

Abstract
Compartmentalization of protein kinases and phosphatases with substrates is a means to increase the efficacy of signal transduction events. The A-kinase anchoring protein, AKAP79, is a multivalent anchoring protein that maintains the cAMP-dependent protein kinase, protein kinase C, and protein phosphatase-2B (PP2B/calcineurin) at the postsynaptic membrane of excitatory synapses where it is recruited into complexes with N-methyl-d-aspartic acid or α-amino-3-hydroxy-5-methyl-isoxazole-4-propionic acid (AMPA)-subtype glutamate receptors. We have used cellular targeting of AKAP79 truncation and deletion mutants as an assay to map the PP2B-binding site on AKAP79. We demonstrate that residues 315–360 are necessary and sufficient for AKAP79-PP2B anchoring in cells. Multiple determinants contained within this region bind directly to the A subunit of PP2B and inhibit phosphatase activity. Peptides spanning the 315–360 region of AKAP79 can antagonize PP2B anchoring in vitro and targeting in transfected cells. Electrophysiological experiments further emphasize this point by demonstrating that a peptide encompassing residues 330–357 of AKAP79 attenuates PP2B-dependent down-regulation of GluR1 receptor currents when perfused into HEK293 cells. We propose that the structural features of this AKAP79-PP2B-binding domain may share similarities with other proteins that serve to coordinate PP2B localization and activity.
The efficient transmission of cellular signals often involves the orientation of signaling proteins in relation to their upstream activators and downstream targets. This is often achieved through association with anchoring and scaffolding proteins that compartmentalize signaling enzymes in distinct subcellular environments (1–3). For example, A-kinase anchoring proteins (AKAPs)1 bind the regulatory (R) subunit of the cAMP-dependent protein kinase (PKA) to localize this broad specificity enzyme to discrete subcellular environments (4, 5). Each AKAP contains a conserved amphipathic helix that binds to the R subunit dimer with high affinity and targeting domains that direct the PKA-AKAP complex to specific subcellular compartments (6, 7). A likely consequence of these protein-protein interactions is that AKAP-PKA complexes are maintained in the vicinity of selected phosphoproteins and substrates for the kinase. Another important role of AKAPs is to serve as scaffolds for the assembly of multiprotein complexes that include PKA, other protein kinases, phosphodiesterases, and a variety of protein phosphatases (8). The simultaneous anchoring of kinases and phosphatases provides an efficient means to confer bi-directional control on the phosphorylation status of substrate proteins (9, 10).
A number of studies (11) have demonstrated that anchoring of kinases and phosphatases ensures the efficient regulation of ion channels and neurotransmitter receptors. One prominent mediator of this process is the multivalent anchoring protein AKAP79 that anchors PKA, protein kinase C (PKC), and protein phosphatase-2B (PP2B/calcineurin) (12–14). Precise orientation of the AKAP79 signaling complex toward substrates such as ionotropic glutamate receptors at the postsynaptic densities of neurons involves additional protein-protein interactions between the anchoring protein and membrane-associated guanylate kinase proteins. This intricate molecular architecture facilitates PKA phosphorylation at a site in the cytoplasmic tail of the GluR1 glutamate receptor channel as well as its efficient dephosphorylation by PP2B (10, 15).
Previous studies (13, 16, 17) have shown that the catalytic subunit of PP2B directly binds to sites in the C-terminal two-thirds of AKAP79 and that interaction with the anchoring protein inhibits phosphatase activity toward peptide substrates. In this report we describe experiments that map the PP2B-binding site to a region of ∼45 amino acids that contains multiple binding sites and inhibitory determinants. Furthermore, we show that a peptide from this region disrupts phosphatase targeting and attenuates PP2B-dependent down-regulation of homomeric GluR1 glutamate receptor currents inside cells.
Materials and Methods
Construction of cDNA Expression Plasmids
The cDNA for human AKAP79 was expressed from pCDNA3 (Invitrogen) constructed as described previously (16). The coding sequences for AKAP79 full-length, the C-terminal truncations shown (residues 1–360, 1–315, and 1–108), or the 321–360-binding peptide fragment were amplified by PCR using specific synthetic oligonucleotide primers. The PCR products introduced a 5′ HindIII site, consensus ribosome-binding site, an ATG initiation codon, and a BamHI site at the 3′ end of the coding region. Each fragment was subcloned as a HindIII-BamHI fragment into pEGFPN1 (Clontech) to generate C-terminal fusions to EGFP. Internal deletions of AKAP79-(Δ109–315), -(Δ109–360), -(Δ151–315), -(Δ151–360), -(Δ315–360), -(Δ328–338), -(Δ348–360), and –(Δ328–360) were generated by the PCR-based Quickchange method (Stratagene) to generate deletions using pEGFPN1-AKAP79 as the template DNA.
The coding region of the A subunit of mouse PP2B was amplified by PCR using specific oligonucleotide primers. This introduced a BamHI at the 5′ end and a 3′ EcoRI site in place of the stop codon. A fragment with BamHI and EcoRI ends was subcloned into pCDNA3.1MycHis(+)A (Invitrogen) to generate a C-terminal Myc epitope fusion.
COS7 Cell Transfection
COS7 cells at 20–50% confluency (24–48 h after plating) were transfected by calcium phosphate precipitation with pcDNA3-AKAP79 or various pEGFP-AKAP79 cDNA expression constructs and pCDNA3-PP2BAMycHis (1–2 μg each plasmid per glass coverslip in 6-well plates for immunocytochemistry and 5–10 μg of each plasmid for 10-cm plates for immunoprecipitation) for 4–6 h at 5% CO2, 37 °C. Cells were then washed with phosphate-buffered saline (PBS), fed with normal growth medium (Dulbecco's modified Eagle's medium, 10% fetal bovine serum, 1% penicillin/streptomycin, Invitrogen), and grown 24–48 h prior to fixation for immunocytochemistry or preparation of cell extracts for immunoprecipitation.
