

Summary
Mycoplasma arthritidis -derived mitogen (MAM) is a superantigen that can activate large fractions of T cells bearing particular TCR Vβ elements. Here we report the crystal structure of MAM complexed with a major histocompatibility complex (MHC) antigen, HLA-DR1, loaded with haemagglutinin peptide 306–318 (HA). The structure reveals that MAM has a novel fold composed of two α-helical domains. This fold is entirely different from that of the pyrogenic superantigens, consisting of a β-grasped motif and a β barrel. In the complex, the N-terminal domain of MAM binds orthogonally to the MHC α1 domain and the bound HA peptide, and to a lesser extent to the MHC β1 domain. Two MAM molecules form an asymmetric dimer and cross-link two MHC antigens to form a plausible, dimerized MAM-MHC complex. These data provide the first crystallographic evidence that superantigens can dimerize MHC molecules. Based on our structure, a model of the TCR2MAM2MHC2 complex is proposed.
Introduction
Superantigens (SAgs) are immunoregulatory proteins generally produced by bacteria and viruses (Kotzin et al., 1993; Li et al., 1999). Upon their binding to the MHC class II molecules, SAgs are recognized by TCR in a Vβ-restricted fashion, resulting in polyclonal activation of a large pool of T lymphocytes (up to 20%). Their discovery offered hopes for effective T cell-based therapies, provided insight into some diseases caused by SAgs and, more importantly, proved to be an effective tool for studying interactions within the trimolecular complex: MHC class II/peptide/TCR.
The best-characterized group of SAgs belongs to the pyrogenic toxin SAg family from Staphylococcus aureus and Streptococcus pyogenes, which include staphylococcal enterotoxins (SEs), staphylococcal toxic shock syndrome toxin-1 (TSST-1), and streptococcal pyrogenic exotoxins (SPEs). Although the crystal structures of several SAgs were reported (see reviews, Li et al., 1999; Mitchell et al., 2000; Sundberg et al., 2002a), our knowledge is limited to the pyrogenic toxins. These SAgs share very similar three-dimensional structures, composed of a β-grasped motif and a β barrel. It is currently unclear whether other SAgs that share no sequence homology with the pyrogenic toxins bear the same scaffold of three-dimensional structure.
Despite carrying the common structural fold, the pyrogenic SAgs bind to the MHC molecules in considerably diversified modes. Currently, two SAg binding sites, including a low-affinity and a zinc-coordinated, high-affinity binding site, have been identified on MHC (Hudson et al., 1995). The SAgs SEA (Petersson et al., 2002), SEB (Jardetzky et al., 1994), SEC3 (Sundberg et al., 2002b), and TSST-1 (Kim et al., 1994) share a partially overlapping low-affinity binding site on the MHC α1 domain. SEA, SEB, and SEC3 do not contact the bound peptides in the “groove” of the MHC class II molecules, whereas TSST-1 seems to cover a portion of the C-terminal part of the bound peptide. In contrast, SPEC and SEH bind, each via a zinc ion, in slightly varying orientations to the high-affinity binding site on the MHC β chain (Li et al., 2001; Petersson et al., 2001) and make extensive contacts with the N-terminal portion of the bound peptides. In addition, some SAgs can form zinc-dependent homodimers (Al-Daccak et al., 1998; Langlois et al., 2003), which could in turn dimerize the MHC class II molecules on the APC surface (Al-Daccak et al., 1998). It has been proposed that dimerization or oligomerization of the MHC antigens by SAgs is critical for both T cell activation and T cell-independent cytokine expression on various human APCs (Mehindate et al., 1995). Although a hypothetical model of a SEA1-MHC2 complex was recently proposed on the basis of the crystal structure of a SEA mutant in complex with MHC (Petersson et al., 2002), a dimerized SAg-MHC complex has not yet been described crystallographically.
Mycoplasma arthritidis-derived mitogen (MAM) is produced by Mycoplasma arthritidis (Cole, 1991). MAM can induce spontaneous chronic arthritis in genetically susceptible strains of rodents that resembles human rheumatoid arthritis. Although MAM functions like a conventional SAg, it does not share significant global sequence homology with other SAgs (Cole et al., 1996). Like other SAgs, MAM interacts with TCR in a Vβ-restricted fashion. However, in contrast to other SAgs, MAM binding is influenced by the TCR CDR3 (Hodtsev et al., 1998), suggesting that MAM represents a new type of ligand for TCR, distinct from both conventional peptide antigens and other known SAgs. It was recently observed that MAM preferentially binds to HLA-DR1 with high affinity comparable to murine MHC class II (Etongue-Mayer et al., 2002).
In this study, we report the crystal structure, at 2.6 Å resolution, of MAM complexed with a MHC class II molecule, HLA-DR1, loaded with haemagglutinin peptide 306–318 (HA). Our results show that MAM adopts a novel fold composed of two completely α-helical domains, and thus represents a new family of SAgs. In the complex, the N-terminal domains of MAM interact orthogonally with the antigen-presenting domains of the MHC molecules. Two MAM molecules form an asymmetric dimer through the C-terminal domains of MAM. The MAM dimer cross-links two HLA-DR1/HA complexes to form a dimerized MAM-MHC complex, providing the first crystallographic evidence that superantigens can dimerize MHC molecules.
Results and Discussion
Overall Structure of the MAM/HA/ HLA-DR1 Complexes
The structure was solved by a combination of molecular replacement and single-wavelength anomalous diffrac- tion (SAD) phasing methods (Figure 1A; Table 1). All parts of the complex structure were well ordered. A representative electron density map is shown (Figure 1D). In the final refined model, two MAM/HA/HLA-DR1 complexes form an asymmetric dimer (Figure 1A). Each MAM/HA/DR1 complex comprises residues 1–181 of the DR1 α chain, 1–190 of the DR1 β chain, 306–318 of the HA peptide, 1–213 of MAM, a serine residue from the GST tag, and two phosphate groups. In addition, 106 water molecules were included in the final model.
Figure 1. Overall Structure of the HLA-DR1/HA/MAM Complex.

(A) A dimerized MAM-MHC complex in the asymmetric unit. Two pairs of phosphate groups are shown in ball-and-stick representation at each complex interface.
(B) Ribbon diagram of MAM dimer. The N- and C termini of MAM-2 are labeled with N and C, respectively. The elements contributing to the dimer interface are labeled, including α5 (1), loop α5-α6 (2), α8 (4), and loop α8-α9 (3) from MAM-1, and α2 (7), α4 (2), α8 (2) and α9 (2) from MAM-2. The number in parentheses indicates the number of residues contributed to the dimer interface.
(C) A single HLA-DR1/HA/MAM complex, showing the overall geometry between HLA-DR1 and MAM. The α1, α2, β1, and β2 domains of HLA-DR1 are labeled. A pair of phosphate groups is shown in ball-and-stick representation at the interface. Each component is colored as follows: MAM, red; the DR1 α domain, cyan; the DR1 β domain, blue; the HA peptide, magenta. The N-terminal loop of MAM (1–25) is colored orange.
(D) Stereoview of a representative (2Fo-Fc) electron density map, contoured at 1.0 σ, at the complex interface, showing the HA peptide (magenta), MAM (green), phosphate groups (yellow). Here and in the following figures (unless otherwise specified), atoms are colored as follows: carbon, yellow; nitrogen, blue; oxygen, red; phosphate, green. Residues are labeled for the HA peptide (magenta) and MAM (black).
(E) Stereoview ribbon diagram of MAM monomer showing the overall fold and secondary structure elements of MAM with every tenth residue labeled. The ten α helices, including α1 (25–35), α2 (41–65), α3 (71–93), and α4 (96–125) from the N-terminal domain, and α5 (126–146), α6 (157–168), α7 (170–175), α8 (176–195), α9 (199–203), and α10 (204–211) from the C-terminal domain, are labeled. The N- and C termini are labeled as N and C, respectively. Asn125, which is shared by α4 and α5 helices and covalently connects the N- and C-terminal domains, is labeled. The N-terminal loop (1–25) is colored orange.
