

Background: PP5 binds HSP90 and regulates cell growth and stress-induced signaling pathways.
Results: PP5 associates with ERKs in a manner facilitated, in part, by HSP90. Rac1 and Ras regulate PP5·ERK1/2 complexes.
Conclusion: PP5 and ERK play a role in the feedback phosphorylation of PP5-associated Raf1.
Significance: Differential responsiveness of PP5-ERK1 and PP5-ERK2 interactions to oncogenic small G proteins may contribute to particular tumor traits.
Keywords: ERK, MAP Kinases (MAPKs), Rac, Raf, Ras, S100 Proteins, MEK, PP5, Feedback Phosphorylation, Small G Protein
Abstract
Serine/threonine protein phosphatase 5 (PP5, PPP5C) is known to interact with the chaperonin heat shock protein 90 (HSP90) and is involved in the regulation of multiple cellular signaling cascades that control diverse cellular processes, such as cell growth, differentiation, proliferation, motility, and apoptosis. Here, we identify PP5 in stable complexes with extracellular signal-regulated kinases (ERKs). Studies using mutant proteins reveal that the formation of PP5·ERK1 and PP5·ERK2 complexes partially depends on HSP90 binding to PP5 but does not require PP5 or ERK1/2 activity. However, PP5 and ERK activity regulates the phosphorylation state of Raf1 kinase, an upstream activator of ERK signaling. Whereas expression of constitutively active Rac1 promotes the assembly of PP5·ERK1/2 complexes, acute activation of ERK1/2 fails to influence the phosphatase-kinase interaction. Introduction of oncogenic HRas (HRasV12) has no effect on PP5-ERK1 binding but selectively decreases the interaction of PP5 with ERK2, in a manner that is independent of PP5 and MAPK/ERK kinase (MEK) activity, yet paradoxically requires ERK2 activity. Additional studies conducted with oncogenic variants of KRas4B reveal that KRasL61, but not KRasV12, also decreases the PP5-ERK2 interaction. The expression of wild type HRas or KRas proteins fails to reduce PP5-ERK2 binding, indicating that the effect is specific to HRasV12 and KRasL61 gain-of-function mutations. These findings reveal a novel, differential responsiveness of PP5-ERK1 and PP5-ERK2 interactions to select oncogenic Ras variants and also support a role for PP5·ERK complexes in regulating the feedback phosphorylation of PP5-associated Raf1.
Introduction
Protein phosphatases play crucial roles in regulating the amplitude and duration of mitogen-activated protein kinase (MAPK) signaling, which, in turn, dictates the nature of the cellular response(s) to various stimuli (1, 2). Serine/threonine protein phosphatase 5 (PP52, PPP5C) is one of several phosphatases that regulate the extracellular signal-regulated kinase (ERK) subgroup of MAPKs (3). By dephosphorylating an activating phosphoserine on Raf1, PP5 is known to suppress downstream signaling to MAPK/ERK kinase (MEK) and ERK (4). Unlike other members of the phosphoserine/threonine protein phosphatase (PPP) superfamily, the regulatory, substrate targeting, and catalytic domains of PP5 are expressed as a single polypeptide chain. When PP5 is in solution as a monomer, it assumes an autoinhibitory conformation generated by the spontaneous association of its N-terminal region with a unique C-terminal J-helix, which blocks substrate access to the catalytic site. This conformation renders “free” PP5 in an inactive conformational state with very low “basal” phosphatase activity (5, 6). The N-terminal region of PP5 also contains tetratricopeptide repeat (TPR) domains that mediate protein-protein interactions (6). The interaction of PP5 with a protein via its TPR domains produces a conformational change that allows substrate access to the catalytic site, which is located in a shallow groove on the surface of the catalytic domain of PP5 (7). This “activates” PP5 catalytic activity (8, 9). For example, the association of activated Rac1, heat shock protein 90 (HSP90), Gα12 and Gα13, and select Ca2+-bound S100 proteins with the TPR domains of PP5 has been shown to increase PP5 catalytic activity (6, 10–12). Furthermore, PP5 can become activated by treatment with polyunsaturated fatty acids (e.g. arachidonic acid) that probably also disrupt the autoinhibitory conformation (13).
The association of PP5 with many different proteins that affect signal transduction networks indicates that PP5 may have diverse biological activity. Interestingly, PP5 is known to associate with a number of protein kinases, including Raf1, ASK1, ATM, ATR/Chk1, DNA-PKcs, eIF2α kinase, and IKKβ (4, 14–20). Nonetheless, the role of PP5 in the regulation of these kinases is unclear and in some instances controversial. Many protein kinases that associate with PP5 are HSP90 clients (21), and it has been proposed that PP5 may affect kinase function indirectly via its interactions with HSP90, possibly via modulating the chaperone function of HSP90 that then alters the maturation and activity of the associated client kinase (19, 22, 23). Conversely, given that HSP90 binding enhances PP5-mediated dephosphorylation of the co-chaperone Cdc37, PP5 could alter kinase function by regulating the phospho-state of the associated kinase (22).
Historically, the cellular functions attributed to ERK1 and ERK2 were initially viewed as redundant because 1) at the level of their primary amino acid sequences, ERK1 and ERK2 share ∼83% identity in humans, 2) both ERK1 and ERK2 display parallel activation in response to a variety of stimuli, 3) they share common mechanisms for activation as well as similar kinase activity following activation, and 4) ERK1 and ERK2 exhibit comparable spatiotemporal expression patterns during development (24–26). Although ERK1 and ERK2 do indeed possess many overlapping properties, genetic studies have shown that their functions are not developmentally interchangeable. Notably, ERK1−/− mice are viable with deficits in thymocyte maturation (27), whereas the genetic disruption of ERK2 is lethal. ERK2−/− mice display embryonic lethality before embryonic day 8.5 due to defects in placental development and mesoderm differentiation (26, 28). Therefore, the preferential roles for ERK1 or ERK2 in the regulation of cell differentiation, proliferation, and growth are probably the result of distinct ERK-regulated gene targets and non-overlapping ERK-interacting proteins (28–30). Consistent with this idea, knockdown studies of ERK1/2 in zebrafish revealed uniquely regulated ERK1 and ERK2 genes and demonstrated that select genes are regulated in a differential manner (i.e. increased expression following ERK1 knockdown but decreased following ERK2 knockdown) (31). Moreover, the identification of proteins that uniquely associate with ERK1 (e.g. αvβ3 integrin and MEK partner 1) or ERK2 (e.g. NIPA, Bmf, and Sec16) provides additional support that ERK1 and ERK2 possess distinct functions (32–36).
In this report, we show that ERKs form stable complexes with PP5 and demonstrate that these PP5-kinase interactions are facilitated, in part, via HSP90. Analyses of the PP5·ERK1 and PP5·ERK2 complexes reveal that the assembly of these complexes in unstimulated cells is independent of both phosphatase and kinase activity. Interestingly, the PP5-ERK interactions are regulated by constitutively active variants of the small G proteins Rac1 and Ras. Whereas Rac1L61 enhances the assembly of both PP5·ERK1 and PP5·ERK2 complexes, oncogenic HRasV12 and KRasL61 decreases PP5-ERK2 interactions, without affecting PP5-ERK1 interactions. The selective alteration in PP5-ERK2 binding induced by HRasV12 is independent of both MEK and PP5 activity but paradoxically requires ERK2 kinase activity. Our studies also support a novel role for PP5·ERK complexes in regulating the feedback phosphorylation of Raf1 at Ser-289, Ser-296, and/or Ser-301.
EXPERIMENTAL PROCEDURES
Plasmids, Antibodies, and Other Reagents
FLAG-K97A-PP5/pcDNA3 (FLAG-PP5HBD) and FLAG-H304Q-PP5/pcDNA3 (FLAG-PP5PD) were generated using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA), mismatched primers (Integrated DNA Technologies, Coralville, IA), and wild type FLAG-PP5/pcDNA3 (FLAG-PP5WT) as the template (a gift from Dr. Hidenori Ichijo, Tokyo Medical and Dental University, Tokyo, Japan). HA-ERK1/pCEP4 was a gift from Dr. Melanie Cobb (University of Texas Southwestern Medical Center, Dallas, TX). Wild type HA-ERK1/pcDNA3 (HA-ERK1WT) was generated by excising the HA-ERK1 insert from HA-ERK1/pCEP4 with NotI and subsequent ligation into NotI-digested pcDNA3. HA-K71R-ERK1/pcDNA3 (HA-ERK1KD) was generated by site-directed mutagenesis of HA-ERK1/pCEP4 followed by excision with NotI and subsequent ligation of the HA-K71R-ERK1 insert into NotI-digested pcDNA3. HA-K52R-ERK2/pcDNA3 (HA-ERK2KD) was generated by site-directed mutagenesis of wild type HA-ERK2/pcDNA3 (HA-ERK2WT) (a gift from Dr. Vsevolod Gurevich, Vanderbilt University, Nashville, TN). Dr. Melanie Cobb provided G12V-HaRas/pRc/CMV (HRasV12), which was mutated to generate wild type HaRas/pRc/CMV (HRasWT). Wild type HA-KRas4B/pEF hybrid (HA-KRasWT) (a gift from Dr. Richard Marais, Institute of Cancer Research, London, UK) was used as a template to create the HA-G12V-KRas4B/pEF hybrid (HA-KRasV12) and HA-Q61L-KRas4B/pEF hybrid (HA-KRasL61) by site-directed mutagenesis. HA-ERK1b/pcDNA3 and HA-ERK1c/pcDNA3 were from Dr. Rony Seger (Weizmann Institute of Science, Rehovot, Israel), whereas Q61L-Rac1/pcDNA IIIB (Rac1L61) and Myc-T17N-Rac1/pcDNA3.1 (Myc-RacN17) were from Dr. Ann Richmond (Vanderbilt University). Dr. Joseph Avruch (Harvard Medical School, Boston, MA) provided Myc-Raf1/pMT2. Proper construction of all plasmids was verified by automated DNA sequencing (Vanderbilt University VANTAGE Sequencing Core).
The mouse anti-FLAG M2 antibody, rabbit anti-FLAG antibody, mouse anti-FLAG M2 affinity gel, and FLAG peptide were from Sigma-Aldrich. The rat IgG1 anti-HA (Clone 3F10) antibody and rat anti-HA (Clone 3F10) affinity matrix were from Roche Applied Science. Protein G-Sepharose 4B conjugate was from Invitrogen. Microcystin-agarose was from EMD Millipore Corp. (Billerica, MA). The mouse anti-HSP90α/β (F-8), goat anti-ERK1 (C-16-G), goat anti-ERK2 (C-14-G), rabbit anti-ERK2 (K-23), normal rabbit IgG, normal goat IgG, and rabbit anti-Rac1 (C-11) antibodies were from Santa Cruz Biotechnology, Inc. The rabbit anti-Ras (catalog no. 3965), rabbit anti-phospho-p44/42 MAPK (ERK1/2; pThr-202/pTyr-204), rabbit anti-phospho-Raf1 (pSer-289/pSer-296/pSer-301), and rabbit anti-phospho-Raf1 (pSer-338) (clone 56A6) antibodies were from Cell Signaling Technology, Inc. (Danvers, MA). The mouse anti-PP5 and mouse anti-Raf1 antibodies were from BD Biosciences. The rabbit anti-PP5 antibody was from Bethyl Laboratories, Inc. (Montgomery, TX). The Alexa Fluor 680-conjugated goat anti-mouse IgG, donkey anti-goat IgG, and goat anti-rat IgG secondary antibodies were from Molecular Probes, Inc. (Eugene, OR). The donkey anti-rabbit IRDye 800CW secondary antibody was from LI-COR (Lincoln, NE).
