn.
Liping Jia, Email: jialiping306@163.com.
Hua Jing, Email: jinghua6379@163.com.
Hu Xia, Email: xiahu306@163.com.
Yi Yang, Email: yangyi8835@126.com.
Baoli Zhu, Email: zhubaoli@im.ac.cn.
Yongfei Hu, Email: huyf@im.ac.cn.
Yan Cui, Email: dryancui@aliyun.com.
Acknowledgments
This work was supported in part by the National Natural Science Foundation of China (NSFC) (31270168) and the Beijing Municipal Science & Technology Development Program (Z131102002813063).
References
- Chatterjee SN, Chaudhuri K. Lipopolysaccharides of Vibrio cholerae. I. Physical and chemical characterization. Biochim Biophys Acta. 2003;1639:65–79. doi: 10.1016/j.bbadis.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Ramamurthy T, Nair GB. Evolving identity of epidemic Vibrio cholerae: past and the present. Sci Cult. 2010;76:153–159. [Google Scholar]
- Albert MJ, Siddique AK, Islam MS, Faruque AS, Ansaruzzaman M, Faruque SM, Sack RB. Large outbreak of clinical cholera due to Vibrio cholerae non-O1 in Bangladesh. Lancet. 1993;341:704. doi: 10.1016/0140-6736(93)90481-u. [DOI] [PubMed] [Google Scholar]
- Ramamurthy T, Garg S, Sharma R, Bhattacharya SK, Nair GB, Shimada T, Takeda T, Karasawa T, Kurazano H, Pal A. et al. Emergence of novel strain of Vibrio cholerae with epidemic potential in southern and eastern India. Lancet. 1993;341:703–704. doi: 10.1016/0140-6736(93)90480-5. [DOI] [PubMed] [Google Scholar]
- Qu M, Xu J, Ding Y, Wang R, Liu P, Kan B, Qi G, Liu Y, Gao S. Molecular epidemiology of Vibrio cholerae O139 in China: polymorphism of ribotypes and CTX elements. J Clin Microbiol. 2003;41:2306–2310. doi: 10.1128/JCM.41.6.2306-2310.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin EJ, Lin W, Mekalanos JJ, Waldor MK. Replication and integration of a Vibrio cholerae cryptic plasmid linked to the CTX prophage. Mol Microbiol. 1998;28:1247–1254. doi: 10.1046/j.1365-2958.1998.00889.x. [DOI] [PubMed] [Google Scholar]
- Safa A, Nair GB, Kong RYC. Evolution of new variants of Vibrio cholerae O1. Trends Microbiol. 2009;18:46–54. doi: 10.1016/j.tim.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Waldor MK, Tschäpe H, Mekalanos JJ. A new type of conjugative transposon encodes resistance to sulfamethoxazole, trimethoprim, and streptomycin in Vibrio cholerae O139. J Bacteriol. 1996;178:4157–4165. doi: 10.1128/jb.178.14.4157-4165.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel AK, Jain M, Kumar P, Jiang SC. Molecular characterization of Vibrio cholerae outbreak strains with altered El Tor biotype from southern India. World J Microbiol Biotechnol. 2010;26:281–287. doi: 10.1007/s11274-009-0171-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin CS, Sorenson J, Harris JB, Robins WP, Charles RC, Jean-Charles RR, Bullard J, Webster DR, Kasarskis A, Peluso P, Paxinos EE, Yamaichi Y, Calderwood SB, Mekalanos JJ, Schadt EE, Waldor MK. The origin of the Haitian cholera outbreak strain. N Engl J Med. 2011;364:33–42. doi: 10.1056/NEJMoa1012928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen BM, Lee JH, Cuong NT, Choi SY, Hien NT, Anh DD, Lee HR, Ansaruzzaman M, Endtz HP, Chun J, Lopez AL, Czerkinsky C, Clemens JD, Kim DW. Cholera outbreaks caused by an altered Vibrio cholerae O1 El Tor Biotype strain producing classical cholera toxin B in Vietnam in 2007 to 2008. J Clin Microbiol. 2009;47:1568–1571. doi: 10.1128/JCM.02040-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MP, Peterson DA, Biggs PJ, Solexa QA. At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinforma. 2010;11:485. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23:673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M. et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA. The COG database: an updated version includes eukaryotes. BMC Bioinforma. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Pop M. ARDB–Antibiotic Resistance Genes Database. Nucleic Acids Res. 2009;37:D443–D447. doi: 10.1093/nar/gkn656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 1000 Genome Project Data Processing Subgroup. The sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agren J, Sundström A, Håfström T, Segerman B. Gegenees: fragmented alignment of multiple genomes for determining phylogenomic distances and genetic signatures unique for specified target groups. PLoS One. 2012;7:e39107. doi: 10.1371/journal.pone.0039107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak RA, Fouts DE, Spagnoletti M, Colombo MM, Ceccarelli D, Garriss G, Déry C, Burrus V, Waldor MK. Comparative ICE genomics: insights into the evolution of the SXT/R391 family of ICEs. PLoS Genet. 2009;5:e1000786. doi: 10.1371/journal.pgen.1000786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Yang X, Qin J, Lu N, Cheng G, Wu N, Pan Y, Li J, Zhu L, Wang X, Meng Z, Zhao F, Liu D, Ma J, Qin N, Xiang C, Xiao Y, Li L, Yang H, Wang J, Yang R, Gao GF, Wang J, Zhu B. Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat Commun. 2013;4:2151. doi: 10.1038/ncomms3151. [DOI] [PubMed] [Google Scholar]