Table 1.
Data Collection, SAD Phasing, and Refinement Statistics
| Data collection and SAD phasing
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Data | Wavelength (Å) | Resolution (Å) | Redundancy | Completeness (%) | Average I/σ(I) | Rsym (%) | Rano (%) | Se Sites | FOM |
| Native | 1.03 | 3.3 | 2.1 | 81.3 (74.0)a | 7.4 (3.9) | 11 (43.9) | |||
| SAD1 | 0.978 | 3.4 | 4.7 | 99.5 (99.9) | 12.7 (5.3) | 13.9 (33.4) | 6.8 | 6 | 0.09 |
| SAD2 | 0.979 | 2.6 | 3.6 | 95.9 (80.4) | 15 (2.1) | 8.0 (48.9) | |||
| Refinement
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Resolution (Å) | No. of Reflections | Rwork | Rfree | Non-H Atoms
|
Average B | Rmsd
|
|||
| Protein | Water | PO43− | Distance | Angle | |||||
| 100–2.6 | 65311 | 23.8% | 24.6% | 9888 | 106 | 4 | 45.7 Å2 | 0.008 Å | 1.4° |
Values in parentheses are those for the highest resolution shell.
The Novel Superantigen Fold
As shown in Figure 1E, MAM displays a completely α-helical structure that consists of ten α helices. The overall structure of MAM monomer is L-shaped and is composed of two domains. Topology analysis, using the SCOP database (Murzin et al., 1995) and the Web servers TOP (Lu, 2000) and VAST (Gibrat et al., 1996), showed that there is no published fold similar to the overall structure of MAM. However, the structures of individual N- and C-terminal domains are similar to the known folds. The central feature of the N-terminal domain (residues 1–125) is a four-α-helical bundle (Figure 1E), which is similar to many functionally unrelated four- helical proteins. Database searches indicated that the bundle is most similar to that of bacteriorhodopsin (Luecke et al., 1998) (root-mean-square deviation [rmsd] of 2.7 Å for 80 Cα pairs) and to that of E. coli L-aspartate ammonia-lyase (Shi et al., 1997) (rmsd of 2.9 Å for 79 Cα pairs). Beyond the bundle, the N terminus of MAM is a long loop of 25 residues (1–25), which wraps around the α-helical bundle (Figures 1C, 1E, and 2C). The C-terminal domain (126–213) contains six helices (Figure 1E). The scaffold of the C-terminal domain is most similar to that of the N-terminal domain of bacteriophage procapsid protein gpD (Dokland et al., 1997), in which one central helix is surrounded by six others. Three pairs of the helices, including α5, α6, and α8 in MAM and α1, α3, and α4 in gpD, can be superimposed, resulting in an rmsd of 2.2 Å for 40 Cα pairs.
Figure 2. Details of MAM Dimer Interface and MAM/HA/DR1 Interface Shown as Stereoviews.

In all three panels, the letter before the residue number represents the chain name in the crystal structure.
(A) MAM homodimer interface. MAM-1 is colored gray, and MAM-2 is colored green. Key residues are depicted as ball-and-stick representations with bonds and carbon atoms colored as follows: MAM-1, yellow; MAM-2, green. Residues are color-labeled black and red for MAM-1 and MAM-2, respectively. The four hydrogen bonds, including Arg154 Nη2-Arg192 Nη1, Tyr193 Oη-Lys51 Nζ, Gly196 O-His58 Nδ1, and Asp197 O-Gln54 Nε2, are shown as orange dashed lines.
(B and C) Detailed views of HLA-DR1/HA/MAM interfaces, showing hydrogen bonds and hydrophobic interactions. The ribbon diagram of each component in the complex is colored as follows: MAM, light blue; peptide, magenta; HLA-DR1, gray. Key residues are displayed as ball-and-stick representations with bonds and carbon/phosphate atoms colored as follows: MAM, green; the HA peptide, magenta; HLA-DR1, yellow; PO43−, cyan. Hydrogen bonds are shown as orange dashed lines. Residues are color-labeled as follows: MAM, black; HLA-DR1, blue; peptide, magenta; phosphate, red.
Although MAM is a two-domain structure, each domain is tightly packed against the other (Figure 1E). The helix α5 from the C-terminal domain folds back toward the N-terminal helical bundle, making α5 almost perpendicular to α4. The domain association buries a total of 1201 Å2 of solvent-accessible surface (SAS) area (calculated using a probe of 1.4 Å), with equal contributions from both domains. At the domain interface, there are 12 hydrogen bonds including two salt bridges (data not shown), suggesting that MAM forms a rigid structure.
The crystal structure of the MAM/HA/DR1 complex clearly reveals that MAM adopts a novel fold that is entirely different from those of other pyrogenic SAgs. To date, more than a dozen SAgs have been structurally characterized. Except Urtica dioica agglutinin, a plant lectin with superantigenic activity (Saul et al., 2000), all other SAgs of known structure are from the pyrogenic SAg family. The typical structure of a pyrogenic SAg is composed of two domains, defined as large and small. The large domain consists of a central helix resting against a four-stranded β sheet, which is known as a β-grasped motif (Mitchell et al., 2000). The small domain is a five-stranded β barrel of oligonucleotide/oligosaccharide binding fold (Mitchell et al., 2000). In contrast, the structure of MAM is completely α-helical, representing a novel SAg fold and a new type of ligand for TCR.
The MAM Homodimer and Dimerized MAM/MHC Complex
Although size-exclusion chromatography showed that MAM exists as a monomer in solution at micromolar concentration (data not shown), cross-linking experiments have suggested that MAM can form a zinc-dependent dimer in solution (Langlois et al., 2003). Indeed, there are two HLA-DR1/HA/MAM complexes in the asymmetric unit in the crystal. Two MAM molecules form a dimer bridging two MHC molecules (Figure 1A). Although the MAM dimer is formed through a nearly perfect pseudo 2-fold symmetry (177° rotation), the two MAM molecules, termed MAM-1 and MAM-2, interact asymmetrically (Figure 1B). Overall, the C-terminal domains of MAM molecules play a major role in dimer formation. In particular, the C-terminal domain of MAM-1 interacts with both the N- and C-terminal domains of MAM-2, whereas the C-terminal domain of MAM-2 only contacts the C-terminal domain of MAM-1.
At the dimer interface, the C-terminal domain of MAM-1 fits into a V-shaped pocket between the two domains of MAM-2 (Figure 1B). There are 24 residues from both molecules involved in the dimer interface. However, only one residue, Asp 197, is used in common, but involving different interactions. Asp 197 from MAM-1 forms a hydrogen bond with His58 of MAM-2, whereas Asp197 from MAM-2 forms van der Waals contacts with Arg154 of MAM-1 (Figure 2A). For MAM-1, all 11 residues at the dimer interface are from the C-terminal domain. In contrast, 13 residues from MAM-2 involved in the dimer association are contributed by both domains, al- though the N-terminal domain plays the major role by contributing two-thirds of the interactions. Most of the residues at the dimer interface are polar residues, but only four hydrogen bonds occur (Figure 2A). The main hydrophobic contacts are contributed by Ile144, Tyr193, and Tyr194 from MAM-1, and Leu50 from MAM-2. Other interactions include mainly side chain stacking contacts through the aliphatic portions of the polar residues (Figure 2A).