The MEK1/2 inhibitor U0126 was from Cell Signaling Technology, Inc. (Danvers, MA). Epidermal growth factor (EGF) and phorbol 12-myristate 13-acetate (PMA) were from Invitrogen and Sigma-Aldrich, respectively. The phosphatase substrate, 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP), was from Molecular Probes. Odyssey Blocking Buffer was from LI-COR. TransIT-293 transfection reagent was from Mirus (Madison, WI), and X-tremeGENE 9 DNA transfection reagent was from Roche Applied Science. Bio-Safe Coomassie G-250 Stain was from Bio-Rad. Arachidonic acid was from Nu-Chek Prep, Inc. (Elysian, MN). All other reagents were purchased from Sigma-Aldrich or Thermo Fisher Scientific Inc.
Cell Culture, Transfection, and Treatments
HEK-293FT cells were maintained at 37 °C and 5% CO2 in DMEM supplemented with 10% fetal calf serum. Cells (6-well plates) were transfected with the indicated plasmids using the Mirus TransIT-293 transfection reagent (2.5 μl:1 μg ratio of reagent to DNA) according to the manufacturer's protocol unless otherwise noted. For reciprocal immunoprecipitation experiments (anti-FLAG and anti-HA), cells (6-cm plates) were transfected with 500 ng of wild type FLAG-PP5 or FLAG-PP5HBD, 500 ng of wild type HA-ERK1 or HA-ERK2, and pcDNA3, as needed, to equalize the amount of DNA. For all of the other FLAG immunoprecipitation experiments, cells (6-well plates) were transfected with the following constructs: 350 ng each of wild type FLAG-PP5, FLAG-PP5HBD, FLAG-PP5PD, wild type HA-ERK1, HA-ERK1KD, HA-ERK1b, HA-ERK1c, wild type HA-ERK2, or HA-ERK2KD; 200 ng each of wild type HRas, HRasV12, wild type HA-KRas, HA-KRasV12, or HA-KRasL61; 500 ng each of Rac1L61 or Myc-Rac1N17; and the appropriate amount of pcDNA3 to ensure equivalent amounts of total DNA across samples within experiments. For cell stimulation studies, at 40–48 h post-transfection, cells were treated with 50 ng/ml EGF for 5 min, 100 nm PMA for 20 min, or an equivalent volume of DMSO for 20 min prior to lysis. In some experiments, cells were treated with U0126 (50 μm) or an equivalent volume of DMSO for 30 min prior to cell harvesting. To isolate FLAG-PP5·endogenous ERK complexes, cells (6-cm plates) were transfected with a combination of 700 ng of wild type FLAG-PP5, 1000 ng Rac1L61, and the appropriate amount of pcDNA3 to equalize amounts of DNA. Cells were subsequently treated with 100 ng/ml EGF (for Rac1L61-expressing cells) or an equivalent volume of solvent for 5 min prior to lysis. To isolate endogenous PP5·HA-ERK2 complexes, cells (10-cm plates) were transfected with a combination of 2100 ng of wild type HA-ERK2, 3000 ng of Rac1L61, and the appropriate amount of pcDNA3 to equalize the amounts of DNA in the transfections. X-tremeGENE 9 DNA was used as the transfection reagent (3 μl:1 μg ratio of reagent to DNA). To examine Raf1 feedback phosphorylation, cells (6-well plates) were co-transfected with 1000 ng of Myc-Raf1, 350 ng of wild type FLAG-PP5 or FLAG-PP5PD, 350 ng of wild type HA-ERK2 or HA-ERK2KD, 500 ng of Rac1L61 or 200 ng of HRasV12, and the appropriate amount of pcDNA3 to equalize amounts of DNA using X-tremeGENE 9 DNA; EGF-dependent feedback phosphorylation of Raf1 was monitored in cells transfected for 40–48 h and then treated with solvent lacking or containing 100 ng/ml EGF for 30 min prior to lysis.
Cell Extraction, Immunoprecipitations, and Pull-downs
Cells were washed twice with ice-cold PBS and then lysed using ice-cold Buffer B (20 mm Tris-HCl, pH 8.0, 137 mm NaCl, 10% glycerol, and 1% Igepal CA-630) or RIPA buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Igepal CA-630, 0.5% sodium deoxycholate, and 0.1% SDS) containing inhibitors (17 μg/ml aprotinin, 10 μm leupeptin, 1 mm phenylmethylsulfonyl fluoride, and 1 mm Na3VO4). For Raf1 phosphorylation experiments, cells were lysed in Phospho-Buffer (20 mm Tris-HCl, pH 7.2, 2 mm EGTA, 5 mm EDTA, 30 mm sodium fluoride, 20 mm sodium pyrophosphate, and 0.5% Igepal CA-630) containing inhibitors (40 mm β-glycerophosphate, 1 mm Na3VO4, 3 mm benzamidine, 5 μm pepstatin A, 10 μm leupeptin, and 1 mm phenylmethylsulfonyl fluoride). Clarified lysates were generated following centrifugation at 16,300 × g for 10 min at 4 °C. For FLAG and HA immunoprecipitations, the clarified lysates were gently rotated with 10 μl of a 50% slurry of resin for 4 h at 4 °C. Immunoprecipitations of endogenous ERK (400 ng of goat anti-ERK2 and 1000 ng of goat anti-ERK1 antibody) and endogenous PP5 (800 ng of rabbit anti-PP5) were performed by incubating antibodies with the clarified lysates overnight at 4 °C with rotation followed by subsequent incubation with protein G-Sepharose 4B (15 μl of a 50% slurry), rotating for 1 h at 4 °C. Microcystin-agarose (20 μl of a 50% slurry) pull-downs were performed by incubating the resin with the clarified lysates overnight at 4 °C with rotation. Bound proteins were washed five times with 350 μl of Buffer B/RIPA buffer lacking inhibitors or Phospho-Buffer containing inhibitors and eluted with 25 μl (FLAG and HA immune complexes), 15 μl (ERK and PP5 immune complexes), or 18 μl (microcystin pull-downs) of 2× SDS sample buffer.
Expression and Purification of Recombinant S100A1 and S100B Proteins
Recombinant rat S100A1 and bovine S100B proteins were overexpressed in Escherichia coli as described previously (37, 38). Recombinant S100A1 was purified under reducing conditions using a modification of the procedure described by Landar et al. (37). Following removal of cellular debris by centrifugation, bacterial lysates were incubated for 30 min with a 10% solution of streptomycin sulfate, and DNA precipitates were removed by centrifugation. Heat-labile proteins were removed by heating to 60 °C, boiling for 4 min, and centrifugation at 15,000 × g for 45 min at 4 °C. After the addition of 0.1 m NaCl, the heat-stable fraction was fractionated on a Diethylaminoethyl Fast Flow (HiPrep DEAE FF 16/10) column (GE Healthcare). S100A1-containing fractions (260 ± 90 mm NaCl) were dialyzed and loaded on a phenyl-Sepharose 6 Fast Flow 16/10 column (GE Healthcare). S100A1-containing fractions were eluted with 10 mm EDTA (>95% purity by SDS-PAGE), concentrated (<5 ml), and fractionated on a Superdex S200-PG size exclusion column equilibrated with Chelex-treated buffer (10 mm Tris-HCl, pH 7.5, 50 mm NaCl, 1 mm DTT). Yields of S100A1 protein (>99% pure) were typically 40–50 mg of purified protein/liter of bacterial culture. Recombinant S100B was purified using a modification of the procedure described by Amburgey et al. (38). Following the removal of cellular debris by centrifugation, bacterial lysates were incubated for 30 min with a 10% solution of streptomycin sulfate, and DNA precipitates were removed by centrifugation. The S100B-containing fraction (80% supernatant) from ammonium sulfate precipitation was dialyzed and loaded on a DEAE Fast Flow column. After concentration (<5 ml), the S100B-containing fraction (0.3 m NaCl) (>95% purity by SDS-PAGE) was fractionated on a Superdex S200-PG size exclusion column equilibrated with Chelex-treated buffer. Yields of S100B protein (>99% pure) were typically 20–30 mg/liter of bacterial culture. The identity and purity of recombinant S100A1 and S100B were confirmed by amino acid analysis and electrospray mass spectrometry.
Purification and Quantification of PP5
HEK-293FT cells (10-cm plate) were transfected with 3 μg of wild type FLAG-PP5 or FLAG-PP5HBD. At 40–48 h post-transfection, cells were washed twice with 4 ml of ice-cold PBS and subsequently lysed with 1 ml of Buffer B plus inhibitors. Clarified lysates from two plates were pooled and then incubated with 40 μl of a 50% slurry of anti-FLAG resin and rotated overnight at 4 °C. Bound proteins were washed three times with 800 μl of Buffer B lacking inhibitors and once with 1 ml of PP5 Storage Buffer (20 mm Tris-HCl, pH 7.5, 0.1% β-mercaptoethanol, 0.2 mm MnCl2) and then eluted from the beads by washing three times with 90 μl of PP5 Activity Buffer containing 100 μg/ml FLAG peptide; for each elution, the beads were gently rotated for 60–90 min at 4 °C. The pooled eluates were combined with an equivalent volume of 100% glycerol and stored at −20 °C. The amount of PP5 protein was determined by SDS-PAGE and Colloidal Blue staining (Invitrogen) using serial dilutions of bovine serum albumin (Pierce) as standards. Quantification was accomplished using the Odyssey infrared imaging system and Odyssey application software version 3.0 (LI-COR).