Immunoprecipitation
COS7 cell extracts were prepared by lysis in ice-cold buffer, 20 mM HEPES, pH 7.4, 100 mM NaCl, 5 mM EDTA, 0.5% Triton X-100 (w/v), 1 mM dithiothreitol, 2 μg/ml leupeptin and pepstatin, 1 mM benzamidine, 1 mM 4-(2-aminoethyl)benzenesulfonyl fluoride. Centrifugation at 16,000 × g for 15 min clarified each lysate. AKAP79 immune complexes were isolated by incubation with 5 μg of affinitypurified rabbit polyclonal anti-AKAP79 (918I) or 2 μg of mouse monoclonal anti-Myc (9E10) antibody (Santa Cruz Biotechnology) for 4 h at 4 °C. Control experiments were performed by incubating lysates with a non-related IgG at the same concentration for 4 h at 4 °C. Immune complexes were separated from the reaction mixtures by incubation with protein A-Sepharose (25 μl, 50% slurry equilibrated in lysis buffer) for 1 h at 4 °C. After microcentrifugation, the supernatant was removed, and immune complexes were eluted with SDS-PAGE sample buffer. Co-purification of components in each signaling complex was analyzed by immunoblotting with anti-AKAP79, anti-Myc, or anti-PP2BA. Immunoblots were then visualized using enhanced chemiluminescence (ECL; Pierce) and Kodak X-Omat AR film.
Immunocytochemistry and Fluorescence Microscopy
Transfected COS7 cells (24–48 h post-transfection) on glass coverslips were washed twice in PBS, fixed in 3.7% formaldehyde/PBS, and permeabilized with 0.2% Triton X-100 in PBS for 5 min. Permeabilized cells were washed with PBS and blocked in PBS + 1.0% bovine serum albumin (BSA) for 30 min. Each sample was incubated in primary antibodies (1:250 anti-Myc or 1:1000 anti-AKAP79) for 1 h at room temperature. After further washing in PBS + BSA, coverslips were incubated with excess secondary antibodies (goat anti-mouse Texas Red, goat anti-rabbit antibody-FITC, or goat anti-rabbit-Cy5, Molecular Probes) for 1 h at room temperature. The washed coverslips were mounted on glass slides and analyzed using a either a Zeiss Axiovert TV-135 inverted microscope (63×plan-apo, oil, 1.4 NA) equipped with a digital CCD camera (Sensys) and Meta-GFP imaging software (Universal Imaging) or a Nikon TE-300 inverted microscope (100×plan-apo, oil, 1.4 NA) equipped with a digital CCD camera (Princeton Instruments) and Slidebook 3.0 imaging software (Intelligent Imaging Innovations).
Bacterial expression and purification of recombinant AKAP79 was performed as described previously (14).
PP2B Activity Assays
PP2B phosphatase activity was assayed using a PKA-phosphorylated RII peptide as described (18).
AKAP Peptide, PP2B Binding Assay
N-Biotinylated AKAP79-(338–357), AKAP79-(330–357), AKAP79-(318–338), or control peptides (10 μg) were incubated with streptavidin-agarose (Sigma) (20 μl of packed beads) in hypotonic buffer (10 mM HEPES, pH 7.4, 10 mM KCl, 1.5 mM MgCl2, 0.1% Nonidet P-40, 0.1% BSA, 1 mM dithiothreitol, 1 mM benzamidine, 1 mM leupeptin, and 1 mM 4-(2-aminoethyl)benzenesulfonyl fluoride) for 4 h at 4 °C. The slurry was centrifuged (3000 × g), and the unbound peptide was removed. The beads were then incubated with purified recombinant PP2B (1 μg) in hypotonic buffer in a final volume of 100 μl for 15 h at 4 °C. Protein complexes were pelleted by centrifugation (3000 × g), washed 5 times with hypotonic buffer, 1 M NaCl and another 5 times with hypotonic buffer alone. The materials were eluted with SDS-PAGE sample buffer. Binding of the phosphatase was detected by immunoblot using anti-PP2B A subunit antibodies.
Electrophysiology
HEK293 cells were plated on 35-mm dishes at ∼50,000 cells/ml. Cells were transfected with 1 μg of GluR1 with or without 1 μg of AKAP79-GFP by the calcium phosphate method as described previously (10). The CD4 cell surface marker was also included as a transfection marker. Cells were visually selected for recording by adherence of CD4 antibody-coated beads 24 h after transfection.
GFP epifluorescence was used to confirm expression of AKAP79 in these cells. Whole-cell recordings were made with an Axopatch 200B amplifier (Axon Instruments). Patch pipettes (2–4 megohms) contained 140 mM cesium methanesulfonate, 10 mM HEPES, 5 mM adenosine triphosphate (sodium salt), 5 mM MgCl2, 0.2 mM CaCl2, and 1 1,2-bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid (pH 7.4). The 330–357-residue peptide was added to the pipette solution from frozen concentrated stocks. The extracellular solution contained 150 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 10 mM HEPES, 10 mM glucose (pH 7.4). Solution exchanges were accomplished through a series of flow pipes. Flow pipes were controlled by solenoid valves and moved into position by a piezoelectric bimorph. HEK293 cells were lifted off the bottom of the dish to speed the solution exchange time. GluR1 receptor currents were evoked by a 500-ms application of glutamate in the presence of cyclothiazide (100 μM) at 30-s intervals. Currents were digitized at 5 kHz and filtered at 1 kHz with a Digidata 1200B board and Clampex 7 software (Axon Instruments). Series resistance (85–95%) and whole-cell capacitance compensation were used and routinely monitored throughout the experiments. Only HEK293 with series resistance <5 megohms were analyzed. All experiments were initiated within 1 min of establishing whole-cell configuration and performed at a holding potential of −60 mV at 20 °C.