The total buried surface area of 1818 Å2 at the dimer interface falls within the average range of 1600 ± 400 Å2 observed for protein-protein interactions (Lo Conte et al., 1999). Each MAM monomer contributes the buried SAS area roughly equivalently (MAM-1 [53%] and MAM-2 [47%]) of about 900 Å2, representing 7% of the total surface area of 12,642 Å2 for a MAM monomer. Although 46 residues contribute to the buried surface area, only 7, including Arg154, Arg192, Tyr194, Glu195, and Asp197 from MAM-1, and Leu50 and Lys201 from MAM-2, make large contributions, totaling about 43% of the buried surface area.
Although it was reported that zinc ion may be involved in MAM/MHC interaction or in the formation of a zinc-dependent MAM homodimer (Etongue-Mayer et al., 2002; Langlois et al., 2003), no zinc ion could be located at the current structure. In addition, a cross-linking experiment using disuccinimidyle glutarate (DSG) suggested that a portion of MAM exists as a dimer regardless of the presence of zinc ions (data not shown). Therefore, the exact role of zinc ion for MAM function needs further investigation.
As discussed above, the interactions at the dimer interface are asymmetric. The asymmetric association of the dimer is not due to the internal domain flexibility of MAM. Although the two domains of MAM were refined with two independent groups of noncrystallographic symmetry (NCS) restraints, the two MAM molecules in the asymmetric unit are almost identical, with an rmsd of 0.09 Å for all 213 Cα pairs. Through crystal packing, each MAM molecule interacts identically with two MHC molecules (Figure 4A). Therefore, except at the dimer interface, the two MAM monomers face essentially the same environment, implying that crystal-packing interactions may not be the determinant factor in affecting the dimer association. In addition, the shape correlation statistic (Sc) (Lawrence and Coleman, 1993) of 0.64 for the dimer interface is comparable to the value for antibody-antigen complexes (0.64–0.75) (Li et al., 2003), indicating a good shape complementarity between the two monomers. These data suggest that the dimer association in the crystal structure represents a physiological form of MAM dimer existing in solution, although crystal-packing effects may not be completely ruled out.
Figure 4. Model of Dimerized TCR-MAM-MHC Complex.
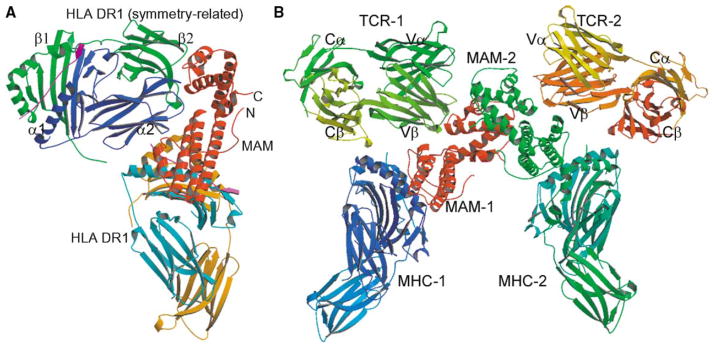
(A) Overall geometry of MAM interaction with the HLA-DR1/HA complexes. The α1, α2, β1, and β2 domains of the symmetry-related HLA-DR1 molecule are labeled.
(B) A model of the TCR2-MAM2-MHC2 complex. The model was built by superposition of the Vβ17 domain of the JM22 TCR onto the Cα-reversed α2 domain of the symmetry-related HLA-DR1/HA complex in the crystal structure of the MAM/HA/DR1 complex.
In the crystal the MAM homodimer bridges two MHC molecules to form a dimerized MAM-MHC complex (Figure 1A). This dimerized complex may represent a very plausible arrangement under physiological condition because the C termini of the two MHC molecules in the complex dimer are oriented in the same direction, indicating that they can be anchored on the surface of the same APC. Thus our crystal structure provides the first crystallographic evidence that the SAgs can indeed dimerize MHC class II molecules.
The MAM/HA/DR1 Interface: Overall View of the Complex
Each MAM molecule of the MAM dimer interacts with one class II MHC HLA-DR1/HA complex in the same fashion (Figure 1A). In fact, structure superposition of the two MAM/HA/HLA-DR1 complexes in the asymmetric unit results in an rmsd of 0.36 Å for 597 Cα pairs. Therefore, although the results and further discussion will be based on the conformation of complex 1 (Figure 1C), the conclusions will be generally applicable to complex 2 as well.
In the complex structure, the MAM/HA/DR1 interaction buries 2208 Å2 of SAS area by an equal contribution from MAM and the HLA-DR1/HA complex. This total buried surface area is the largest observed among the known SAg/MHC complexes (Jardetzky et al., 1994; Kim et al., 1994; Li et al., 2001; Petersson et al., 2001, 2002). The interface is also larger than that in the TCR/peptide/ MHC complexes (~1800 Å2) (Garcia et al., 1999) or that in most antibody-antigen interfaces (1600 ± 400 Å2) (Lo Conte et al., 1999). The Sc value of 0.69 for the MAM/ HA/DR1 interface is similar to that for other SAg/MHC complexes (0.62–0.65) and is higher than that for most TCR/peptide/MHC complexes (0.46–0.70) (Li et al., 1999). It reflects a good shape complementarity. Consistently, MAM binds to HLA-DR1 in high affinity of nanomolar range (Etongue-Mayer et al., 2002).
The contacting interface is composed of 27 residues from MAM, 20 from the DR1 α chain, 6 from the DR1 β chain, and 9 from the bound HA peptide (Table 2). Six of the residues from MAM and DR1 molecules, including Lys39α, Leu60α, Gln12m, His14m, Phe15m, and Val85m, individually make large contributions of more than 80 Å2 to the buried SAS area. (Here and in the following unless otherwise specified, suffixes after residue numbers signify as follows: α, the DR1 α chain; β, the DR1 β chain; and m, MAM). These results are consistent with the finding that synthetic MAM peptides composed of residues 14–31 or 11–38 can inhibit MAM binding to the HLA-DR1 molecule (Etongue-Mayer et al., 2002; Knudtson et al., 1997). In addition, a double mutant of MAM, in which His14 and Asp31 were both changed to alanines, failed to trigger the activation of T cell hybridomas ex- pressing Vβ8.1 and Vβ6 (Langlois et al., 2003). The double-mutant MAM can activate the Kmls hybridoma ex- pressing Vβ15.8, although it does so less efficiently than does the wild-type. This implies that other regions of MAM must play a role in MAM-DR1 recognition. Indeed, although individually contributing less buried surface area, 16 residues cover up more than 70% of their SAS area upon complex formation (Table 2). These results are consistent with mutagenesis and biochemical data. For example, synthetic peptides encompassing MAM residues 76–90 or 71–95 can inhibit the binding between MAM and HLA-DR1 (Etongue-Mayer et al., 2002; Knudtson et al., 1997). On the other hand, triple mutations at the DR1 residues Ile63α, Val65α, and Ala68α resulted in the loss of presentation of MAM to T cells (Etongue-Mayer et al., 2002), while two other mutations at Val42α and Glu46α, which are not at the MAM/HA/DR1 interface, do not affect the presentation of MAM to T cells (Etongue-Mayer et al., 2002).
Table 2.