Phosphatase Assay
PP5 activity was measured by adapting a previously described protocol (39). Briefly, purified wild type FLAG-PP5 or FLAG-PP5HBD (25 nm) was incubated in 80 μl of PP5 Activity Buffer (100 mm MOPS, pH 7.0, 0.1% β-mercaptoethanol, 0.1 mg/ml BSA) lacking or containing 250 μm arachidonic acid or 5 μm S100B/S100A1 plus 1 mm CaCl2 for 30 min at 4 °C, with agitation every 5 min, before being transferred to a 96-well assay plate (Corning/Costar 3691 black with clear, flat bottom). DiFMUP substrate (167 μm) was freshly prepared in PP5 Activity Buffer; 1 mm CaCl2 (final concentration) was included for the assays containing S100B/S100A1. DiFMUP substrate (125 μl) was transferred to a 96-well compound plate (Falcon Microtest U-Bottom 353077, BD Biosciences). Both 96-well plates were placed in a FLEXstation III (Molecular Devices, Sunnyvale, CA), and samples were allowed to equilibrate to 30 °C for 5 min prior to the assay. The substrate (120 μl) and phosphatase (80 μl) mixtures were combined and triturated by the FLEXstation III, resulting in final concentrations of 10 nm phosphatase, 100 μm arachidonic acid, 200 nm S100B/S100A1, 1 mm CaCl2, and 100 μm DiFMUP. The assays were carried out at 30 °C for 15 min, and the conversion of DiFMUP to 6,8-difluoro-4-methylumbelliferyl (DiFMU) was monitored as previously described, with the PMT sensitivity setting set to low (40). The data were recorded and analyzed by SoftMax Pro software version 5.4 (Molecular Devices, Sunnyvale, CA).
S100 Displacement of ERK from PP5 Complexes
HEK-293FT cells (10-cm plates) were untransfected or transfected with 1400 ng of wild type HA-ERK2 using X-tremeGENE 9 DNA. At 40–48 h post-transfection, cells were lysed in Buffer B containing inhibitors and clarified by centrifugation. The cell lysates were incubated with 30 μl of a 50% slurry of microcystin-agarose overnight with rotation at 4 °C. Bound proteins were washed five times with 350 μl of Buffer B lacking inhibitors and then once with 350 μl of S100 Binding Buffer (20 mm Tris-HCl, pH 7.5, 100 mm KCl, 0.01% Tween 20, and 1 mm CaCl2). The beads were then split in half into separate tubes and incubated in a final volume of 50 μl of S100 Binding Buffer lacking or containing 20 μg of S100A1 for 1 h at room temperature with agitation. Proteins immobilized on the resin were washed four times with 350 μl of S100 Binding Buffer and eluted with 15 μ1 of 2× SDS sample buffer.
Western Analysis
Equal volumes of SDS-solubilized cell lysates and immune complexes were subjected to SDS-PAGE (10% Tris-glycine gels), transferred to 0.2- or 0.45-μm Optitran BA-S 85 reinforced nitrocellulose (Whatman, Dassel, Germany), blocked with Odyssey Blocking Buffer, and immunoblotted with the indicated primary antibodies and corresponding secondary antibodies. Phospho-specific antibodies were diluted 1:1000 in Odyssey Blocking Buffer containing 0.1% Tween 20. All other primary and secondary antibodies were diluted in Tris-buffered saline containing 0.5% BSA and 0.1% Tween 20; secondary antibodies were diluted 1:10,000. Visualization and quantification of the immunolabeled proteins was accomplished using the Odyssey infrared imaging system (LI-COR) and Odyssey application software version 3.0.
Statistics
Statistical comparisons were performed using GraphPad Prism version 4.03 (GraphPad Software, San Diego, CA).
RESULTS
PP5 Forms Stable Complexes with ERKs
Our Western analysis of FLAG-PP5 immune complexes isolated from HEK-293FT cells revealed a number of novel PP5-interacting proteins, which included ERK2 (data not shown). Therefore, we performed additional experiments to determine if PP5 could interact with ERK2, ERK1, and two ERK1 splice variants (rodent ERK1b and primate ERK1c) (41, 42). Western analysis of FLAG immune complexes from HEK-293FT cells co-expressing FLAG-PP5 and HA-ERK1, HA-ERK1b, HA-ERK1c, or HA-ERK2 revealed that PP5 is capable of interacting with each of the MAPKs (Fig. 1, A and B). The binding of these MAPKs to PP5 does not appear to involve the extreme C terminus of the kinases, because this region is very divergent among the ERKs (41, 42). No kinase was detected in the control immunoprecipitations, and endogenous HSP90 was found to co-precipitate with FLAG-PP5. Western analysis of reciprocal immunoprecipitations (i.e. HA-ERK) not only confirmed the presence of PP5·ERK1 and PP5·ERK2 complexes but also revealed the existence of PP5-ERK interactions in the presence of little or no HSP90.
FIGURE 1.

Analysis of PP5-ERK interactions and PP5 activity. A, HEK-293FT cells were transfected with FLAG-PP5, HA-ERK1, or HA-ERK2 alone or in combination, as indicated. Western analysis of FLAG immune complexes (FLAG IPs), HA immune complexes (HA IPs), and cell lysates were performed using FLAG, HA, and HSP90 antibodies. *, IgG heavy chain. B, HEK-293FT cells were transfected with FLAG-PP5, HA-ERK1b, or HA-ERK1c alone or in combination, as indicated. FLAG IPs and cell lysates were analyzed by Western as in A. C, HEK-293FT cells were transfected with wild type FLAG-PP5 or FLAG-PP5HBD alone or together with HA-ERK1 or HA-ERK2; cells were also transfected with the indicated HA-tagged kinase and pcDNA3 (Vector). FLAG IPs and cell lysates were analyzed as described in A. A significant reduction in the binding of HA-ERK1 (88.21 ± 3.01%, p < 0.0001) and HA-ERK2 (77.3 ± 8.97%, p = 0.0003) to FLAG-PP5HBD was found when ERK signals were normalized to levels of mutant PP5 in the IPs and compared with the corresponding values in the wild type FLAG-PP5 conditions, which were set to 100. The results represent the means ± S.E. analyzed by one-sample t test using two-tailed p values. D, approximately 118 ng (10 nm) of purified wild type FLAG-PP5 (WT) or HSP90 binding-deficient mutant of FLAG-PP5 (HBD) were continuously assayed over 900 s for phosphatase activity toward DiFMUP (relative fluorescent units (RFU)) in the presence of only buffer (Basal), 100 μm arachidonic acid (AA), or 200 nm S100B plus 1 mm CaCl2 (S100B). Background fluorescence (i.e. samples containing only DiFMUP + arachidonic acid or DiFMUP + S100B + CaCl2) was measured and subtracted from the corresponding fluorescent values of the phosphatase-containing samples. Levels of fluorescence in WT + arachidonic acid, HBD + arachidonic acid, and HBD + S100B preparations were virtually identical. The results represent the means ± S.E. from six independent experiments, three experiments performed with duplicates from each of two separate purifications of WT and HBD. S.E. bars are obscured by the symbols for most data points. E, quantification of phosphatase activity at the 900 s time point. Two-way analysis of variance identified a statistically significant genotype versus activator interaction (F(5,30) = 61.83, p < 0.0001). Tukey post-tests are shown as follows: ***, versus basal, p < 0.0001; ^^^, WT versus HBD, p < 0.0001. Error bars, S.E. F, HEK-293FT cells expressing FLAG-PP5 and HA-ERK2 were lysed in Buffer B (B) or RIPA buffer (R) (Lysates). FLAG immunoprecipitations (FLAG IPs) were performed from the cell lysates and washed (IP Wash) in either Buffer B or RIPA buffer, as indicated. The FLAG IPs and corresponding cell lysates were analyzed by Western using HSP90, HA, and FLAG antibodies. The data are representative of experiments performed three (A), three (B), six (C), six (D), and two (F) independent times with similar results.
Because the binding of PP5 with interacting proteins is often mediated by HSP90 (43), we next examined the ability of the ERKs to interact with a mutated form of PP5 containing a single point mutation that disrupts the association of PP5 with HSP90 (HSP90 binding-deficient; FLAG-PP5HBD) (44). Under conditions where both the expression of the kinases and the levels of immunopurified phosphatase were comparable, we observed that the amount of ERK1/2 bound to PP5HBD was dramatically reduced relative to wild type PP5 (Fig. 1C), indicating that HSP90 plays a role in facilitating these interactions. However, in PP5HBD immune complexes that contain no HSP90, ERK1/2 was still detected, suggesting that not all PP5·ERK complexes require the presence of HSP90.
To ensure the functionality of the FLAG-PP5HBD mutant, we analyzed its activity in the presence of two different activators of PP5, arachidonic acid and S100 family members (6, 10). Both wild type PP5 and PP5HBD exhibited comparable basal and arachidonic acid-stimulated activities (Fig. 1, D and E). In addition, both S100B and S100A1 induced the activation of PP5HBD to a level comparable with the activation observed following treatment with arachidonic acid (Fig. 1, D and E) (data not shown). Studies in our laboratory have established that FLAG-PP5 immunopurified from RIPA buffer-solubilized cell lysates lacks HSP90. To further substantiate the ability of PP5 to bind ERK1/2 in the absence of HSP90, we examined the association of HA-ERK2 with wild type FLAG-PP5 immune complexes isolated from cells that were lysed in different buffers (i.e. Buffer B or RIPA buffer). As shown in Fig. 1F, RIPA buffer solubilization maintains the interaction of ERK2 with an HSP90-uncoupled, monomeric form of PP5. Collectively, these data indicate that PP5 binding to ERK can occur in the absence of HSP90.
To determine if PP5-ERK1/2 interactions are dependent on kinase or phosphatase activity, we performed binding experiments using catalytically inactive forms of these enzymes. Western analysis of FLAG immune complexes from lysates of unstimulated cells co-expressing wild type FLAG-PP5 and kinase-dead ERK1 (HA-ERK1KD) or ERK2 (HA-ERK2KD) (45) revealed that catalytically inactive kinases retained their ability to associate with wild type PP5 (Fig. 2A). Likewise, analysis of FLAG immune complexes from lysates of cells co-expressing phosphatase-dead PP5 (FLAG-PP5PD) (7, 11, 46) and wild type HA-ERK1 or HA-ERK2 revealed that PP5-ERK1/2 interactions were maintained in the absence of PP5 activity (Fig. 2B). Together, these findings demonstrate that neither kinase nor phosphatase activity is required for the assembly of PP5·ERK1 and PP5·ERK2 complexes in unstimulated cells.
FIGURE 2.

The interaction of PP5 with ERK1/2 is independent of kinase and phosphatase activity. A, FLAG immunoprecipitations (FLAG IPs) were performed from lysates of HEK-293FT cells transfected with wild type FLAG-PP5, kinase-dead ERK1 (HA-ERK1KD), or kinase-dead ERK2 (HA-ERK2KD) alone or in combination, as indicated. The cell lysates and FLAG IPs were subjected to Western analysis using FLAG, HA, and HSP90 antibodies. B, FLAG IPs were performed from lysates of HEK-293FT cells transfected with phosphatase-dead PP5 (FLAG-PP5PD), wild type HA-ERK1, or wild type HA-ERK2 alone or in combination, as indicated. The cell lysates and FLAG IPs were analyzed as described in A. *, IgG heavy chain. The data are representative of experiments performed three (A) and three (B) independent times with similar results.