Results
AKAP79 Mediates Plasma Membrane Targeting of PP2B in Transfected COS7 Cells
As a prelude to detailed mapping studies, we tested whether expression of recombinant AKAPs could target PP2B to the plasma membrane. In COS7 cells, the cellular distribution of PP2B and selected AKAPs was visualized by immunocytochemical techniques (Fig. 1). Control experiments show that transient expression of a C-terminal Myc-tagged A subunit of PP2B results in a cytoplasmic distribution of the phosphatase (Fig. 1, A–C). In contrast, expression of AKAP79 results in an accumulation of the anchoring protein at the plasma membrane in cortical ruffles (Fig. 1D) as reported previously (16). Most important, co-expression of AKAP79 results in the accumulation of PP2B at the plasma membrane in cortical ruffles (Fig. 1, E and F). Similar results were obtained upon expression of an AKAP79-green fluorescent protein fusion protein (GFP), suggesting that the addition of the fluorescent moiety did not affect cellular targeting of the anchoring protein or perturb interaction with the phosphatase (Fig. 1, G–I). Further controls demonstrated that expression of another membrane-targeted anchoring protein, AKAP18-GFP, had no effect on the cytoplasmic localization of PP2B (Fig. 1, J–L). Collectively, these experiments demonstrate that AKAP79 participates in directing the subcellular location of PP2B in COS7 cells.
Fig. 1. AKAP79 influences the subcellular location of PP2B in COS7 cells.

COS7 cells were transfected with myc-PP2B catalytic A subunit and a variety of anchoring protein constructs. A–C, cells were fixed and immunostained for Myc-PP2B (anti-Myc, Texas Red). D–F, double immunofluorescence staining of cells to detect myc-PP2B (red) and AKAP79 (green). G–I, double immunofluorescence detection of myc-PP2B (red) and AKAP79-GFP (green). J–L, double immunofluorescence detection of myc-PP2B (red) and AKAP18-GFP (green). Analysis was performed on an inverted microscope with a digital CCD camera. Overlap of the green and red signals is seen as yellow in the composite images.
Mapping the PP2B-binding Site on AKAP79
Previous studies (16, 17) have demonstrated that the C-terminal two-thirds of AKAP79 (residues 108–427) are sufficient to bind PP2B in vitro and in COS7 cells. In order to map further the PP2B-binding site on AKAP79, we sequentially deleted C-terminal regions of the anchoring protein, and we tested the ability of these recombinant GFP fusion proteins to interact with the phosphatase in COS7 cells (Fig. 2A). Only the longest fragment, AKAP79-(1–360), retained the ability to interact with the phosphatase (Fig. 2, B–D). Two shorter fragments, AKAP79-(1–315) and AKAP79-(1–108), did not alter the subcellular location of the phosphatase (Fig. 2, E–G and H–J). These results imply that residues 315–360 of the anchoring protein are necessary for interaction with PP2B. This notion was further supported by cellular analysis of additional AKAP79 constructs where internal sections of the anchoring protein were removed (Fig. 2A). Deletion of residues 151–315 generated an AKAP79 form that retained the ability to target the phosphatase (Fig. 2, K–M), whereas removal of residues 151–360 abolished interaction with PP2B (Fig. 2, N–P). These experiments confirmed that residues 315–360 of AKAP79 are necessary for interaction with PP2B. In order to determine whether this region was sufficient for phosphatase binding, a further construct, AKAP79-(Δ315–360), was generated (Fig. 2A). This internally truncated version of the anchoring protein was unable to alter PP2B location when expressed in COS7 cells (Fig. 2, Q–S) suggesting that a 45-amino acid segment between residues 315 and 360 of the anchoring protein is required for PP2B targeting.
Fig. 2. Residues 315–360 of AKAP79 are necessary for targeting of PP2B in COS7 cells.
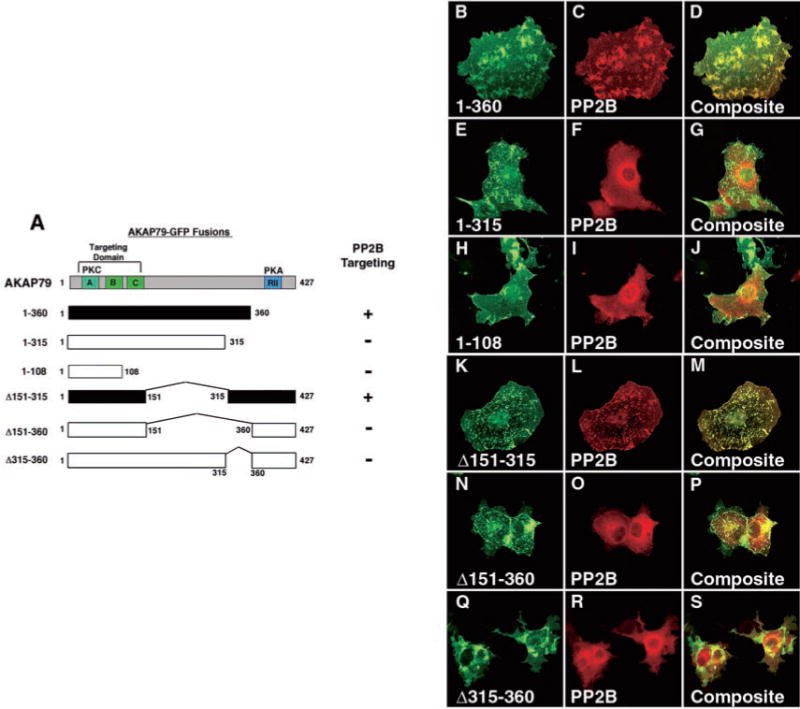
A, schematic diagram of the AKAP79 fragments used to analyze PP2B localization in COS7 cells. The first and last residue of each construct and the positions of internal deletions are indicated. GFP was fused at the C terminus of each fragment. The approximate location of the PKA (blue) and PKC (cyan)-binding sites and subcellular targeting domains (green) are indicated. Constructs capable of targeting PP2B (filled boxes) are indicated. B–S, COS7 cells transfected with myc-PP2B catalytic subunit and a variety of anchoring protein constructs. Cells were fixed, immunostained, and imaged to visualize the location of each AKAP79 fragment (green) and Myc-PP2B (red). Analysis was performed on an inverted microscope with a digital CCD camera. Overlap of the green and red signals is seen as yellow in the composite images. B–D, characterization of the AKAP79-(1–360) fragment. E–G, characterization of the AKAP79-(1–315) fragment. H–J, characterization of the AKAP79-(1–108) fragment. K–M, characterization of the AKAP79-(Δ151–315) fragment. N–P, characterization of the AKAP79-(Δ151–360) fragment. Q–S, characterization of the AKAP79-(Δ315–360) fragment.