Interactions between HLA-DR1/HA and MAM
| MAM
|
||||
|---|---|---|---|---|
| Hydrogen Bondsa | Van der Waals Contactsb | |||
| HLA-DR1 | ||||
| Tyr13αc (7, 88%)d | Oη | Asp88 (58, 91%) | Oδ2 | Asp88 |
| Asp17α (20, 30%) | O | Lys92 (48, 30%) | Nζ | |
| Gln18α (68, 61%) | Oε1 | Arg91 (28, 25%) | Nη1 | Asp88, Arg91, Lys92 |
| Nε2 | Arg91 | Nη1 | ||
| Nε2 | Asp88 | Oδ1 | ||
| O | Lys92 | Nζ | ||
| Ala37α (69, 73%) | O | Gln99 (29, 85%) | Nε2 | Thr89 (40, 78%), Gln99 |
| Lys38α (47, 36%) | Arg102 (57, 43%) | |||
| Lys39α (91, 67%) | Nζ | Glu109 (56, 49%) | Oε1 | Ile106 (64, 90%), Glu109 |
| Gln57α (49, 39%) | Oε1 | Lys82 (57, 79%) | Nζ | Lys82, Glu109, Lys113 (19, 27%) |
| Leu60α (81, 86%) | Val85 (80, 91%), Ile106 | |||
| Ala61α (45, 83%) | Met78 (17, 57%) | |||
| Asn62α (5, 100%) | His14 (100, 91%) | |||
| Ile63α (11, 85%) | Val85 | |||
| Ala64α (55, 98%) | Leu81 (42, 76%), Lys82, Val85, | |||
| Val65α (41, 98%) | His14, Met78 | |||
| Lys67α (51, 65%) | Nζ | Ser84 (22, 55%) | Oγ | Ser84, Asp88 |
| Nζ | Asp88 | |||
| Nζ | Ser84 | O | ||
| Ala68α (36, 72%) | Leu81 | |||
| Tyr60β (43, 43%) | Oη | Gln17 (7, 12%) | Oε1 | |
| Gln64β (23, 31%) | Phe15 (131, 96%) | |||
| Asp66β (38, 30%) | Lys13 (32, 33%) | |||
| Leu67β (51, 73%) | Phe15 | |||
| HA peptide | ||||
| P3 Lys (26, 44%) | Nζ | Gln12 (84, 61%) | Oε1 | Gln12 |
| P5 Asn (59, 94%) | Nδ2 | Lys13 | O | Gln12, Lys13, His14 |
| P6 Thr (11, 100%) | O | His14 | Nδ1 | His14 |
| P7 Leu (21, 72%) | Lys13, His14, Phe15 | |||
| P8 Lys (104, 97%) | N | His14 | O | His14, Phe15, Asn18, |
| Nζ | Asn18 (56, 48%) | Oδ1 | Leu19 (29, 40%) | |
| Nζ | Asn18 | Nδ2 | Val 16 (18, 100%) | |
|
| ||||
| HLA-DR1/HA | MAM | |||
|
| ||||
| First PO43− | ||||
| O1 | His14 | Nε2 | ||
| Gln12 | O | |||
| O2 | Lys113 | Nζ | ||
| Asp117 | Oδ1 | |||
| O3 | P3 Lys Nζ | |||
| O4 | Gln12 | N | ||
| Asp117 | Oδ1 | |||
| Second PO43− | ||||
| O1 | P3 Lys Nζ | |||
Hydrogen bonds were calculated using a 2.5–3.4 Å donor-acceptor distance.
Van der Waals contacts <4 Å.
Residues that bury more than 80 Å2 or that bury more than 70% of their SAS area upon complex formation are indicated in bold face.
Values in parentheses indicate the buried solvent-accessible surface area (Å2) and the percentile.
One pair of phosphate groups was found at equivalent positions of the edge of each complex interface (Experimental Procedures). The phosphate groups in the crystal structure may contribute to the stabilization of the local structure of the complex (Table 2), although the exact function of the phosphate is unclear.
Upon complex formation, there are no major structural changes at the interface in the structure of MHC, which is in agreement with the findings for other complexes between SAg and MHC, SAg and TCR, and TCR and MHC (Li et al., 1999), suggesting that conformational change is not a mechanism for MAM to trigger T cell activation.
The MAM/HA/DR1 Interface: Interactions between MAM and HLA-DR1
The MAM/HA/DR1 complex is formed through contacts between the N-terminal domain of MAM and the antigen-presenting domain of HLA-DR1 (Figure 1C). The site on HLA-DR1/HA recognized by MAM is composed of four components: (1) the loops between strands β1 and β2 (L1) and between β3 and β4 (L3) of the DR1 α chain; (2) the α1 helix of the DR1 α chain; (3) the HA peptide; and (4) the β1 helix of the DR1 β chain. The structural elements of MAM at the MAM/HA/DR1 interface mainly involve the N-terminal loop (1–25) and helices α3 and α4. With the tip of the MAM helical bundle pointing away from the MHC groove, helices α3 and α4 of the bundle interact orthogonally with the α1 helix of the DR1 α chain (Figure 1C). The DR1 helix α1 evidently acts as a roller bearing to support the MAM helices. In addition, helices α3 and α4 of MAM contact the MHC L1 and L3 loop regions (Figure 1C). Although helices α3 and α4 are distant from the peptide binding groove, the N-terminal loop of MAM fills the space between the HA peptide and MAM helices α3 and α4 (Figure 1C). The middle portion of the loop, composed of MAM residues 12–18, is located directly above and runs nearly parallel to the HA peptide, creating both hydrogen bonds and van der Waals contacts with the peptide. The loop further extends interactions with the β1 helix of the DR1 β chain (Figures 1C and 2C). The C-terminal domain of MAM is oriented upward and away from the MHC class II molecule, consistent with the observation that a synthetic peptide composed of MAM C-terminal residues 197–211 did not inhibit MAM binding to the HLA-DR1 molecule (Etongue-Mayer et al., 2002). In addition, the structure agrees very well with results of truncation studies (Langlois et al., 2000). Two C-terminal-truncated MAM molecules, terminated at 132 and 176, respectively, kept their ability to bind the MHC class II molecules, although they failed to stimulate the activation of T cell hybridomas.
Table 2 lists the interactions between MAM and HLA-DR1/HA at the complex interface. Of the 26 residues that either make large surface contributions or bury a large percentage of the SAS area upon complex formation, 15, including P7 Leu from the HA peptide, are hydrophobic and 11 are hydrophilic. This implies that hydrophobic interactions play a major role in the formation of the MAM/HA/DR1 complex. The hydrophobic interface can be divided into three “patches” (Figures 2B and 2C). In the first hydrophobic patch, MAM residue Phe15m fits into a small hydrophobic pocket created by hydrophobic residues Tyr60β, Trp61β, and Leu67β, and the aliphatic part of Gln64β of the DR1 β chain (Figure 2C). In addition, the HA residue, P7 Leu, is part of the hydrophobic base for Phe15m (P7 refers the seventh residue from the first anchor residue [P1] for the bound peptide in the MHC “groove”). The second hydrophobic patch is above the α1 helix of the DR1 α chain, facing the peptide binding groove. Residues involved in this patch include Val16m, Leu19m, Met78m, and Leu81m of MAM, and Ala61α, Ala64α, Val65α, Ala68α, and Ile72α of the DR1 α1 helix (Figure 2C). The first and second hydrophobic patches are separated by five hydrogen bonds between MAM and the HA peptide (Figure 2C; Table 2). The second hydrophobic patch extends beyond the DR1 α1 helix to form the third hydrophobic patch. The interactions at the third patch involve the MAM residues Val85, Val86, and Ile106, and the DR1 residues Tyr13α, Met36α, Leu60α, Ile63α, and Ala64α (Figure 2B). The second and third patches are connected through residue Ala64α of HLA-DR1 and together form a large, curved hydrophobic surface.
The three hydrophobic patches (Figures 2B and 2C) are clustered at the center of the complex interface. They are surrounded by 19 hydrogen bonds, involving both main chain and side chain atoms (Table 2). Most of the hydrogen bonds, including those in which only MAM residues are completely buried upon complex formation (Lys82m Nζ-Gln57α Oε1, Asp88m Oδ1-Gln18α Nε2, Asp88m Oδ1-Lys67α Nζ, and Asp88m Oδ2-Tyr13α Oη), are at the periphery of the interface (Figures 2B and 2C).