Active Rac1 Promotes Assembly of PP5·ERK1 and PP5·ERK2 Complexes
Activated Rac1 has been shown to promote the translocation of PP5 to the plasma membrane (11), one of several subcellular sites where the compartmentalization of ERK1/2 forms spatially distinct pockets of ERK activity that are important in creating diversity in cellular responses to stimuli (47). To determine if PP5-ERK1/2 interactions could be altered in the presence of active Rac1, we evaluated the binding of PP5 to ERK1/2 in cells co-expressing constitutively active Rac1 (Rac1L61). Significantly enhanced binding of PP5 to both ERK1 and ERK2 (5.2- and 2.8-fold increases, respectively) was observed in cells expressing Rac1L61, relative to control cells (Fig. 3, A and B). Moreover, the interaction of PP5 with ERK1/2 was unaffected by expression of a dominant negative form of Rac1 (Myc-Rac1N17) (Fig. 3, A and B). Given that Rac1L61 augmented the interaction of the ectopic proteins, we next tested whether Rac1L61 could promote increased interaction of endogenous-ectopic proteins. Indeed, the expression of Rac1L61 enhanced the interaction of endogenous ERK1/2 with FLAG-PP5 (Fig. 3C). Likewise, the expression of Rac1L61 led to increased interaction of endogenous PP5 with HA-ERK2, as visualized by Western analysis of both endogenous PP5 immune complexes and microcystin-agarose pull-downs (an affinity resin for protein serine/threonine phosphatases, such as PP5) (Fig. 3D). To test whether endogenous PP5 interacts with endogenous ERK, we performed microcystin-agarose pull-downs from lysates of untransfected cells. As shown in Fig. 3E (left), endogenous ERK1/2 co-purified with endogenous PP5 on this phosphatase affinity resin. To demonstrate that the binding of ERK1/2 to microcystin-agarose is due to its interaction with PP5, we performed an in vitro displacement assay using S100A1, a PP5 regulatory protein that competitively inhibits the association of PP5 with other interacting partners (10). S100A1 significantly displaced endogenous ERK1/2 from the resin without affecting the binding of endogenous PP5 (Fig. 3E, left). S100A1 also disrupted the interaction of HA-ERK2 with endogenous PP5 (Fig. 3E, right), thus indicating that our overexpression system recapitulates the native condition. The co-purification of PP5 and ERK with microcystin-agarose also supports our observations that phosphatase activity is not required for the PP5-ERK interaction because microcystin is a potent inhibitor of PP5 activity (48). Moreover, analysis of microcystin pull-downs from a variety of other cell types (e.g. MCF7, MDCK, and MDA-MB-435) also revealed co-purification of endogenous ERK1/2 and PP5 (data not shown).
FIGURE 3.

Active Rac1 promotes assembly of PP5·ERK1 and PP5·ERK2 complexes. A, HEK-293FT cells were co-transfected with FLAG-PP5 and either HA-ERK1 or HA-ERK2 together with pcDNA3 (Vector), constitutively active Rac1 (Rac1L61), or dominant negative Rac1 (Myc-Rac1N17), as indicated. Western analysis of FLAG immune complexes (FLAG IPs) and cell lysates were performed using the HA, FLAG, HSP90, and Rac1 antibodies. B, quantification of the percentage of maximal HA-ERK binding normalized to the FLAG-PP5 signal in FLAG IPs, with binding in the vector samples set to 100. One-way analysis of variance identified a significant increase in PP5-ERK1 (F(2,6) = 6.222, p = 0.0344) and PP5-ERK2 (F(2,6) = 17.28, p = 0.0032) association in the presence of Rac1L61. Tukey post-tests are shown as follows: *, p < 0.05; **, p < 0.01. Data are mean ± S.E. No significant differences in the expression levels of FLAG-PP5, HA-ERK1, or HA-ERK2 were detected following normalization to HSP90 levels. C, HEK-293FT cells were transfected with pcDNA3 (Vector) or FLAG-PP5 in the absence (−) or presence (+) of constitutively active Rac1 (Rac1L61). Cells transfected with Rac1L61 were also treated with 100 ng/ml EGF for 5 min prior to lysis. Endogenous ERK1/2 immune complexes (ERK1/2 IPs) and cell lysates were analyzed by Western using phospho-ERK1/2 (p-ERK1/2), ERK2, PP5, and FLAG antibodies. D, HEK-293FT cells were co-transfected with HA-ERK2 and pcDNA3 (−) or Rac1L61 (+). Western analysis of cell lysates and proteins purifying with normal rabbit IgG (IgG IPs), rabbit anti-PP5 antibody (PP5 IPs), and microcystin-agarose (MC PDs) were performed using ERK2 and PP5 antibodies. E, lysates from untransfected (left) and HA-ERK2-expressing (right) HEK-293FT cells were incubated with microcystin-agarose, and bound proteins were extensively washed prior to splitting the resin into separate tubes, which were then incubated with buffer lacking (−) or containing (+; 20 μg) purified S100A1. A fraction of the reaction mixture was collected and analyzed by SDS-PAGE (15% Tris-glycine gels), and stained with Coomassie G-250 to detect S100A1. Following incubation, bound proteins were extensively washed and eluted for analysis by Western blotting with antibodies detecting the ERK1/2 and PP5 proteins. Unpaired, one-tailed t tests identified a significant decrease in the levels of bound ERK following incubation with S100A1 (left, *, p = 0.0117; right, *, p = 0.0279). Error bars, S.E. The data are representative of experiments performed three (A), three (C), two (D), three (E, left), and two (E, right) independent times with similar results.
Oncogenic Ras Alters the Interaction of PP5 with ERK2 but Not ERK1
To address whether activation of the Ras-Raf-MEK-ERK signaling cascade influences PP5-ERK1/2 interactions, we treated cells with a variety of stimuli and monitored binding by co-immunoprecipitation assays coupled with Western analysis. Acute treatment of cells with EGF potently stimulated this pathway, as visualized in the cell lysates using a phospho-ERK1/2 antibody, but failed to alter PP5-ERK1/2 interactions (Fig. 4A). Likewise, acute treatment of cells with PMA, which activates ERK in a Ras-independent manner via activation of PKC (49), also failed to impact the PP5·ERK1/2 complexes. Interestingly, introduction of constitutively active HRas (HRasV12), which chronically activates ERK signaling, led to a loss of PP5-ERK2 binding without influencing the association of PP5 and ERK1. To determine if the HRasV12-induced effect was specific to the gain-of-function G12V mutation, we compared the ability of wild type HRas (HRasWT) and HRasV12 to influence the PP5-ERK2 interaction. Although HRasWT and HRasV12 expression led to comparable increases in ERK1/2 phosphorylation, relative to cells lacking Ras expression, only the oncogenic variant was capable of decreasing the association of PP5 and ERK2 (Fig. 4B). These observations prompted us to examine another Ras isoform, namely KRas4B, and two of its oncogenic variants (i.e. KRasV12 and KRasL61) for their ability to modulate the interaction of PP5 with ERK2. Introduction of wild type KRas4B (KRasWT), KRasV12, and KRasL61 each resulted in enhanced levels of ERK phosphorylation, and, analogous to HRasV12, KRasL61 was fully capable of modifying this phosphatase-kinase interaction (Fig. 4B). Surprisingly, despite similar levels of activated ERK in cells expressing KRasL61, oncogenic KRasV12 did not decrease the PP5-ERK2 interaction (Fig. 4B). These findings reveal novel stimulus-induced differences in PP5-ERK1/2 binding and indicate that the PP5·ERK2 complex is selectively regulated in response to specific oncogenic Ras-initiated signaling events.
FIGURE 4.

Specific oncogenic Ras variants selectively decrease PP5-ERK2, but not PP5-ERK1, interactions. A, HEK-293FT cells were transfected with HA-ERK1, HA-ERK2, or FLAG-PP5 alone or in combination and treated with nothing (Ø), 50 ng/ml EGF (5 min), 100 nm PMA (20 min), or an equivalent volume of DMSO (20 min) as a vehicle control. HEK-293FT cells transfected with FLAG-PP5 and HA-ERK1 or HA-ERK2 in combination with pcDNA3 (Vector) or HRasV12 were not treated prior to lysis. Western analysis of the FLAG immune complexes (FLAG IPs) and cell lysates was done using antibodies recognizing phospho-ERK1/2 (p-ERK1/2), Ras, HSP90, HA, and FLAG. Note that phospho-HA-ERK2 (p-HA-ERK2) and endogenous phospho-ERK1 (endog. p-ERK1) co-migrate. B, HEK-293FT cells transfected with (+) or without (−) HA-ERK2 or FLAG-PP5 were co-transfected with HRasV12, wild type HRas (HRasWT), HA-KRasV12, HA-KRasL61, or wild type HA-KRas (HA-KRasWT). FLAG IPs and cell lysates were analyzed by Western blot as in A. The data are representative of experiments performed four (A) and three (B) independent times with similar results.
Kinase Activity, but Not Phosphatase Activity, Is Required for HRasV12-induced Alteration of PP5·ERK2 Complexes
We next performed experiments to determine if the HRasV12-induced reduction in PP5-ERK2 interactions is dependent on the enzymatic activity of PP5 and/or ERK2. The impact of phosphatase activity on this event was assessed by immunoblotting for ERK2 in FLAG immune complexes from cells co-expressing FLAG-PP5PD and wild type HA-ERK2 in the absence or presence of HRasV12. As shown in Fig. 5A, HRasV12 decreased the interaction between PP5PD and wild type ERK2, similar to what was observed for the wild type PP5-ERK2 interaction (Fig. 4A), thus demonstrating that phosphatase activity was not required for this effect. To ascertain if ERK2 activity was important for the HRasV12-induced changes in the PP5·ERK2 complex, we evaluated the binding of both wild type HA-ERK2 and HA-ERK2KD to wild type FLAG-PP5 in the absence or presence of HRasV12. As predicted by previous observations, HRasV12 expression diminished the interaction of PP5 and wild type ERK2; however, the interaction of wild type PP5 and ERK2KD was maintained in HRasV12-expressing cells (Fig. 5B). Consistent with the results shown in Figs. 2 and 4A, the PP5-ERK1 interaction was independent of phosphatase and kinase activity in both the absence and presence of HRasV12. Collectively, these data demonstrate that the association of PP5 with ERK1/2 is independent of both phosphatase and kinase activity, whereas the HRasV12-mediated reduction of PP5-ERK2 binding requires ERK2 activity.
FIGURE 5.

Kinase activity, but not phosphatase activity, is required for HRasV12-dependent disruption of the PP5·ERK2 complex. A, HEK-293FT cells were transfected with wild type HA-ERK1 or wild type HA-ERK2 together with (+) or without (−) phosphatase-dead FLAG-PP5 (FLAG-PP5PD) and HRasV12. FLAG immune complexes (FLAG IPs) and cell lysates were analyzed by Western blotting using antibodies recognizing the indicated proteins. B, wild type HA-ERK1 (HA-ERK1WT), kinase-dead HA-ERK1 (HA-ERK1KD), wild type HA-ERK2 (HA-ERK2WT), and kinase-dead HA-ERK2 (HA-ERK2KD) were transfected into HEK-293FT cells alone or together with wild type FLAG-PP5 in the absence or presence of HRasV12. FLAG IPs and cell lysates were analyzed as described in A. *, IgG heavy chain. The data are representative of experiments performed three (A) and five (B) independent times with similar results.