Independent biochemical support for our cellular mapping studies was obtained by co-immunoprecipitation of AKAP79-PP2B complexes from COS7 cells (Fig. 3). Cells were transfected with Myc-tagged A subunit of PP2B and selected AKAP79 forms (Fig. 3A). Efficient expression of each recombinant AKAP form was confirmed by immunoblotting cell extracts with anti-AKAP79 rabbit polyclonal antisera (Fig. 3B). The ability of each AKAP79-fusion protein to associate with the Myc-tagged A subunit of PP2B was evaluated by immunoprecipitation with anti-Myc monoclonal antibody. As expected, AKAP79 and the 1–360-residue fragment co-precipitated with PP2B (Fig. 3B, left and center left panels), whereas the 1–315- and the Δ315–360-residue forms did not (Fig. 3B, center right and right panels). Control experiments confirmed that none of the AKAP79 forms were present in immune complexes isolated with an unrelated IgG (Fig. 3B). These data use an independent approach to imply further that determinants between residues 315–360 of AKAP79 are necessary for PP2B binding.
Fig. 3. AKAP79 residues 315–360 are necessary for binding to PP2B in vitro.

A, schematic diagram of the AKAP79 fragments used for biochemical analysis of PP2B binding. The first and last residues of each construct and the positions of internal deletions are indicated. GFP was fused to the C terminus of each fragment. The approximate locations of the PKA, PKC, and PP2B-binding sites and subcellular targeting domains are indicated. AKAP79 fragments capable of binding PP2B (filled bars) are indicated. B, each AKAP79 construct was assayed for co-immunoprecipitation (IP) of PP2B following a protocol technique outlined under “Materials and Methods.” Each anchoring protein fragment (indicated above each panel) was detected in PP2B immune complexes by immunoblotting (IB) with anti-AKAP79 polyclonal antisera. The level of each AKAP79 fragment in cell extracts (Extract) and analysis of control immune complexes (IgG) are indicated. Representative examples of three independent experiments are presented.
Direct in Vitro Binding of PP2B to an AKAP79-(330–357) Peptide
A series of three overlapping peptides spanning the 315–360-region of AKAP79 were synthesized to establish whether these residues were responsible for direct binding and inhibition of phosphatase activity (Fig. 4A). In vitro pulldown assays were performed to test whether each peptide bound to recombinant A subunit of PP2B (Fig. 4B). Each peptide bound to the phosphatase, whereas control experiments performed with a 24-residue peptide of similar amino acid composition were negative (Fig. 4B). Because the AKAP79-(318–338) and the AKAP79-(338–357) peptides encompass different segments of the PP2B-binding site, it is suggested that phosphatasebinding determinants are dispersed throughout this region of the anchoring protein (Fig. 4B).
Fig. 4. Multiple PP2B inhibitory determinants are located between residues 315 and 360 of AKAP79.
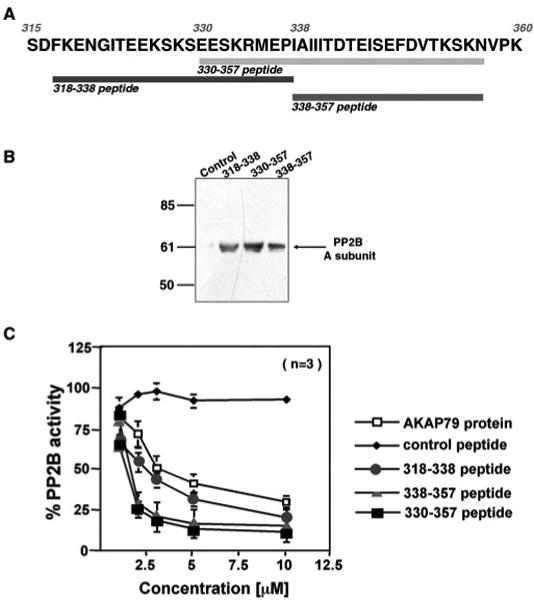
A, the sequence of residues 315–360 of AKAP79 is presented in the one-letter code. The regions encompassed by each peptide and the first and last residues of the peptide are indicated. B, each AKAP79 peptide binds to PP2B in vitro. Biotinylated AKAP79 peptides were immobilized to streptavidin-agarose and used to capture recombinant A subunit of PP2B by in vitro pulldown assays. The bound phosphatase was eluted from the column and detected by immunoblot using polyclonal antibodies against PP2B. Samples were analyzed from pulldown experiments using lane 1 (control peptide), lane 2 (AKAP79-(318–338) peptide), lane 3 (AKAP79-(330–357) peptide), and lane 4 (AKAP79-(338–357)) peptide. The migration position of the A subunit of PP2B and molecular weight markers are indicated. C, each AKAP79 peptide inhibits PP2B activity. Phosphatase activity was measured by the procedure described under “Materials and Methods” in the presence of AKAP79, control, or AKAK79 peptides over a concentration range from 1 to 10 μM. Doseresponse curves were obtained to measure the inhibitory potency of AKAP79 protein (open squares), the AKAP79-(318–338) peptide (circles), the AKAP79-(330–357) peptide (squares), the AKAP79-(338–357) peptide (triangles), and control peptide (diamonds). The data presented in the amalgamated results are from three independent experiments.