The MAM/HA/DR1 Interface: Interactions between MAM and the HA Peptide
The MAM and HLA-DR1 molecules together contribute about 89% of the total buried surface area, and the remaining 11% is contributed by the bound HA peptide (Figure 2C; Table 2). Upon complex formation, 246 Å2 of the SAS area of the HA peptide is buried. Nearly half of this (104 Å2) is contributed by one residue, P8 Lys (Table 2). In addition to P8 Lys, P5 Asn, P6 Thr, and P7 Leu bury more than 70% of their SAS area upon complex formation (Table 2).
At the interface, a substantial number of contacts exist between the bound HA peptide and MAM. The HA peptide contributes 43 contacts to the interaction. These account for about one-third of the total contacts between MAM and the HLA-DR1/HA molecules. In addition to the hydrophobic contribution of P7 Leu, the HA peptide forms six hydrogen bonds, including two salt bridges, with MAM (Table 2). The hydrogen bonds between His14m O-P8 Lys N and His14m Nδ1-P6 Thr O (Table 2; Figure 2C) are the only two completely buried ones out of the 19 hydrogen bonds at the interface. Indeed, mutation of MAM residue His14 disrupts MAM binding to class II MHC molecules (Etongue-Mayer et al., 2002). The results suggest that MAM is, at least in part, a peptide-dependent SAg.
Structural Comparison with Other SAg-MHC Complexes
Although they have very different structures, MAM and the pyrogenic SAgs share similar binding sites on the MHC α chain (Figures 3A, 3C, and 3D). The binding site on the HLA-DR1 molecule for MAM is most similar to that for TSST-1 (Figures 3A and 3D), although the total buried surface area in the MAM/DR1 complex is nearly double of that in the TSST-1/DR1 complex. On the other hand, the surface property of MAM and TSST-1 as well as SEB for binding to MHC is very similar. For instance, at one portion of the hydrophobic interface, the TSST-1 residues are Ile42, Leu44, Ile81, and Phe83, which correspond to the MAM residues Leu19, Leu81, Leu16, and Met78, respectively. At another portion of the hydrophobic interface, SEB residues Phe44 and Tyr89 are replaced by Val85 and Ile106 in MAM, respectively. The Tyr89→Ile106 replacement results in the loss of a hydrophilic pocket accommodating the MHC residue, Lys39α, in the SEB/DR1 and TSST-1/DR1 complexes, in which Lys39α is deeply buried at the interface and is involved in buried hydrogen bonds. Consistently, the lysine residue is very important for the binding of both SEB and TSST-1 to MHC. In contrast, the pocket in the MAM-DR1 complex is filled with Ile106 and Val86. As a result, the side chain of Lys39α rotates away from the interface but still forms a salt bridge with MAM residue Glu109 at the surface (Figure 2B). Such a salt bridge may be less important for complex formation than is the buried one, since it may be replaced by water-mediated hydrogen bonds, as demonstrated in the antibody-antigen complex (Dall’Acqua et al., 1998). Indeed, mutation of Lys39α to alanine affects neither the binding affinity between MAM and HLA-DR1 nor the ability to stimulate the activation of T cells (Etongue-Mayer et al., 2002). Nevertheless, the binding site on HLA-DR1 for MAM overlaps those for the pyrogenic SAgs (Figures 3A, 3C, and 3D). Consistently, preincubation of THP-1 cells with SEB, TSST-1, or SEAD227A can completely abolish MAM-induced cytokine expression (Bernatchez et al., 1997). In addition, another SEAF47A mutant, which only binds to the DR1 β chain, can also block MAM’s activity (Bernatchez et al., 1997). This can be explained by the facts that the binding site on MHC for MAM partially overlaps that for SPEC (Li et al., 2001) (Figures 3A and 3E) and that SEA shares with SPEC a single high-affinity binding site on MHC (Li et al., 2001). When the antigen-presenting domains in the SPEC/MHC and MAM/MHC complexes are optimally superimposed, SPEC partially over- laps MAM (Figures 3A and 3E), to an extent sufficient to sterically hinder MAM from accessing its binding site on the MHC α chain.
Figure 3. Surface Representation of the Binding Sites for Representative SAgs and TCRs on MHC/Peptide Complexes.

Surface representation of the binding sites for representative SAgs and TCRs on MHC in complexes, including (A) MAM/HA/HLA-DR1; (B) TCR/HA/HLA-DR1; (C) SEB/HLA-DR1; (D) TSST-1/HLA-DR1; (E) SPEC/MBP/HLA-DR2a. The antigen-presenting domains (α1 and β1) of MHC are optimally superimposed. The buried surface area is colored green. The bound peptides within or out of the binding sites are colored using atomic colors as follows: carbon, yellow; oxygen, red; nitrogen, blue. The N- and C termini and the P5 position of the bound peptides are labeled.
Comparison with Conventional TCR/MHC Complexes: Role of the Bound Peptide
Although MAM and TCR have completely different three-dimensional structures, structural comparison of the HLA-DR1/HA/MAM and HLA-DR1/HA/TCR (Hennecke et al., 2000) complexes reveals that the binding sites for MAM and the TCR HA1.7 on the HLA-DR1 molecule are very similar (Figures 3A and 3B). In the HLA-DR1/HA/ TCR complex, the TCR HA1.7 interacts with residues 39, 57, 58, 61, 65, 67, and 68 of the DR1 α chain, all of which contact MAM in the HLA-DR1/HA/MAM complex. On the DR1 β chain, MAM contacts with residues 60, 61, 64, 66, 67, and 70, while the TCR HA1.7 interacts with residues 64, 66, 69, 70, 77, and 81.
Both MAM and TCR contact the bound HA peptide at a comparable level (Figures 3A and 3B). In fact, the buried surface area by the HA peptide in the MAM/HA/ DR1 complex represents about 22% of the total area contributed by the DR1/HA complex and is comparable to that by the antigenic peptides (~30%) in the conventional TCR/peptide/MHC complexes (Garcia et al., 1999).
Structural comparison reveals that MAM and TCR similarly contact the central portion of the bound HA peptide. This is in contrast to other SAg-MHC complexes of known structures, in which TSST-1, SPEC, or SEH binds either to the C-terminal or to the N-terminal region of the bound peptides, respectively (Kim et al., 1994; Li et al., 2001; Petersson et al., 2001) (Figures 3D and 3E). Indeed, the TCR HA1.7 “spans” the HA peptide between residues P-1 and P8, while MAM contacts the HA residues from P3 to P8 (Figures 3A and 3B). Therefore, both MAM and TCR bind to the central region, at or around the P5 position in the peptide (Figures 3A and 3B). P5 plays a major role in the conventional TCR- peptide/MHC recognition (Garcia et al., 1999). In fact, the HA peptide has very similar conformations in both MAM/MHC and TCR/MHC complexes. Particularly, the side chain conformation of P5 Asn of the HA peptide is nearly identical in both MAM/MHC and TCR/MHC complexes (data not shown), whereas it is different from that in the unbound form of HLA-DR1/HA. These data imply that the bound peptide plays a significant role in the MAM/MHC recognition. On the other hand, MAM may mimic the peptide dependence in conventional an- tigen presentation. Therefore, the nature of the peptide may strongly influence MAM presentation. Consistently, immunological data indicated that MAM binding to MHC is influenced by the bound peptide (Etongue-Mayer et al., 2002). When Hela cells were transfected with HLA-DR1 covalently linked with the HA307-318 peptide, they presented MAM poorly. Triple mutations on the HA peptide, in which P5 Asn, P8 Lys, and P11 Thr were mutated to alanine, significantly increased MAM binding.