Disruption of the PP5-ERK2 Interaction by HRasV12 Is Independent of MEK Activity
Because the data indicate that ERK2 activity is necessary for the HRasV12-induced decrease in PP5-ERK2 interactions, we wanted to determine if MEK1/2 plays a role in this process. To address this question, cells co-expressing wild type HA-ERK2, wild type FLAG-PP5, and oncogenic HRas were treated with the MEK1/2 inhibitor, U0126, or solvent as a control. Western analysis of cell lysates and FLAG immune complexes revealed that MEK inhibition potently abrogated ERK phosphorylation but did not influence the loss of PP5-ERK2 binding caused by HRasV12 (Fig. 6A). Quantification of these data revealed that expression of HRasV12 produced a dramatic reduction in the association of PP5 and ERK2, regardless of the activation state of MEK (reductions of 86.08% ± 1.62 and 81.95% ± 2.26% in the absence or presence of U0126, respectively) (Fig. 6B). Therefore, it appears that acute inhibition of ERK, in the context of HRasV12 signaling, does not allow reassembly of PP5·ERK2 complexes. Collectively, these data support the idea that the HRasV12/KRasL61-mediated regulation of the PP5·ERK2 complex is independent of the activation state of ERK yet paradoxically requires ERK2 activity.
FIGURE 6.
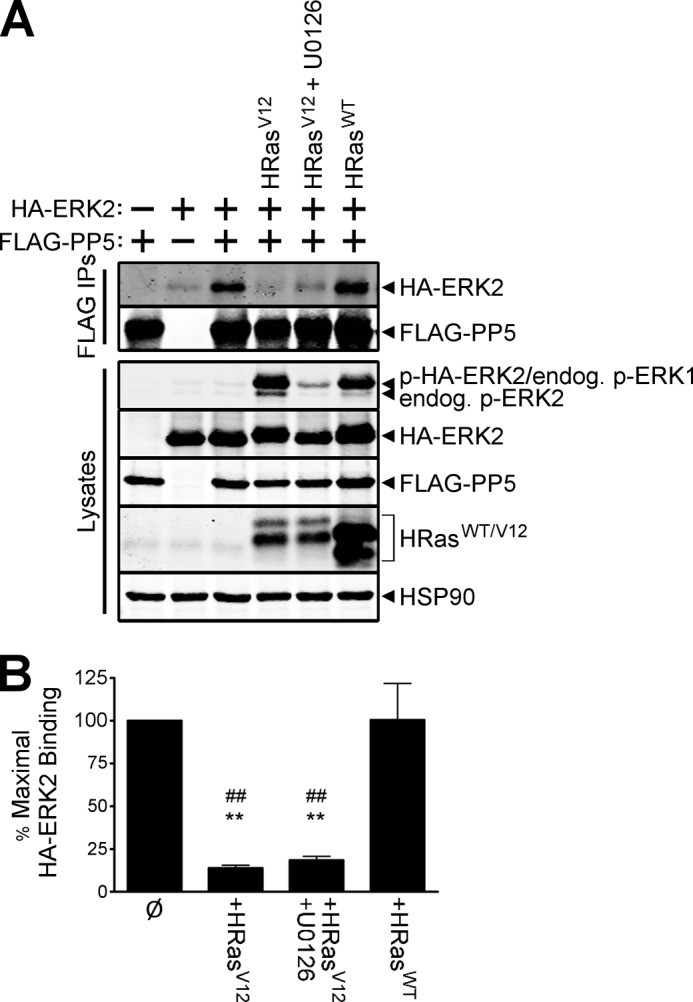
HRasV12 induces disruption of the PP5·ERK2 complex independently of the activation state of ERK2. A, HEK-293FT cells were transfected with (+) or without (−) HA-ERK2 and FLAG-PP5 together with HRasV12 or wild type HRas (HRasWT); cells were treated with the MEK inhibitor U0126 or DMSO for 30 min prior to lysis. FLAG immune complexes (FLAG IPs) and cell lysates were analyzed by Western using antibodies recognizing the indicated proteins. B, percentage of maximal binding of HA-ERK2 to FLAG-PP5. ERK2 binding signals, quantified for cells in A that co-expressed HA-ERK2 and FLAG-PP5, were normalized to levels of PP5 in the FLAG IPs and compared with the corresponding values in the absence of any Ras expression (Ø), which were set to 100. Significant reductions in PP5 binding to ERK2 were observed in the presence of HRasV12 (86.08 ± 1.62%) and following acute treatment with U0126 (81.95 ± 2.26%), whereas HRasWT expression failed to disrupt the interaction. The results represent the means ± S.E. based on one-way analysis of variance (F(3,8) = 20.42, p = 0.0004). Tukey post-tests are shown as follows: **, versus Ø, p < 0.01; ##, versus HRasWT, p < 0.01. Error bars, S.E. The data are representative of experiments performed three independent times with similar results.
PP5·ERK2 Complexes Regulate Raf1 Phosphorylation
Ras-Raf-MEK-ERK signaling pathways acquire their highly adaptive character, in part, from multiple, ERK-mediated feedback phosphorylation loops that act to promote or suppress the activity of upstream components (50, 51). Although several feedback phosphorylation events on Raf1 are mediated by ERK1/2, the mechanism(s) that dictates the recruitment of ERK to Raf1 is unknown (52, 53). Because PP5 dephosphorylation of Raf1 at Ser(P)-338 (4) temporally overlaps with the phosphorylation of Raf1 by ERK (52, 53), we explored the possibility that PP5·ERK2 complexes play a role in the feedback phosphorylation state of Raf1. This question was addressed by examining the phospho-state of PP5-associated Raf1 from unstimulated and EGF-stimulated HEK-293FT cells transfected with combinations of Myc-Raf1, wild type or kinase-dead HA-ERK2, and wild type or catalytically inactive FLAG-PP5. Western analysis of the FLAG immune complexes from cells expressing wild type HA-ERK2 and FLAG-PP5 revealed an EGF-dependent increase in the phosphorylation of ERK-dependent sites (EDS) (i.e. Ser-289/Ser-296/Ser-301) and the activating site (Ser-338) on Raf1 (Fig. 7A, lane 3 versus lane 4). Expression of PP5PD, in lieu of wild type PP5, resulted in increased phosphorylation of EDS and Ser-338 in unstimulated cells (Fig. 7A, lane 3 versus lane 6), which was augmented by treatment with EGF (Fig. 7A, lane 6 versus lane 7). In contrast to wild type ERK2-expressing cells, EGF failed to promote increased phosphorylation of EDS in ERK2KD-expressing cells and in cells lacking wild type HA-ERK2; however, the phosphorylation of Ser-338 remained elevated (Fig. 7A, lane 7 versus lanes 8 and 9). Together, these findings indicate that ERK2 phosphorylates EDS but not Ser-338 and that PP5 activity modulates the phosphorylation state of both EDS and Ser-338.
FIGURE 7.
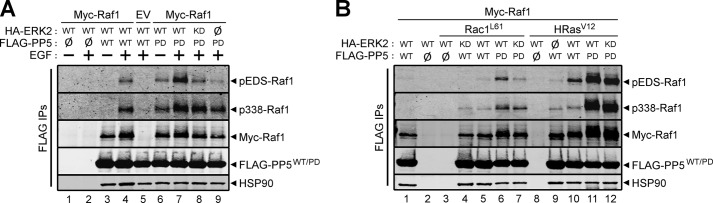
PP5·ERK2 complexes regulate Raf1 feedback phosphorylation, which is elevated in Rac1L61- and HRasV12-expressing cells. A, HEK-293FT cells were co-transfected with Myc-Raf1 or pcDNA3 (EV) together with the indicated combinations of nothing (Ø), wild type (WT) or kinase-dead HA-ERK2 (KD), and wild type (WT) or catalytically inactive FLAG-PP5 (PD). Cells were treated with solvent containing (+) or lacking (−) 100 ng/ml EGF for 30 min prior to lysis. FLAG immune complexes (FLAG IPs) were analyzed by Western using phospho-Ser-289/296/301-Raf1 (pEDS-Raf1), phospho-Ser-338-Raf1 (p338-Raf1), Raf1, HSP90, and FLAG antibodies. B, HEK-293FT cells were co-transfected to express Myc-Raf1, in the presence or absence of Rac1L61 or HRasV12, together with the indicated combinations of nothing (Ø), wild type (WT) or kinase-dead HA-ERK2 (KD), and wild type (WT) or catalytically inactive FLAG-PP5 (PD). FLAG IPs were analyzed as in A. The data are representative of experiments performed two (A) and two (B) independent times with similar results.
To determine if Rac1L61 or HRasV12 influence the phosphorylation of PP5-associated Raf1 by modulating the PP5-ERK2 interaction, we co-expressed each small G protein in combination with wild type or catalytically inactive forms of HA-ERK2 and FLAG-PP5. Western analysis of the FLAG immune complexes revealed very little phosphorylation of either EDS or Ser-338 in the PP5-associated Raf1 in Rac1L61-expressing cells containing wild type PP5 (Fig. 7B, lane 1 versus lane 5). In contrast, the phosphorylation of both EDS and Ser-338 in the PP5-associated Raf1 complexes were drastically elevated in cells expressing catalytically inactive PP5 (Fig. 7B, lane 5 versus lane 6). Comparable levels of Raf1 Ser-338 phosphorylation were observed in cells co-expressing PP5PD and either wild type ERK2 or ERK2KD; however, only the phosphorylation of EDS was altered in ERK2KD-expressing cells relative to wild type ERK2-expressing cells (Fig. 7B, lane 6 versus lane 7). Surprisingly, the co-expression of HRasV12 also promoted phosphorylation of EDS (Fig. 7B, lane 1 versus lane 10), via a mechanism that required ERK2 (Fig. 7B, lane 9 versus lane 10). Consistent with the EGF and Rac1L61 data, loss of PP5 activity dramatically enhanced phosphorylation of both EDS and Ser-338 in HRasV12-expressing cells (Fig. 7B, lane 10 versus lane 11), whereas the additional loss of ERK2 activity partially reduced phosphorylation of EDS but not Ser-338 (Fig. 7B, lane 11 versus lane 12). Together, these data support a dynamic role for PP5·ERK2 complexes in regulating Raf1 phosphorylation.