Previous studies (13, 16) have shown that AKAP79 inhibits PP2B activity in a dose-dependent manner. Therefore, each PP2B-binding peptide was assayed as an inhibitor of the phosphatase using a PKA-phosphorylated substrate peptide (18). Recombinant AKAP79 inhibits PP2B activity in a dose-dependent manner with an IC50 value of 5.3 ± 0.5 μM (n = 3) (Fig. 4C, open squares). All three peptides also inhibit PP2B activity over the same concentration range, with IC50 values of 3.2 ± 0.17 μM (n = 3) for the 318–338-residue peptide (Fig. 4C, circles), 1.5 ± 0.45 μM (n = 3) for the 338–357-residue peptide (Fig. 4C, triangles), and 1.5 ± 0.2 μM (n = 3) for the 330–357-residue peptide (Fig. 4C, squares). Incubation with a 24-residue control peptide of similar amino acid composition did not affect phosphatase activity (Fig. 4C, diamonds). Collectively, these experiments suggest that there are at least two sites between residues 318 and 357 of AKAP79 that bind PP2B and inhibit phosphatase activity. The presence of multiple PP2B-binding determinants in this region was confirmed by analysis of three additional AKAP79 deletion mutants in COS7 cells (data not shown). Removal of residues 328–338 or residue 348s-360 from the anchoring protein resulted in decreased Myc-PP2BA targeting to the plasma membrane, whereas deletion of residues 328–360 completely abolished PP2B anchoring.
The 330–357 Peptides Are Antagonists of PP2B/AKAP79 Interaction
One goal of our mapping studies was to generate reagents that could perturb PP2B localization and function inside cells. A likely candidate was the AKAP79-(330–357) peptide, as it bound strongly to PP2B and was an effective inhibitor of phosphatase activity. Various concentrations of the AKAP79-(330–357) peptide (1–10 μM) were incubated with the phosphatase-anchoring protein complex to antagonize AKAP79/PP2B interaction in vitro (Fig. 5A, top panel). AKAP79 immune complexes were isolated using polyclonal antibodies against the anchoring protein, and displacement of PP2B was measured by quantitative Western blot (Fig. 5A, middle panel, and B). Control experiments confirmed that equivalent amounts of AKAP79 were immunoprecipitated in each reaction mixture (Fig. 5A, bottom panel). Inclusion of the 330–357-residue peptide blocked co-precipitation of PP2B in a dose-dependent manner over a concentration range of 1–10 μM, whereas a control peptide of similar amino acid composition did not affect complex formation (Fig. 5,A, middle panel, and B).
Fig. 5. The PP2B-binding region of AKAP79 antagonizes phosphatase interaction with the anchoring protein.

A, purified AKAP79 and PP2B holoenzymes were incubated in vitro with increasing concentrations (0–10 μM) of 330–357 peptide or unrelated control peptide (10 μM, Ht31 PKA-R-binding peptide). PP2B binding to AKAP79 was then analyzed by immunoprecipitation (IP) with anti-AKAP79. Top panel, co-purification of PP2B in the presence of increasing AKAP79-(330–357) peptide (indicated above each lane) was detected by immunoblotting with polyclonal antibodies against the phosphatase. The migration position of the A subunit of PP2B is indicated. Bottom panel, equal levels of AKAP79 in each experiment were confirmed by immunoblot. The migration position of AKAP79 is indicated. B, immunoblots from four independent experiments were analyzed densitometrically using NIH image software for quantification. Values on the y axis are normalized (set at 1.0) to the amount of PP2B immunoprecipitated with AKAP79 in the absence of peptide. Normalized intensity values are represented as the mean ± S.D. across all experiments. The concentration of the AKAP79-(330–357) peptide is indicated below each column. C, schematic diagram depicting the region of AKAP79 used to generate a soluble GFP fusion protein for cellular expression in COS7 cells. The first and last residues of the anchoring protein fragment are indicated. D–K, cellular expression of the AKAP79-(321–360)-GFP fusion protein is sufficient to disrupt PP2B/AKAP interaction inside cells. COS7 cells were transfected with control plasmid or the AKAP79-(321–360)-GFP construct. Both samples were also transfected with vectors encoding full-length AKAP79 and the A subunit of PP2B. Transfected cells were then fixed, stained, and analyzed by triple-fluorescence microscopy for GFP (green) (D and H), AKAP79-Cy5 (blue) (E and I), and PP2B-Texas Red (red) (F and J). G and K, an overlay of all three fluorescence channels is shown in the composite panel.
In parallel, experiments were performed to test whether expression of the PP2B-binding region alone could displace the phosphatase from interaction with AKAP79 inside cells. A chimeric construct encompassing residues 321–360 of AKAP79 fused to GFP (Fig. 5C) was co-transfected into COS7 cells with AKAP79 and Myc-tagged PP2B (Fig. 5, D–K). The cellular locations of the anchoring protein (Fig. 5, E and I, blue) and the phosphatase (Fig. 5, F and J, red) were assessed by immunocytochemistry. Expression of the PP2B-binding fragment altered the subcellular distribution of PP2B (Fig. 5J) but did not affect the targeting of AKAP79 (Fig. 5I). This is most clearly demonstrated in the composite image (Fig. 5K). In contrast, compartmentalization of the anchoring protein and the phosphatase was unaltered in control cells (Fig. 5, D–G). These findings indicate that overexpression of the untargeted AKAP79-(321–360) peptide disrupts plasma membrane anchoring of PP2B, suggesting that this region of the anchoring protein is sufficient to disrupt the cellular targeting of the phosphatase inside cells.