Although the peptide dependence of MAM appears inconsistent with the general concept that SAgs activate T cells by optimally crosslinking TCR to MHC, resulting in the bypass of the normal constrains of the TCR specificity and MHC restriction, there is increasing evidence to suggest that the bound peptide in the MHC groove may play a crucial role in modulating SAg activity (Kozono et al., 1995; Wen et al., 1996, 1997). For instance, it has been found that TSST-1 can be presented to T cells 5000-fold stronger by certain peptides than by others. Such a peptide dependence may allow SAgs to distinguish between different types of APCs, which are likely to display distinct arrays of peptides (Woodland et al., 1997). Thus, SAgs may be targeted preferentially to dendritic cells or macrophages and away from cells that express more restricted peptide repertoires. The possibility is supported by experimental data showing that SAgs can be presented differently by identical MHC molecules on different cell types (Yagi et al., 1994). Furthermore, TSST-1 does not compete with SEB for binding to HLA-DR1 on APCs (Thibodeau et al., 1994), even though structural studies demonstrate that both SAgs bind to overlapping regions on HLA-DR1 (Kim et al., 1994). The peptide dependence of SAgs may also allow SAgs to bind MHC in a high-affinity but low-density fashion so as to mimic the low antigen densities in conventional T cell activation triggered by serial engagement of TCRs (Woodland et al., 1997). Such a low-density binding can ensure that SAgs appropriately activate T cells at an optimal level, which may be necessary to avoid the induction of T cell apoptosis resulting from the binding of high ligand densities (Lenardo, 1991; Woodland et al., 1997).
Hypothetical Model of a TCR2-MAM2-MHC2 Complex
The interaction between MAM and the symmetry-related MHC molecule results in an alternative MAM/HA/DR1 complex (Figure 4A). MAM interacts with the α2 domain of the symmetry-related DR1 molecule. The tip of the α2 domain binds into a large curved pocket created by the two domains of MAM. At the cell surface, the tip of the DR1 α2 domain should be closely associated with the cell membrane. The interactions between MAM and the symmetry-related HLA-DR1 are therefore due to crystal packing. Although it does not represent a binding site for DR1 on MAM, the site on MAM may represent the binding site for TCR. This proposal is because the MHC α2 domain carries an immunoglobulin fold in structure similar to that of a TCR. Indeed, superposition of the Vβ domain of a human JM22 TCR (Vα10.2Vβ17) (Stewart-Jones et al., 2003) onto the Cα-reversed α2 domain of the symmetry-related DR1 gives an rmsd of 2.82 Å for 79 Cα pairs. Therefore, a hypothetical model among MAM, HLA-DR1, and JM22 TCR can be established (Figure 4B). In this model no overlap exists between any parts of the three interacting molecules. Several features of the model are consistent with immunological and mutagenesis data. In the model, MAM predominantly interacts with the TCR β chain, although there is possible interaction between the TCR α chain and MAM. This is consistent with the ability of the TCR β chain alone to bind MAM (Cole, 1991). The TCR contacts both the N- and C-terminal domains of MAM, which agrees well with results of the truncation studies in which two C-terminal deletion mutants of MAM failed to trigger T cell activation (Langlois et al., 2000). In addition, the CDR3 loop of the TCR β chain makes contacts with MAM. Consistent with this, it is suggested that the CDR3 region influences the binding of MAM to TCR (Hodtsev et al., 1998).
A MAM homodimer could cross-link two MHC and two TCR molecules to form a TCR2-MAM2-MHC2 complex (Figure 4B). In the model, the TCR2-MAM2-MHC2 complex is mediated by a MAM homodimer. The C termini of the TCRs are directed in opposite directions. This may suggest that MAM could utilize its high-affinity binding to the MHC molecules to simultaneously recruit multiple T cells, so as to achieve highly efficient T cell activation. Nevertheless, although SAgs and TCR are engaged very differently in class II MHC-mediated T cell-signaling complexes, it is remarkable that they achieve a common end result: highly efficient T cell activation.
Experimental Procedures
Protein Production and Crystallization
Soluble MAM was expressed as a GST-fusion protein as described previously (Langlois et al., 2000). The HLA-DR1/HA complex was prepared using a refolding protocol as described (Frayser et al., 1999). The selenomethionine-substituted (Se-Met) version of the DR1 α chain was purified and then refolded together with the DR1 β chain and the HA peptide (PKYVKQNTLKLAT). Purified MAM was concentrated to about 8.8 mg/ml with a buffer of 10 mM Tris-HCl (pH 8.0), 100 mM NaCl, 1 mM DTT; the HA/HLA-DR1 complex was concentrated to 5.4 mg/ml with a buffer of 20 mM HEPES (pH 7.5), 100 mM NaCl, 1 mM DTT. For crystallization, the HLA-DR1/HA complex and MAM were mixed in an equimolar ratio. Initial crystallization conditions were established using sparse matrix screens. Crystals were grown at room temperature in hanging drops by mixing 2 μl of protein solution in the presence of 1 mM Zn(OAc)2 with an equal volume of reservoir solution containing 1.7 M potassium sodium phosphate, 0.1 M HEPES (pH 7.5). Microseeding was used to produce large crystals for both the native and Se-Met complexes for X-ray data collection.
X-Ray Data Collection, Structure Determination, and Refinement
The crystals belong to space group I222, with unit cell parameters a = 137.35 Å, b = 178.18 Å, c = 179.56 Å. There are two MAM/HA/ DR1 complexes per asymmetric unit. Prior to data collection, all crystals were transferred to a reservoir solution containing 20% glycerol and then flash cooled under a nitrogen stream at 100 K and stored in liquid nitrogen. Native data were collected to 3.2 Å resolution at 100 K using a small crystal at beamline 19-BM of the Advanced Photon Source (APS) (Argonne National Laboratory). All of the data were processed and scaled using HKL2000 (Otwinowski and Minor, 1997). With the crystal structure of the HLA-DR1/HA complex (Stern et al., 1994) as a search model, clear solutions for the HLA-DR1 molecules were obtained using the native data by the molecular replacement (MR) program AMoRe (Navaza, 1994). The resultant electron density maps showed some densities beyond the boundary of the DR1/HA molecules, but were insufficient to enable of tracing the MAM molecule. Therefore, a set of single anomalous diffraction (SAD) data for the Se-Met complex crystal was collected to 3.4 Å resolution using a synchrotron X-ray source at beamline X12C (λ = 0.978 Å) of the National Synchrotron Light Source (NSLS) (Brookhaven National Laboratory). High-resolution SAD data up to 2.5 Å resolution were later collected at beamline X25 of NSLS.
In combining of the phase contribution from the MR solutions of the MHC molecules, we performed a phased SAD calculation using the 3.4 Å SAD data with the program SOLVE (Terwilliger and Berendzen, 1999). The positions for six Se atoms were clearly defined. With the combined MR and SAD phase information, noncrystallographic symmetry (NCS) averaging using the program RESOLVE (Terwilliger, 2001) was able to produce an interpretable map. Fragments containing about 50% of the MAM residues could be automatically traced by the program RESOLVE. The model of a MAM monomer was finally completed by manually fitting the electron density map with the MAM sequence using the program TURBO FRODO (Roussel and Cambillau, 1989). The second MAM molecule was then generated using the NCS symmetry. The 2.5 Å resolution data set, with 7% of the data (4640 reflections) set aside as the test data for the Rfree cross-validation, was used to complete the final refinement using CNS (Brunger et al., 1998). Each domain in the MAM/HA/HLA- DR1 complexes was refined as an independent rigid body at the early stage of refinement using the rigid-body refinement option in CNS. Iterative cycles of simulated annealing, positional, tortion angle, and temperature factor (B) refinement were then carried out, interspersed with model rebuilding into σA-weighted (Fo-Fc) and (2Fo-Fc) electron density maps. Omit maps for groups of MAM residues were frequently calculated to check the correct tracing and conformations. In addition, NCS restraints defined by pairs of individual domain were used throughout the refinement. During structure refinement and model rebuilding, four peaks over 8 σ were observed in the (Fo-Fc) electron density maps. The peaks form two pairs. Each peak is at least 3.7 Å away from the closest protein ligand; therefore it is impossible for these peaks to represent zinc ions that have average distance of 2.09 Å between a zinc ion and its ligand. These densities could be fitted perfectly as phosphate groups (PO43−), which are present at a high concentration (1.7 M) in the crystallization buffer. Although the crystals were grown in the presence of 1 mM ZnCl2, zinc ion could not be found at any part of the complex structure, including both the MAM dimer and MAM-DR1 interfaces. Consistently, an anomalous signal corresponding to the zinc absorption edge could not be detected for the crystals using a synchrotron X-ray beam (data not shown), implying that zinc ion may not be coordinated into the crystals.