DISCUSSION
PP5·ERK Complexes and HSP90
Our studies confirm the association of PP5 with HSP90 and Raf1 (4, 43) and reveal novel interactions between PP5 and ERKs. Analysis of HSP90-bound PP5 and monomeric PP5 (i.e. completely devoid of HSP90) indicate that the association of PP5 with ERK1/2 can occur in the presence or absence of HSP90 (Fig. 1C). We observed decreased binding of ERK1/2 to monomeric PP5, relative to HSP90·PP5 complexes (Fig. 1C). This may indicate that the autoinhibitory conformation of monomeric PP5 yields an encounter complex with a reduced capacity to interact with the kinases (54). Alternatively, kinase binding sites on HSP90, which regulate HSP90-Akt interactions (55), may facilitate the association of PP5 and ERK1/2 when HSP90 is present. Putative ERK docking domains and reverse D domains (56) exist in PP5, and our data indicate that monomeric PP5 can directly interact with ERK1/2 in the absence of HSP90. This suggests that at least some ERK1/2 binding determinants are located on PP5. Future studies will be needed to determine the precise role of HSP90 in the assembly, maintenance, or maturation of PP5·ERK complexes.
Regulation of PP5·ERK Complexes and Raf1 Feedback Phosphorylation
Phosphatase·kinase complexes can be regulated by intramolecular reversible phosphorylation (57–60), and we observed that the HRasV12-induced reduction in PP5-ERK2 binding required ERK2 activity (Fig. 5B). It is unlikely that ERK1/2 phosphorylates PP5 directly, because PP5 lacks both the proline-directed serine/threonine (PX(S/T)) motif and the secondary DEF (docking site for ERK, FXFP) domain found in many ERK1/2 substrates (61). In addition, in vitro phosphorylation assays failed to detect ERK1/2-mediated phosphorylation of PP5 (data not shown). Nonetheless, our studies clearly demonstrate that the formation of PP5·ERK1/2 complexes can occur in a phosphatase and kinase activity-independent manner in unstimulated cells (Fig. 2). Thus, although PP5 activity is not required for its interaction with ERK1/2, PP5-mediated dephosphorylation could regulate the activity of the associated kinase, either directly or indirectly via HSP90. Consistent with a prior study (4), we did not detect any direct dephosphorylation of residues within the T-loop of ERK1/2 in our in vitro assays containing arachidonic acid-activated PP5 (data not shown). However, our data do not rule out the possibility that PP5 mediates dephosphorylation of residues outside the T-loop of ERK, which may directly impact kinase function. Moreover, as first reported for HSP90·heme-regulated elF2α kinase (19) and also observed for Cdc37·HSP90·protein kinase complexes (22), PP5 may function to alter HSP90 chaperone activity, which in turn may regulate kinase maturation/activity.
The formation of macromolecular complexes containing protein kinases and phosphatases is important for maintaining the proper phosphorylation state of target proteins within key cell signaling pathways (62), including the Ras-Raf-MEK-ERK cascade. Recent investigations indicate that Raf1 activity is modulated via its association with two different protein phosphatases, PP2A and PP5. The extant data indicate that PP2A functions as a positive regulator of Raf1, acting to dephosphorylate an inhibitory phospho-Ser-259 site (60). In contrast, PP5 acts as a negative regulator of Raf1 by dephosphorylating a site (phospho-Ser-338) that helps maintain Raf1 activity (4). In addition to Ser-259 and Ser-338 phosphorylation, Raf1 is subject to ERK-mediated feedback phosphorylation at several sites, including Ser-289, Ser-296, and Ser-301 (collectively termed EDS), which can either enhance or inhibit Raf1 function (52, 53). Given the similar temporal relationship between ERK phosphorylation of EDS (52, 53) and PP5 dephosphorylation of Ser(P)-338 (4), PP5·ERK complexes may be involved in coordinating these processes. Our current observations validate both the role of ERK in the regulation of EDS phosphorylation and the role of PP5 in the dephosphorylation of Raf1 at Ser(P)-338 (Fig. 7). In addition, our data reveal a novel role for PP5 in modulating the phosphorylation state of Raf1 at EDS (Fig. 7). PP2A has also been implicated in the dephosphorylation of pEDS (52), suggesting both phosphatases may act in concert to fine tune Raf1 activity. Interestingly, both PP5 and PP2A appear to play a similar role in the activation and feedback regulation of the dual specificity phosphatase CDC25 (20, 63). Collectively, these findings suggest that PP5·ERK complexes provide an additional control mechanism that regulates the phosphorylation status of Raf1 (Fig. 8).
FIGURE 8.
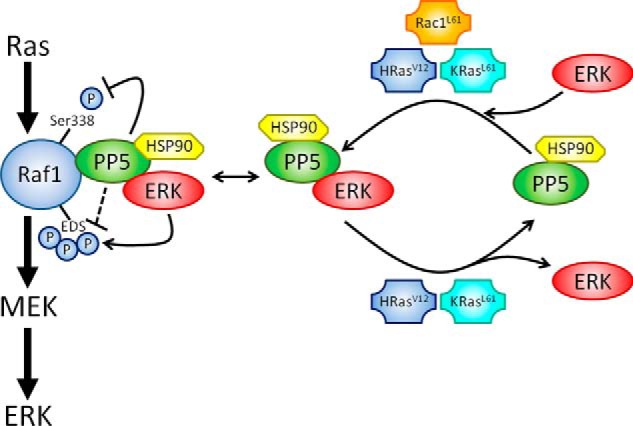
A model depicting the role and regulation of PP5·ERK complexes. PP5 suppresses Raf1 signaling by dephosphorylating Ser-338, a site important in making Raf1 permissive to further phosphorylation for full activation. Our studies support an additional role for PP5 in regulating the phosphorylation state of several EDS on Raf1 and suggest that PP5·ERK complexes coordinate Raf1 feedback phosphorylation events. Furthermore, we find that PP5-ERK interactions are modulated by active small G proteins. Active Rac1 (Rac1L61) promotes PP5-ERK1/2 interactions. In contrast, active HRas (HRasV12) and KRas4B (KRasL61), but not KRasV12, promote rapid turnover of PP5·ERK2 complexes without affecting PP5·ERK1 complexes.
Small G Protein-dependent Regulation of PP5·ERK Complexes and Their Potential Involvement in Oncogenic Processes
The identification of PP5·ERK1/2 complexes in unstimulated cells prompted us to examine the possibility that PP5-ERK1/2 interactions can be altered in response to cell stimulation. Acute activation of the Ras-Raf-MEK-ERK signaling cascade with EGF did not affect the binding of PP5 with ERK1/2. However, chronic activation of this pathway, via expression of constitutively active small G proteins, drastically altered the levels of PP5·ERK1/2 complexes (Figs. 3 and 4). Expression of Rac1L61 promoted the interaction of PP5 with both ERK1 and ERK2 (Fig. 3), whereas the expression of HRasV12 led to decreased PP5-ERK2 binding without affecting PP5-ERK1 binding (Fig. 4). These observations suggest that the two small G proteins probably utilize separate mechanisms and/or signaling pathways to modulate the assembly of PP5·ERK complexes in cells.
The expression of HRasV12 and KRasL61 greatly suppressed PP5-ERK2 binding, whereas the expression of HRasWT or KRasWT had no apparent affect (Fig. 4B). This indicates that the diminishment in PP5-ERK2 interactions produced by HRasV12 and KRasL61 may not simply be the result of Ras proteins serving as a scaffold; rather it may reflect a consequence of the oncogenic activity of these Ras variants. Interestingly, KRasV12 expression does not alter the binding between ERK2 and PP5, despite its potent activation of ERK (Fig. 4B). This observation, together with the MEK inhibitor data (Fig. 6), indicates that chronic activation of the Ras-Raf-MEK-ERK pathway is not sufficient to alter the PP5-ERK2 interaction and suggests a different Ras effector pathway (e.g. PI3K, Tiam1, PLCϵ, or RalGEF) may be involved in modulating PP5-ERK2 interactions. Although it has been known for some time that the various Ras isoforms demonstrate bias in their capacity to activate downstream effectors (64), recent work has revealed that variants of the same Ras isoform are also capable of differentially activating downstream effectors (65). Thus, it is unclear if the HRasV12- and KRasL61-dependent loss of PP5-ERK2 binding occurs through an undefined, shared effector pathway that is unaffected by KRasV12 signaling or through unique pathways that eventually converge on PP5·ERK2, but not PP5·ERK1, complexes.
Active Rac1 promotes both the assembly of PP5·ERK2 complexes (Fig. 3, A and B) and PP5- and ERK2-coordinated regulation of Raf1 phosphorylation (Fig. 7). Surprisingly, oncogenic HRasV12 expression also enhanced phosphorylation of EDS (Fig. 7B, lane 1 versus lane 10), which seemingly contradicts our data showing that HRasV12 decreased PP5-ERK2 binding (Fig. 4). Because co-immunoprecipitation assays only capture a single moment in a dynamic equilibrium, one possible explanation for the apparent decrease in the PP5-ERK2 interaction is that HRasV12 promotes enhanced turnover of the PP5·ERK2 complex (Fig. 8), creating a false impression that the binding is decreased. A hallmark of protein kinase complexes subject to rapid turnover is increased phosphorylation of substrates (66). Consistent with this idea, we observe high levels of EDS phosphorylation on the PP5-associated Raf1 in cells expressing HRasV12 (Fig. 7).
The ERK1/2 pathway integrates various cytosolic signals to regulate cellular proliferation, differentiation, growth, and apoptosis. Thus, it is no surprise that abnormal ERK signaling contributes to the formation and development of a variety of tumors and RASopathies (67, 68). Although nearly 30% of all human tumors contain a gain-of-function mutation in a ras gene (69), several recent reports demonstrate that ERK2, and not ERK1, predominantly, if not exclusively, regulates several oncogenic properties of certain human tumors (70–73). We observed a striking difference in the responsiveness of PP5·ERK2 complexes to various oncogenic Ras proteins (Fig. 4). This not only highlights the contextually kaleidoscopic nature of the ERK interactome, it also hints at a potential mechanism through which oncogenic Ras mutants regulate signaling selectivity via the ERK2 isoform. Given that PP5 has been implicated in tumor development (74, 75), it is possible that particular PP5 and ERK2 properties, such as their binding with one another, are dysregulated in the context of aberrant Ras signaling and contribute to particular oncogenic characteristics that define certain human tumors.
In summary, our studies identify novel PP5-ERK interactions and uncover a new role for small G proteins in regulating PP5·ERK1/2 complexes. Future studies of the mechanism(s) by which Rac1 and Ras modulate PP5-ERK1/2 interactions may shed light on the selective regulation of PP5·ERK2 complexes by constitutively active HRasV12 and KRasL61 and their role in tumor development. Raf1 was recently identified as one of the top 10 essential genes in a model for murine embryonic HRasV12-mediated epidermal hyperplasia (76). Our data support a role for PP5·ERK1/2 complexes in modulating the feedback phosphorylation of Raf1. As such, characterization of the ERK-mediated hyperphosphorylation of Raf1 in tumors harboring oncogenic mutations of the ras gene may provide insight on the individual diversity of cancers as well as their susceptibility to signal transduction-modulating drugs. Given that protein phosphatases shape the spatiotemporal dynamics of signaling cascades and have emerged as attractive targets for the treatment of diabetes, obesity, and cancer (77, 78), it may be interesting to investigate the PP5·ERK complex as a therapeutic target of diseases characterized by aberrant ERK signaling.