The AKAP79-(330–357) Peptide Disrupts PP2B Function Inside Cells
Recent data suggest that AKAP79 is likely to coordinate the phosphorylation state and activity of the AMPA-type glutamate receptor ion channels (10, 15). Co-expression of AKAP79 with GluR1 subunit of the AMPA receptor leads to a time-dependent down-regulation (rundown) of recombinant GluR1 receptor currents expressed in HEK293 cells. This effect is contingent on a rise in intracellular calcium and PP2B activity (10). For these reasons we chose to test the effects of the AKAP79-(330–357) peptide on homomeric GluR1 receptor currents expressed in HEK293 cells (Fig. 6). Whole-cell patch clamp recording techniques were used to recapitulate the AKAP79-dependent rundown of GluR1 receptor currents (Fig. 6). Expression of AKAP79 promoted a reduction to 64.8 ± 4.0, n = 7, p < 0.01 GluR1 current over a 15-min period (Fig. 6, open triangles) when compared with controls (Fig. 6, open circles, 86.2 ± 5.8% of initial current, n = 6). However, when the AKAP79-(330–357) peptide was included in the whole-cell pipette solution, this AKAP79-dependent rundown of GluR1 receptor currents was blunted to 83.5 ± 5.6% of initial current, n = 7 p > 0.3 (Fig. 6, closed triangles). Most important, control cells expressing GluR1 alone were unaffected by the peptide (Fig. 6, closed circles, 90.7 ± 3.9% of initial current, n = 7 p > 0.3). These results indicate that the AKAP79-(330–357) peptide disrupts a cellular function attributed to AKAP79-anchored PP2B inside cells.
Fig. 6. The AKAP79-(330–357) peptide disrupts PP2B-dependent modulation of GluR1 receptor currents by AKAP79.

HEK293 cells were transfected with GluR1 alone (control) or with AKAP79. Whole-cell GluR1 receptor currents were evoked by glutamate (1 mM) in the presence of cyclothiazide (100 μM). GluR1 receptor currents selectively rundown when co-expressed with AKAP79 (open triangles) are compared with control cells (open circles). Whole-cell dialysis of the 330–357 peptide (10 μM) into cells blunts the AKAP79-dependent rundown (filled triangles), whereas it is without effect in control cells (filled circles). Side panels show representative traces, and parentheses denote number of observations for each condition at time 0 and 15 min.
Discussion
We have mapped a region in the neuronally expressed anchoring protein AKAP79 that is necessary for binding and inhibiting the calcium/calmodulin-dependant phosphatase PP2B/calcineurin. A combination of biochemical and enzymatic approaches has identified multiple binding and inhibitory determinants that are located between residues 318 and 357 of the anchoring protein. Our characterization of this PP2B-binding region is the latest in a series of studies that have dissected functional regions within AKAP79 that are responsible for the interaction with distinct signaling enzymes. Residues 391–408 form an amphipathic helix that binds to the R subunit dimer of the PKA holoenzyme with nanomolar affinity (7, 12, 19). Peptides spanning this region have been used by many investigators to disrupt a variety of cAMP-responsive events by uncoupling PKA location inside cells (20–25). A PKC-binding site is located between residues 31 and 52 (14). Peptides spanning this region bind to the kinase with micromolar affinities and inhibit the bound enzyme. This effect is reversed by calcium/calmodulin, which competes for interaction with the 31–52 region and releases the active kinase from the signaling complex at the postsynaptic densities of neurons (26, 27).
In this report, we demonstrate that a family of peptides synthesized to regions of the anchoring protein between residues 318 and 357 disrupt the subcellular location of the phosphatase and inhibit PP2B-dependent phosphorylation events. Another region of AKAP79 within the membrane targeting domain (residues 88–102) may also participate in PP2B anchoring (13, 16, 17), although the inhibitory potency of peptides to this site is ∼50-fold less than the AKAP79-(330–357) peptide, and they cannot displace the phosphatase from its sites of anchoring. Therefore, we propose that the region between residues 318 and 357 is likely to contain most of the high affinity determinants for PP2B anchoring. This postulate is consistent with recent reports (17, 28, 29) that recombinant fragments of AKAP79 that include this region inhibit PP2B signaling events in transfected cells and transgenic mice. Interestingly, the AKAP79-(330–357) and AKAP79-(338–357) peptides are slightly more potent inhibitors of the phosphatase than the full-length anchoring protein. Similarly we have observed that protein fragments of AKAP79 sometimes work more efficiently than the full-length protein. For example, residues 108–427 inhibit PP2B more effectively than the full-length protein (16). Likewise, the first 75 amino acids of the anchoring protein is a more potent inhibitor of PKC activity than the full-length protein (14).
The AKAP79-(318–357) sequence exhibits some similarity to the binding sites of other PP2B-interacting proteins. For example, the organization of serines, threonines, and the clustering of basic amino acids in this region is reminiscent of the 38-residue PP2B-binding region in Cain, (calcineurin inhibitor)/cabin (30, 31). Both proteins are non-competitive inhibitors of PP2B activity that inhibit the phosphatase in the low micromolar range and bind PP2B at a site distinct from the immunosuppressant drugs FK506 and cyclosporin A (13, 17, 32). Another common characteristic is the presence of a loosely conserved “PIXIXIT” motif (where X represents any amino acid). This sequence was first identified in the transcription factor NFATc and has been found in the docking sites of other PP2B interacting partners including Cain/cabin and the muscle-specific protein MCIP1,2 (33–35). Residues 337–343 of AKAP79 (PIAIIIT) form the PIXIXIT motif, and it is totally conserved in the murine and bovine orthologs, AKAP150 and AKAP75 (36–38). Although not formally proven, it is likely that this sequence represents a principle site of contact with the phosphatase, because the AKAP79-(330–357) peptide, which includes this region, inhibits phosphatase activity at micromolar concentrations and competes for binding with the intact anchoring protein. However, it is clearly evident that other regions of AKAP79 contribute to PP2B interactions as peptides upstream of the PIXIXIT motif also bind and inhibit the phosphatase. These findings are certainly consistent with the notion that multiple determinants located in a central segment of the AKAP79 linear sequence act in tandem to anchor PP2B and inhibit phosphatase activity inside cells. Apparently multisite contact between phosphatases and their binding partners is a common theme in the subcellular targeting of this enzyme class, as a second PP2B-binding site has been defined within the regulatory domain of NFATc which may act synergistically with the PIXIXIT motif to permit a high affinity interaction with the phosphatase (39). Similarly, there are examples of type 1 phosphatase targeting subunits that interact with the catalytic subunit (PP1c) through multiple sites (40, 41).