At 2.6 Å resolution, the current Rcryst is 23.8% with an Rfree of 24.5%. For the nonglycine residues, 99.8% of the main chain torsion angles lie in the most favored or allowed regions of the Ramachandran plot. Only residues Asn33 of the β chain of HLA-DR1 from both complexes in the asymmetric unit fall into the disallowed regions. This residue, clearly defined in the density map, has a conformation very similar to that in the unbound DR1/HA complex (Stern et al., 1994), SEA/HA/DR1 complex (Petersson et al., 2002), or SPEC/MBP/ DR2a (Li et al., 2001) complex. It is located within a mixed γ turn and type II β hairpin, in which unusual main chain torsion angles are frequently observed (Li et al., 2000). The refinement statistics are summarized in Table 1. Atomic coordinates have been deposited in the Protein Data Bank as entry 1R5I.
Acknowledgments
This research is supported by a grant (AI50628) from the National Institute of Health (NIH) (to H.L.). We thank R.A. Mariuzza, G. Winslow, and P. Van Roey for critical readings of the manuscript, L.J. Stern for the gift of HLA-DR1 expression plasmids, and the Peptide Synthesis Core facility at the Wadsworth Center for the synthesis of the HA peptide. We thank R.G. Zhang at APS, and R. Sweet and M. Becker at NSLS for assistance in X-ray data collection. The X-ray diffraction facilities at the APS and NSLS are supported by the Department of Energy and by grants from NIH.
Footnotes
Accession Numbers
Atomic coordinates have been deposited in the Protein Data Bank as entry 1R5I.
References
- Al-Daccak R, Mehindate K, Damdoumi F, Etongue-Mayer P, Nilsson H, Antonsson P, Sundstrom M, Dohlsten M, Sekaly RP, Mourad W. Staphylococcal enterotoxin D is a promiscuous superantigen offering multiple modes of interactions with the MHC class II receptors. J Immunol. 1998;160:225–232. [PubMed] [Google Scholar]
- Bernatchez C, Al-Daccak R, Mayer PE, Mehindate K, Rink L, Mecheri S, Mourad W. Functional analysis of Mycoplasma arthritidis-derived mitogen interactions with class II molecules. Infect Immun. 1997;65:2000–2005. doi: 10.1128/iai.65.6.2000-2005.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Cole BC. The immunobiology of Mycoplasma arthritidis and its superantigen MAM. Curr Top Microbiol Immunol. 1991;174:107–119. doi: 10.1007/978-3-642-50998-8_8. [DOI] [PubMed] [Google Scholar]
- Cole BC, Knudtson KL, Oliphant A, Sawitzke AD, Pole A, Manohar M, Benson LS, Ahmed E, Atkin CL. The sequence of the Mycoplasma arthritidis superantigen, MAM: identification of functional domains and comparison with microbial superantigens and plant lectin mitogens. J Exp Med. 1996;183:1105–1110. doi: 10.1084/jem.183.3.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dall’Acqua W, Goldman ER, Lin W, Teng C, Tsuchiya D, Li H, Ysern X, Braden BC, Li Y, Smith-Gill SJ, Mariuzza RA. A mutational analysis of binding interactions in an antigen-antibody protein-protein complex. Biochemistry. 1998;37:7981–7991. doi: 10.1021/bi980148j. [DOI] [PubMed] [Google Scholar]
- Dokland T, McKenna R, Ilag LL, Bowman BR, Incardona NL, Fane BA, Rossmann MG. Structure of a viral procapsid with molecular scaffolding. Nature. 1997;389:308–313. doi: 10.1038/38537. [DOI] [PubMed] [Google Scholar]
- Etongue-Mayer P, Langlois MA, Ouellette M, Li H, Younes S, Al-Daccak R, Mourad W. Involvement of zinc in the binding of mycoplasma arthritidis-derived mitogen to the proximity of the HLA-DR binding groove regardless of histidine 81 of the beta chain. Eur J Immunol. 2002;32:50–58. doi: 10.1002/1521-4141(200201)32:1<50::AID-IMMU50>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Frayser M, Sato AK, Xu L, Stern LJ. Empty and peptide-loaded class II major histocompatibility complex proteins produced by expression in E. coli, and folding in vitro. Protein Expr Purif. 1999;15:105–114. doi: 10.1006/prep.1998.0987. [DOI] [PubMed] [Google Scholar]
- Garcia KC, Teyton L, Wilson IA. Structural basis of T cell recognition. Annu Rev Immunol. 1999;17:369–397. doi: 10.1146/annurev.immunol.17.1.369. [DOI] [PubMed] [Google Scholar]
- Gibrat JF, Madej T, Bryant SH. Surprising similarities in structure comparison. Curr Opin Struct Biol. 1996;6:377–385. doi: 10.1016/s0959-440x(96)80058-3. [DOI] [PubMed] [Google Scholar]
- Hennecke J, Carfi A, Wiley DC. Structure of a covalently stabilized complex of a human alphabeta T-cell receptor, influenza HA peptide and MHC class II molecule, HLA-DR1. EMBO J. 2000;19:5611–5624. doi: 10.1093/emboj/19.21.5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodtsev AS, Choi Y, Spanopoulou E, Posnett DN. Mycoplasma superantigen is a CDR3-dependent ligand for the T cell antigen receptor. J Exp Med. 1998;187:319–327. doi: 10.1084/jem.187.3.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson KR, Tiedemann RE, Urban RG, Lowe SC, Strominger JL, Fraser JD. Staphylococcal enterotoxin A has two cooperative binding sites on major histocompatibility complex class II. J Exp Med. 1995;182:711–720. doi: 10.1084/jem.182.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardetzky TS, Brown JH, Gorga JC, Stern LJ, Urban RG, Chi YI, Stauffacher C, Strominger JL, Wiley DC. Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature. 1994;368:711–718. doi: 10.1038/368711a0. [DOI] [PubMed] [Google Scholar]
- Kim J, Urban RG, Strominger JL, Wiley DC. Toxic shock syndrome toxin-1 complexed with a class II major histocompatibility molecule HLA-DR1. Science. 1994;266:1870–1874. doi: 10.1126/science.7997880. [DOI] [PubMed] [Google Scholar]
- Knudtson KL, Manohar M, Joyner DE, Ahmed EA, Cole BC. Expression of the superantigen Mycoplasma arthritidis mitogen in Escherichia coli and characterization of the recombinant protein. Infect Immun. 1997;65:4965–4971. doi: 10.1128/iai.65.12.4965-4971.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotzin BL, Leung DY, Kappler J, Marrack P. Superantigens and their potential role in human disease. Adv Immunol. 1993;54:99–166. doi: 10.1016/s0065-2776(08)60534-9. [DOI] [PubMed] [Google Scholar]
- Kozono H, Parker D, White J, Marrack P, Kappler J. Multiple binding sites for bacterial superantigens on soluble class II MHC molecules. Immunity. 1995;3:187–196. doi: 10.1016/1074-7613(95)90088-8. [DOI] [PubMed] [Google Scholar]