Acknowledgments
We thank Ana Carneiro, Ph.D., and Guy Watkins, Ph.D., for expert advice and critical reading of the manuscript. We are also grateful to Ning Wang, Ph.D., and Rey Gomez, M.S., for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants GM051366 (to B. E. W.), CA60750 (to R. E. H.), T32 GM08554 (to M. D. M.), and CA107331 (to D. J. W.). This work was also supported by funds from the Center for Biomolecular Therapeutics at the University of Maryland School of Medicine and the Institute for Bioscience and Biotechnology Research.
- PP5
- serine/threonine protein phosphatase 5
- HSP90
- heat shock protein 90
- PPP
- phospho-serine/threonine protein phosphatase
- TPR
- tetratricopeptide repeat
- HBD
- HSP90 binding-deficient
- PMA
- phorbol 12-myristate 13-acetate
- EDS
- ERK-dependent sites
- DEF
- docking site for ERK, FXFP
- KD
- kinase-dead
- PD
- phosphatase-dead
- IP
- immunoprecipitation
- RIPA
- radioimmune precipitation assay
- DiFMUP
- 6,8-difluoro-4-methylumbelliferyl phosphate.
REFERENCES
- 1. Hornberg J. J., Bruggeman F. J., Binder B., Geest C. R., de Vaate A. J., Lankelma J., Heinrich R., Westerhoff H. V. (2005) Principles behind the multifarious control of signal transduction. ERK phosphorylation and kinase/phosphatase control. FEBS J. 272, 244–258 [DOI] [PubMed] [Google Scholar]
- 2. Ebisuya M., Kondoh K., Nishida E. (2005) The duration, magnitude and compartmentalization of ERK MAP kinase activity. Mechanisms for providing signaling specificity. J. Cell Sci. 118, 2997–3002 [DOI] [PubMed] [Google Scholar]
- 3. Keshet Y., Seger R. (2010) The MAP kinase signaling cascades. A system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 661, 3–38 [DOI] [PubMed] [Google Scholar]
- 4. von Kriegsheim A., Pitt A., Grindlay G. J., Kolch W., Dhillon A. S. (2006) Regulation of the Raf-MEK-ERK pathway by protein phosphatase 5. Nat. Cell Biol. 8, 1011–1016 [DOI] [PubMed] [Google Scholar]
- 5. Sinclair C., Borchers C., Parker C., Tomer K., Charbonneau H., Rossie S. (1999) The tetratricopeptide repeat domain and a C-terminal region control the activity of Ser/Thr protein phosphatase 5. J. Biol. Chem. 274, 23666–23672 [DOI] [PubMed] [Google Scholar]
- 6. Yang J., Roe S. M., Cliff M. J., Williams M. A., Ladbury J. E., Cohen P. T., Barford D. (2005) Molecular basis for TPR domain-mediated regulation of protein phosphatase 5. EMBO J. 24, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Swingle M. R., Honkanen R. E., Ciszak E. M. (2004) Structural basis for the catalytic activity of human serine/threonine protein phosphatase-5. J. Biol. Chem. 279, 33992–33999 [DOI] [PubMed] [Google Scholar]
- 8. Cliff M. J., Williams M. A., Brooke-Smith J., Barford D., Ladbury J. E. (2005) Molecular recognition via coupled folding and binding in a TPR domain. J. Mol. Biol. 346, 717–732 [DOI] [PubMed] [Google Scholar]
- 9. Das A. K., Cohen P. W., Barford D. (1998) The structure of the tetratricopeptide repeats of protein phosphatase 5. Implications for TPR-mediated protein-protein interactions. EMBO J. 17, 1192–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yamaguchi F., Umeda Y., Shimamoto S., Tsuchiya M., Tokumitsu H., Tokuda M., Kobayashi R. (2012) S100 proteins modulate protein phosphatase 5 function. A link between Ca2+ signal transduction and protein dephosphorylation. J. Biol. Chem. 287, 13787–13798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chatterjee A., Wang L., Armstrong D. L., Rossie S. (2010) Activated Rac1 GTPase translocates protein phosphatase 5 to the cell membrane and stimulates phosphatase activity in vitro. J. Biol. Chem. 285, 3872–3882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yamaguchi Y., Katoh H., Mori K., Negishi M. (2002) Gα12 and Gα13 interact with Ser/Thr protein phosphatase type 5 and stimulate its phosphatase activity. Curr. Biol. 12, 1353–1358 [DOI] [PubMed] [Google Scholar]
- 13. Chen M. X., Cohen P. T. (1997) Activation of protein phosphatase 5 by limited proteolysis or the binding of polyunsaturated fatty acids to the TPR domain. FEBS Lett. 400, 136–140 [DOI] [PubMed] [Google Scholar]
- 14. Wechsler T., Chen B. P., Harper R., Morotomi-Yano K., Huang B. C., Meek K., Cleaver J. E., Chen D. J., Wabl M. (2004) DNA-PKcs function regulated specifically by protein phosphatase 5. Proc. Natl. Acad. Sci. U.S.A. 101, 1247–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morita K., Saitoh M., Tobiume K., Matsuura H., Enomoto S., Nishitoh H., Ichijo H. (2001) Negative feedback regulation of ASK1 by protein phosphatase 5 (PP5) in response to oxidative stress. EMBO J. 20, 6028–6036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chiang C. W., Liu W. K., Chiang C. W., Chou C. K. (2011) Phosphorylation-dependent association of the G4–1/G5PR regulatory subunit with IKKβ negatively modulates NF-κB activation through recruitment of protein phosphatase 5. Biochem. J. 433, 187–196 [DOI] [PubMed] [Google Scholar]
- 17. Ali A., Zhang J., Bao S., Liu I., Otterness D., Dean N. M., Abraham R. T., Wang X. F. (2004) Requirement of protein phosphatase 5 in DNA-damage-induced ATM activation. Genes Dev. 18, 249–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang J., Bao S., Furumai R., Kucera K. S., Ali A., Dean N. M., Wang X. F. (2005) Protein phosphatase 5 is required for ATR-mediated checkpoint activation. Mol. Cell. Biol. 25, 9910–9919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shao J., Hartson S. D., Matts R. L. (2002) Evidence that protein phosphatase 5 functions to negatively modulate the maturation of the Hsp90-dependent heme-regulated eIF2α kinase. Biochemistry 41, 6770–6779 [DOI] [PubMed] [Google Scholar]
- 20. Amable L., Grankvist N., Largen J. W., Ortsäter H., Sjöholm Å., Honkanen R. E. (2011) Disruption of serine/threonine protein phosphatase 5 (PP5:PPP5c) in mice reveals a novel role for PP5 in the regulation of ultraviolet light-induced phosphorylation of serine/threonine protein kinase Chk1 (CHEK1). J. Biol. Chem. 286, 40413–40422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Echeverría P. C., Bernthaler A., Dupuis P., Mayer B., Picard D. (2011) An interaction network predicted from public data as a discovery tool. Application to the Hsp90 molecular chaperone machine. PloS One 6, e26044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vaughan C. K., Mollapour M., Smith J. R., Truman A., Hu B., Good V. M., Panaretou B., Neckers L., Clarke P. A., Workman P., Piper P. W., Prodromou C., Pearl L. H. (2008) Hsp90-dependent activation of protein kinases is regulated by chaperone-targeted dephosphorylation of Cdc37. Mol. Cell 31, 886–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Soroka J., Wandinger S. K., Mäusbacher N., Schreiber T., Richter K., Daub H., Buchner J. (2012) Conformational switching of the molecular chaperone Hsp90 via regulated phosphorylation. Mol. Cell 45, 517–528 [DOI] [PubMed] [Google Scholar]
- 24. Boulton T. G., Nye S. H., Robbins D. J., Ip N. Y., Radziejewska E., Morgenbesser S. D., DePinho R. A., Panayotatos N., Cobb M. H., Yancopoulos G. D. (1991) ERKs. A family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 65, 663–675 [DOI] [PubMed] [Google Scholar]
- 25. Lefloch R., Pouysségur J., Lenormand P. (2008) Single and combined silencing of ERK1 and ERK2 reveals their positive contribution to growth signaling depending on their expression levels. Mol. Cell. Biol. 28, 511–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saba-El-Leil M. K., Vella F. D., Vernay B., Voisin L., Chen L., Labrecque N., Ang S. L., Meloche S. (2003) An essential function of the mitogen-activated protein kinase Erk2 in mouse trophoblast development. EMBO Rep. 4, 964–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pagès G., Guérin S., Grall D., Bonino F., Smith A., Anjuere F., Auberger P., Pouysségur J. (1999) Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science 286, 1374–1377 [DOI] [PubMed] [Google Scholar]
- 28. Yao Y., Li W., Wu J., Germann U. A., Su M. S., Kuida K., Boucher D. M. (2003) Extracellular signal-regulated kinase 2 is necessary for mesoderm differentiation. Proc. Natl. Acad. Sci. U.S.A. 100, 12759–12764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li J., Johnson S. E. (2006) ERK2 is required for efficient terminal differentiation of skeletal myoblasts. Biochem. Biophys. Res. Commun. 345, 1425–1433 [DOI] [PubMed] [Google Scholar]
- 30. Vantaggiato C., Formentini I., Bondanza A., Bonini C., Naldini L., Brambilla R. (2006) ERK1 and ERK2 mitogen-activated protein kinases affect Ras-dependent cell signaling differentially. J. Biol. 5, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krens S. F., Corredor-Adámez M., He S., Snaar-Jagalska B. E., Spaink H. P. (2008) ERK1 and ERK2 MAPK are key regulators of distinct gene sets in zebrafish embryogenesis. BMC genomics 9, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Roberts M. S., Woods A. J., Shaw P. E., Norman J. C. (2003) ERK1 associates with αvβ3 integrin and regulates cell spreading on vitronectin. J. Biol. Chem. 278, 1975–1985 [DOI] [PubMed] [Google Scholar]
- 33. Schaeffer H. J. (1998) MP1. A MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science 281, 1668–1671 [DOI] [PubMed] [Google Scholar]
- 34. Shao Y., Aplin A. E. (2012) ERK2 phosphorylation of serine 77 regulates Bmf pro-apoptotic activity. Cell Death Dis. 3, e253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Farhan H., Wendeler M. W., Mitrovic S., Fava E., Silberberg Y., Sharan R., Zerial M., Hauri H. P. (2010) MAPK signaling to the early secretory pathway revealed by kinase/phosphatase functional screening. J. Cell Biol. 189, 997–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Illert A. L., Zech M., Moll C., Albers C., Kreutmair S., Peschel C., Bassermann F., Duyster J. (2012) Extracellular signal-regulated kinase 2 (ERK2) mediates phosphorylation and inactivation of nuclear interaction partner of anaplastic lymphoma kinase (NIPA) at G2/M. J. Biol. Chem. 287, 37997–38005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Landar A., Rustandi R. R., Weber D. J., Zimmer D. B. (1998) S100A1 utilizes different mechanisms for interacting with calcium-dependent and calcium-independent target proteins. Biochemistry 37, 17429–17438 [DOI] [PubMed] [Google Scholar]