One goal of our study was to generate peptide-based antagonists of PP2B function inside cells. Accordingly, we have shown that perfusion of the AKAP79-(330–357) peptide prevents the attenuation of GluR1 type AMPA channels by the AKAP79 signaling complex in HEK293 cells. We have shown previously that AKAP79-mediated anchoring of PP2B confers a calcium-dependent rundown of the channel through the dephosphorylation of serine 845, a site in the cytoplasmic tail of the GluR1 subunit (10). Basal phosphorylation of Ser-845 is maintained by anchored PKA, suggesting that AKAP79 contributes to the regulation of this channel by positioning kinases and phosphates in close proximity to a subset of their substrates (10, 11, 15, 42–45). Our electrophysiological measurements infer that perfusion of the AKAP79-(330–357) peptide shifts the equilibrium in favor of the phosphorylated state of GluR1. Importantly, our control experiments demonstrate that this peptide is only active in the presence of AKAP79, thereby indicating that PP2B targeting ensures efficient modulation of GluR1 currents. Yet our experiments are unable to delineate the mechanism of action of this peptide. Our biochemical analyses would suggest that it must function by displacing PP2B from its site of action and/or as an inhibitor of the anchored phosphatase. Nonetheless, our experiments highlight the utility of this reagent as a cell-based modulator of PP2B. Likewise, Cain has also been used to perturb PP2B function inside cells, as it binds the endocytic machinery through amphiphysin 1 to negatively regulate PP2B/dynamin-dependent synaptic vesicle endocytosis (30, 32). Thus it is tempting to speculate that AKAP79 anchoring and inhibition of PP2B might function in a concerted manner to modulate not only AMPA receptor currents but also to attenuate the phosphorylation events that control receptor endocytosis.
Acknowledgments
We thank Brian Nauert for pCDNA3 PP2B vectors; Iain Fraser for pCDNA AKAP18-GFP; Yvonne Lai (ICOS Corp.) for anti-AKAP79 rabbit polyclonal antisera (918I); and Lisa L. Gomez and Shuvo Alam for technical assistance.
Footnotes
This work was supported in part by Scientist Development Grant AHA-SDG 0130228N from the American Heart Association (to M. L. D.) and National Institutes of Health Grant GM48231 (to J. D. S.).
The abbreviations used are: AKAP, A-kinase anchoring proteins; PKA, cAMP-dependent protein kinase; PKC, protein kinase C; PP2B, protein phosphatase-2B; AMPA, α-amino-3-hydroxy-5-methyl-isoxazole-4-propionic acid; BSA, bovine serum albumin; PBS, phosphatebuffered saline; GFP, green fluorescent protein; EGFP, enhanced GFP.
References
- 1.Pawson T, Scott JD. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- 2.Jordan JD, Landau EM, Iyengar R. Cell. 2000;103:193–200. doi: 10.1016/s0092-8674(00)00112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hunter T. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 4.Colledge M, Scott JD. Trends Cell Biol. 1999;9:216–221. doi: 10.1016/s0962-8924(99)01558-5. [DOI] [PubMed] [Google Scholar]
- 5.Feliciello A, Gottesman ME, Avvedimento EV. J Mol Biol. 2001;308:99–114. doi: 10.1006/jmbi.2001.4585. [DOI] [PubMed] [Google Scholar]
- 6.Carr DW, Stofko-Hahn RE, Fraser IDC, Bishop SM, Acott TS, Brennan RG, Scott JD. J Biol Chem. 1991;266:14188–14192. [PubMed] [Google Scholar]
- 7.Newlon MG, Roy M, Morikis D, Carr DW, Westphal R, Scott JD, Jennings PA. EMBO J. 2001;20:1651–1662. doi: 10.1093/emboj/20.7.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith FD, Scott JD. Curr Biol. 2002;12:R32–R40. doi: 10.1016/s0960-9822(01)00646-7. [DOI] [PubMed] [Google Scholar]
- 9.Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser ID, Langeberg LK, Sheng M, Scott JD. Science. 1999;285:93–96. doi: 10.1126/science.285.5424.93. [DOI] [PubMed] [Google Scholar]
- 10.Tavalin SJ, Colledge M, Hell JW, Langeberg LK, Huganir RL, Scott JD. J Neurosci. 2002;22:3044–3051. doi: 10.1523/JNEUROSCI.22-08-03044.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fraser ID, Scott JD. Neuron. 1999;23:423–426. doi: 10.1016/s0896-6273(00)80795-3. [DOI] [PubMed] [Google Scholar]
- 12.Carr DW, Stofko-Hahn RE, Fraser IDC, Cone RD, Scott JD. J Biol Chem. 1992;24:16816–16823. [PubMed] [Google Scholar]
- 13.Coghlan VM, Perrino BA, Howard M, Langeberg LK, Hicks JB, Gallatin WM, Scott JD. Science. 1995;267:108–112. doi: 10.1126/science.7528941. [DOI] [PubMed] [Google Scholar]
- 14.Klauck TM, Faux MC, Labudda K, Langeberg LK, Jaken S, Scott JD. Science. 1996;271:1589–1592. doi: 10.1126/science.271.5255.1589. [DOI] [PubMed] [Google Scholar]