- Langlois MA, Etongue-Mayer P, Ouellette M, Mourad W. Binding of Mycoplasma arthritidis-derived mitogen to human MHC class II molecules via its N terminus is modulated by invariant chain expression and its C terminus is required for T cell activation. Eur J Immunol. 2000;30:1748–1756. doi: 10.1002/1521-4141(200006)30:6<1748::AID-IMMU1748>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Langlois MA, El Fakhry Y, Mourad W. Zinc-binding sites in the N terminus of Mycoplasma arthritidis-derived mitogen permit the dimer formation required for high affinity binding to HLA-DR and for T cell activation. J Biol Chem. 2003;278:22309–22315. doi: 10.1074/jbc.M300823200. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Coleman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- Lenardo MJ. Interleukin-2 programs mouse alpha beta T lymphocytes for apoptosis. Nature. 1991;353:858–861. doi: 10.1038/353858a0. [DOI] [PubMed] [Google Scholar]
- Li H, Llera A, Malchiodi EL, Mariuzza RA. The structural basis of T cell activation by superantigens. Annu Rev Immunol. 1999;17:435–466. doi: 10.1146/annurev.immunol.17.1.435. [DOI] [PubMed] [Google Scholar]
- Li Y, Li H, Smith-Gill SJ, Mariuzza RA. Three-dimensional structures of the free and antigen-bound Fab from monoclonal antilysozyme antibody HyHEL-63(,) Biochemistry. 2000;39:6296–6309. doi: 10.1021/bi000054l. [DOI] [PubMed] [Google Scholar]
- Li Y, Li H, Dimasi N, McCormick JK, Martin R, Schuck P, Schlievert PM, Mariuzza RA. Crystal structure of a superantigen bound to the high-affinity, zinc-dependent site on MHC class II. Immunity. 2001;14:93–104. doi: 10.1016/s1074-7613(01)00092-9. [DOI] [PubMed] [Google Scholar]
- Li Y, Li H, Yang F, Smith-Gill SJ, Mariuzza RA. X-ray snapshots of the maturation of an antibody response to a protein antigen. Nat Struct Biol. 2003;10:482–488. doi: 10.1038/nsb930. [DOI] [PubMed] [Google Scholar]
- Lo Conte L, Chothia C, Janin J. The atomic structure of protein-protein recognition sites. J Mol Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- Lu G. TOP: a new method for protein structure comparisons and similarity searches. J Appl Crystallogr. 2000;33:176–183. [Google Scholar]
- Luecke H, Richter HT, Lanyi JK. Proton transfer pathways in bacteriorhodopsin at 2.3 angstrom resolution. Science. 1998;280:1934–1937. doi: 10.1126/science.280.5371.1934. [DOI] [PubMed] [Google Scholar]
- Mehindate K, Thibodeau J, Dohlsten M, Kalland T, Sekaly RP, Mourad W. Cross-linking of major histocompatibility complex class II molecules by staphylococcal enterotoxin A superantigen is a requirement for inflammatory cytokine gene expression. J Exp Med. 1995;182:1573–1577. doi: 10.1084/jem.182.5.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DT, Levitt DG, Schlievert PM, Ohlendorf DH. Structural evidence for the evolution of pyrogenic toxin superantigens. J Mol Evol. 2000;51:520–531. doi: 10.1007/s002390010116. [DOI] [PubMed] [Google Scholar]
- Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- Navaza J. AmoRe: an automated package fro molecular replacemnt. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Petersson K, Hakansson M, Nilsson H, Forsberg G, Svensson LA, Liljas A, Walse B. Crystal structure of a superantigen bound to MHC class II displays zinc and peptide dependence. EMBO J. 2001;20:3306–3312. doi: 10.1093/emboj/20.13.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersson K, Thunnissen M, Forsberg G, Walse B. Crystal structure of a SEA variant in complex with MHC class II reveals the ability of SEA to crosslink MHC molecules. Structure. 2002;10:1619–1626. doi: 10.1016/s0969-2126(02)00895-x. [DOI] [PubMed] [Google Scholar]
- Roussel A, Cambillau C. In Silicon Graphics Geometry Partners Directory. Mountain View, CA: Silicon Graphics; 1989. TURBO FRODO; pp. 77–78. [Google Scholar]
- Saul FA, Rovira P, Boulot G, Damme EJ, Peumans WJ, Truffa-Bachi P, Bentley GA. Crystal structure of Urtica dioica agglutinin, a superantigen presented by MHC molecules of class I and class II. Struct Fold Des. 2000;8:593–603. doi: 10.1016/s0969-2126(00)00142-8. [DOI] [PubMed] [Google Scholar]
- Shi W, Dunbar J, Jayasekera MM, Viola RE, Farber GK. The structure of L-aspartate ammonia-lyase from Escherichia coli. Biochemistry. 1997;36:9136–9144. doi: 10.1021/bi9704515. [DOI] [PubMed] [Google Scholar]
- Stern LJ, Brown JH, Jardetzky TS, Gorga JC, Urban RG, Strominger JL, Wiley DC. Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature. 1994;368:215–221. doi: 10.1038/368215a0. [DOI] [PubMed] [Google Scholar]
- Stewart-Jones GB, McMichael AJ, Bell JI, Stuart DI, Jones EY. A structural basis for immunodominant human T cell receptor recognition. Nat Immunol. 2003;4:657–663. doi: 10.1038/ni942. [DOI] [PubMed] [Google Scholar]
- Sundberg EJ, Li Y, Mariuzza RA. So many ways of getting in the way: diversity in the molecular architecture of superantigen-dependent T-cell signaling complexes. Curr Opin Immunol. 2002a;14:36–44. doi: 10.1016/s0952-7915(01)00296-5. [DOI] [PubMed] [Google Scholar]
- Sundberg EJ, Sawicki MW, Southwood S, Andersen PS, Sette A, Mariuzza RA. Minor structural changes in a mutated human melanoma antigen correspond to dramatically enhanced stimulation of a CD4+ tumor-infiltrating lymphocyte line. J Mol Biol. 2002b;319:449–461. doi: 10.1016/S0022-2836(02)00370-4. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC. Maximum-likelihood density modification using pattern recognition of structural motifs. Acta Crystallogr D Biol Crystallogr. 2001;57:1755–1762. doi: 10.1107/S0907444901013737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J. Automated structure solution for MIR and MAD. Acta Crystallogr D Biol Crystallogr. 1999;D55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibodeau J, Cloutier I, Lavoie PM, Labrecque N, Mourad W, Jardetzky T, Sekaly RP. Subsets of HLA-DR1 molecules defined by SEB and TSST-1 binding. Science. 1994;266:1874–1878. doi: 10.1126/science.7997881. [DOI] [PubMed] [Google Scholar]
- Wen R, Cole GA, Surman S, Blackman MA, Woodland DL. Major histocompatibility complex class II-associated peptides control the presentation of bacterial superantigens to T cells. J Exp Med. 1996;183:1083–1092. doi: 10.1084/jem.183.3.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen R, Broussard DR, Surman S, Hogg TL, Blackman MA, Woodland DL. Carboxy-terminal residues of major histocompatibility complex class II-associated peptides control the presentation of the bacterial superantigen toxic shock syndrome toxin-1 to T cells. Eur J Immunol. 1997;27:772–781. doi: 10.1002/eji.1830270328. [DOI] [PubMed] [Google Scholar]
- Woodland DL, Wen R, Blackman MA. Why do superantigens care about peptides? Immunol Today. 1997;18:18–22. doi: 10.1016/s0167-5699(97)80009-6. [DOI] [PubMed] [Google Scholar]
- Yagi J, Uchiyama T, Janeway CA., Jr Stimulator cell type influences the response of T cells to staphylococcal enterotoxins. J Immunol. 1994;152:1154–1162. [PubMed] [Google Scholar]