- 15.Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. Neuron. 2000;27:107–119. doi: 10.1016/s0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 16.Dell'Acqua ML, Faux MC, Thorburn J, Thorburn A, Scott JD. EMBO J. 1998;17:2246–2260. doi: 10.1093/emboj/17.8.2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kashishian A, Howard M, Loh C, Gallatin WM, Hoekstra MF, Lai Y. J Biol Chem. 1998;273:27412–27419. doi: 10.1074/jbc.273.42.27412. [DOI] [PubMed] [Google Scholar]
- 18.Blumenthal DK, Takio K, Hansen RS, Krebs EG. J Biol Chem. 1986;261:8140–8145. [PubMed] [Google Scholar]
- 19.Hausken ZE, Dell'Acqua ML, Coghlan VM, Scott JD. J Biol Chem. 1996;271:29016–29022. doi: 10.1074/jbc.271.46.29016. [DOI] [PubMed] [Google Scholar]
- 20.Rosenmund C, Carr DW, Bergeson SE, Nilaver G, Scott JD, Westbrook GL. Nature. 1994;368:853–856. doi: 10.1038/368853a0. [DOI] [PubMed] [Google Scholar]
- 21.Lester LB, Langeberg LK, Scott JD. Proc Natl Acad Sci U S A. 1997;94:14942–14947. doi: 10.1073/pnas.94.26.14942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fink MA, Zakhary DR, Mackey JA, Desnoyer RW, Apperson-Hansen C, Damron DS, Bond M. Circ Res. 2001;88:291–297. doi: 10.1161/01.res.88.3.291. [DOI] [PubMed] [Google Scholar]
- 23.Feliciello A, Li Y, Avvedimento EV, Gottesman ME, Rubin CS. Curr Biol. 1997;7:1011–1014. doi: 10.1016/s0960-9822(06)00424-6. [DOI] [PubMed] [Google Scholar]
- 24.Sun F, Hug MJ, Bradbury NA, Frizzell RA. J Biol Chem. 2000;275:14360–14366. doi: 10.1074/jbc.275.19.14360. [DOI] [PubMed] [Google Scholar]
- 25.Dodge KL, Carr DW, Sanborn BM. Endocrinology. 1999;140:5165–5170. doi: 10.1210/endo.140.11.7133. [DOI] [PubMed] [Google Scholar]
- 26.Faux MC, Scott JD. J Biol Chem. 1997;272:17038–17044. doi: 10.1074/jbc.272.27.17038. [DOI] [PubMed] [Google Scholar]
- 27.Faux MC, Rollins EN, Edwards AS, Langeberg LK, Newton AC, Scott JD. Biochem J. 1999;343:443–452. [PMC free article] [PubMed] [Google Scholar]
- 28.Taigen T, De Windt LJ, Lim HW, Molkentin JD. Proc Natl Acad Sci U S A. 2000;97:1196–1201. doi: 10.1073/pnas.97.3.1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Windt LJ, Lim HW, Bueno OF, Liang Q, Delling U, Braz JC, Glascock BJ, Kimball TF, del Monte F, Hajjar RJ, Molkentin JD. Proc Natl Acad Sci U S A. 2001;98:3322–3327. doi: 10.1073/pnas.031371998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lai MM, Burnett PE, Wolosker H, Blackshaw S, Snyder SH. J Biol Chem. 1998;273:18325–18331. doi: 10.1074/jbc.273.29.18325. [DOI] [PubMed] [Google Scholar]
- 31.Luo S, Youn HD, Loh C, Stolow M, He W, Liu JO. Immunity. 1998;8:703–711. doi: 10.1016/s1074-7613(00)80575-0. [DOI] [PubMed] [Google Scholar]
- 32.Lai MM, Luo HR, Burnett PE, Hong JJ, Snyder SH. J Biol Chem. 2000;275:34017–34020. doi: 10.1074/jbc.C000429200. [DOI] [PubMed] [Google Scholar]
- 33.Aramburu J, Garcia-Cozar F, Raghavan A, Okamura H, Rao A, Hogan PG. Mol Cell. 1998;1:627–637. doi: 10.1016/s1097-2765(00)80063-5. [DOI] [PubMed] [Google Scholar]
- 34.Aramburu J, Yaffe MB, Lopez-Rodriguez C, Cantley LC, Hogan PG, Rao A. Science. 1999;285:2129–2133. doi: 10.1126/science.285.5436.2129. [DOI] [PubMed] [Google Scholar]
- 35.Rothermel B, Vega RB, Yang J, Wu H, Bassel-Duby R, Williams RS. J Biol Chem. 2000;275:8719–8725. doi: 10.1074/jbc.275.12.8719. [DOI] [PubMed] [Google Scholar]
- 36.Bregman DB, Bhattacharyya N, Rubin CS. J Biol Chem. 1989;264:4648–4656. [PubMed] [Google Scholar]
- 37.Bregman DB, Hirsch AH, Rubin CS. J Biol Chem. 1991;266:7207–7213. [PubMed] [Google Scholar]
- 38.Hirsch AH, Glantz SB, Li Y, You Y, Rubin CS. J Biol Chem. 1992;267:2131–2134. [PubMed] [Google Scholar]
- 39.Park S, Uesugi M, Verdine GL. Proc Natl Acad Sci U S A. 2000;97:7130–7135. doi: 10.1073/pnas.97.13.7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Allen PB, Kwon YG, Nairn AC, Greengard P. J Biol Chem. 1998;273:4089–4095. doi: 10.1074/jbc.273.7.4089. [DOI] [PubMed] [Google Scholar]
- 41.Schillace RV, Voltz JW, Sim AT, Shenolikar S, Scott JD. J Biol Chem. 2001;276:12128–12134. doi: 10.1074/jbc.M010398200. [DOI] [PubMed] [Google Scholar]
- 42.Swope SL, Moss SI, Raymond LA, Huganir RL. Adv Second Messenger Phosphoprotein Res. 1999;33:49–78. doi: 10.1016/s1040-7952(99)80005-6. [DOI] [PubMed] [Google Scholar]
- 43.Kameyama K, Lee HK, Bear MF, Huganir RL. Neuron. 1998;21:1163–1175. doi: 10.1016/s0896-6273(00)80633-9. [DOI] [PubMed] [Google Scholar]
- 44.Lee HK, Kameyama K, Huganir RL, Bear MF. Neuron. 1998;21:1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- 45.Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]