- 38. Amburgey J. C., Abildgaard F., Starich M. R., Shah S., Hilt D. C., Weber D. J. (1995) 1H, 13C and 15N NMR assignments and solution secondary structure of rat Apo-S100 beta. J. Biomol. NMR 6, 171–179 [DOI] [PubMed] [Google Scholar]
- 39. Ni L., Swingle M. S., Bourgeois A. C., Honkanen R. E. (2007) High yield expression of serine/threonine protein phosphatase type 5, and a fluorescent assay suitable for use in the detection of catalytic inhibitors. Assay Drug Dev. Technol. 5, 645–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wegner A. M., McConnell J. L., Blakely R. D., Wadzinski B. E. (2007) An automated fluorescence-based method for continuous assay of PP2A activity. Methods Mol. Biol. 365, 61–69 [DOI] [PubMed] [Google Scholar]
- 41. Aebersold D. M., Shaul Y. D., Yung Y., Yarom N., Yao Z., Hanoch T., Seger R. (2004) Extracellular signal-regulated kinase 1c (ERK1c), a novel 42-kilodalton ERK, demonstrates unique modes of regulation, localization, and function. Mol. Cell. Biol. 24, 10000–10015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yung Y., Yao Z., Hanoch T., Seger R. (2000) ERK1b, a 46-kDa ERK isoform that is differentially regulated by MEK. J. Biol. Chem. 275, 15799–15808 [DOI] [PubMed] [Google Scholar]
- 43. Skarra D. V., Goudreault M., Choi H., Mullin M., Nesvizhskii A. I., Gingras A. C., Honkanen R. E. (2011) Label-free quantitative proteomics and SAINT analysis enable interactome mapping for the human Ser/Thr protein phosphatase 5. Proteomics 11, 1508–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Russell L. C., Whitt S. R., Chen M. S., Chinkers M. (1999) Identification of conserved residues required for the binding of a tetratricopeptide repeat domain to heat shock protein 90. J. Biol. Chem. 274, 20060–20063 [DOI] [PubMed] [Google Scholar]
- 45. Robinson M. J., Harkins P. C., Zhang J., Baer R., Haycock J. W., Cobb M. H., Goldsmith E. J. (1996) Mutation of position 52 in ERK2 creates a nonproductive binding mode for adenosine 5′-triphosphate. Biochemistry 35, 5641–5646 [DOI] [PubMed] [Google Scholar]
- 46. Borthwick E. B., Zeke T., Prescott A. R., Cohen P. T. (2001) Nuclear localization of protein phosphatase 5 is dependent on the carboxy-terminal region. FEBS Lett. 491, 279–284 [DOI] [PubMed] [Google Scholar]
- 47. Wortzel I., Seger R. (2011) The ERK cascade. Distinct functions within various subcellular organelles. Genes Cancer 2, 195–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Swingle M., Ni L., Honkanen R. E. (2007) Small-molecule inhibitors of Ser/Thr protein phosphatases. Specificity, use and common forms of abuse. Methods Mol. Biol. 365, 23–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schönwasser D. C., Marais R. M., Marshall C. J., Parker P. J. (1998) Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol. Cell. Biol. 18, 790–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sturm O. E., Orton R., Grindlay J., Birtwistle M., Vyshemirsky V., Gilbert D., Calder M., Pitt A., Kholodenko B., Kolch W. (2010) The mammalian MAPK/ERK pathway exhibits properties of a negative feedback amplifier. Sci. Signal. 3, ra90. [DOI] [PubMed] [Google Scholar]
- 51. Shin S. Y., Rath O., Choo S. M., Fee F., McFerran B., Kolch W., Cho K. H. (2009) Positive- and negative-feedback regulations coordinate the dynamic behavior of the Ras-Raf-MEK-ERK signal transduction pathway. J. Cell Sci. 122, 425–435 [DOI] [PubMed] [Google Scholar]
- 52. Dougherty M. K., Müller J., Ritt D. A., Zhou M., Zhou X. Z., Copeland T. D., Conrads T. P., Veenstra T. D., Lu K. P., Morrison D. K. (2005) Regulation of Raf-1 by direct feedback phosphorylation. Mol. Cell 17, 215–224 [DOI] [PubMed] [Google Scholar]
- 53. Balan V., Leicht D. T., Zhu J., Balan K., Kaplun A., Singh-Gupta V., Qin J., Ruan H., Comb M. J., Tzivion G. (2006) Identification of novel in vivo Raf-1 phosphorylation sites mediating positive feedback Raf-1 regulation by extracellular signal-regulated kinase. Mol. Biol. Cell 17, 1141–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Uversky V. N., Dunker A. K. (2013) The case for intrinsically disordered proteins playing contributory roles in molecular recognition without a stable 3D structure. F1000 Biol. Rep. 5, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sato S., Fujita N., Tsuruo T. (2000) Modulation of Akt kinase activity by binding to Hsp90. Proc. Natl. Acad. Sci. U.S.A. 97, 10832–10837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Garai Á., Zeke A., Gógl G., Törő I., Fördős F., Blankenburg H., Bárkai T., Varga J., Alexa A., Emig D., Albrecht M., Reményi A. (2012) Specificity of linear motifs that bind to a common mitogen-activated protein kinase docking groove. Sci. Signal. 5, ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Westphal R. S., Anderson K. A., Means A. R., Wadzinski B. E. (1998) A signaling complex of Ca2+-calmodulin-dependent protein kinase IV and protein phosphatase 2A. Science 280, 1258–1261 [DOI] [PubMed] [Google Scholar]
- 58. Hériché J. K., Lebrin F., Rabilloud T., Leroy D., Chambaz E. M., Goldberg Y. (1997) Regulation of protein phosphatase 2A by direct interaction with casein kinase 2α. Science 276, 952–955 [DOI] [PubMed] [Google Scholar]
- 59. Kray A. E., Carter R. S., Pennington K. N., Gomez R. J., Sanders L. E., Llanes J. M., Khan W. N., Ballard D. W., Wadzinski B. E. (2005) Positive regulation of IkappaB kinase signaling by protein serine/threonine phosphatase 2A. J. Biol. Chem. 280, 35974–35982 [DOI] [PubMed] [Google Scholar]
- 60. Adams D. G., Coffee R. L., Jr., Zhang H., Pelech S., Strack S., Wadzinski B. E. (2005) Positive regulation of Raf1-MEK1/2-ERK1/2 signaling by protein serine/threonine phosphatase 2A holoenzymes. J. Biol. Chem. 280, 42644–42654 [DOI] [PubMed] [Google Scholar]
- 61. Fantz D. A., Jacobs D., Glossip D., Kornfeld K. (2001) Docking sites on substrate proteins direct extracellular signal-regulated kinase to phosphorylate specific residues. J. Biol. Chem. 276, 27256–27265 [DOI] [PubMed] [Google Scholar]
- 62. Szomolay B., Shahrezaei V. (2012) Bell-shaped and ultrasensitive dose-response in phosphorylation-dephosphorylation cycles. The role of kinase-phosphatase complex formation. BMC Syst. Biol. 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Leung-Pineda V., Ryan C. E., Piwnica-Worms H. (2006) Phosphorylation of Chk1 by ATR is antagonized by a Chk1-regulated protein phosphatase 2A circuit. Mol. Cell. Biol. 26, 7529–7538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yan J., Roy S., Apolloni A., Lane A., Hancock J. F. (1998) Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J. Biol. Chem. 273, 24052–24056 [DOI] [PubMed] [Google Scholar]
- 65. Buhrman G., Kumar V. S., Cirit M., Haugh J. M., Mattos C. (2011) Allosteric modulation of Ras-GTP is linked to signal transduction through RAF kinase. J. Biol. Chem. 286, 3323–3331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dhillon A. S., Meikle S., Yazici Z., Eulitz M., Kolch W. (2002) Regulation of Raf-1 activation and signalling by dephosphorylation. EMBO J. 21, 64–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Deng Y., Atri D., Eichmann A., Simons M. (2013) Endothelial ERK signaling controls lymphatic fate specification. J. Clin. Invest. 123, 1202–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tidyman W. E., Rauen K. A. (2009) The RASopathies. Developmental syndromes of Ras/MAPK pathway dysregulation. Curr. Opin. Genet. Dev. 19, 230–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fernández-Medarde A., Santos E. (2011) Ras in cancer and developmental diseases. Genes Cancer 2, 344–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Radtke S., Milanovic M., Rossé C., De Rycker M., Lachmann S., Hibbert A., Kermorgant S., Parker P. J. (2013) ERK2 but not ERK1 mediates HGF-induced motility in non-small cell lung carcinoma cell lines. Journal of cell science 126, 2381–2391 [DOI] [PubMed] [Google Scholar]
- 71. Shin S., Dimitri C. A., Yoon S. O., Dowdle W., Blenis J. (2010) ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol. Cell 38, 114–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Aceves-Luquero C. I., Agarwal A., Callejas-Valera J. L., Arias-González L., Esparís-Ogando A., del Peso Ovalle L., Bellón-Echeverria I., de la Cruz-Morcillo M. A., Galán Moya E. M., Moreno Gimeno I., Gómez J. C., Deininger M. W., Pandiella A., Sánchez Prieto R. (2009) ERK2, but not ERK1, mediates acquired and “de novo” resistance to imatinib mesylate. Implication for CML therapy. PloS One 4, e6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. von Thun A., Birtwistle M., Kalna G., Grindlay J., Strachan D., Kolch W., von Kriegsheim A., Norman J. C. (2012) ERK2 drives tumour cell migration in three-dimensional microenvironments by suppressing expression of Rab17 and liprin-β2. J. Cell Sci. 125, 1465–1477 [DOI] [PubMed] [Google Scholar]
- 74. Golden T., Aragon I. V., Rutland B., Tucker J. A., Shevde L. A., Samant R. S., Zhou G., Amable L., Skarra D., Honkanen R. E. (2008) Elevated levels of Ser/Thr protein phosphatase 5 (PP5) in human breast cancer. Biochim. Biophys. Acta 1782, 259–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Golden T., Aragon I. V., Zhou G., Cooper S. R., Dean N. M., Honkanen R. E. (2004) Constitutive over expression of serine/threonine protein phosphatase 5 (PP5) augments estrogen-dependent tumor growth in mice. Cancer Lett. 215, 95–100 [DOI] [PubMed] [Google Scholar]
- 76. Beronja S., Janki P., Heller E., Lien W. H., Keyes B. E., Oshimori N., Fuchs E. (2013) RNAi screens in mice identify physiological regulators of oncogenic growth. Nature 501, 185–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nguyen L. K., Matallanas D., Croucher D. R., von Kriegsheim A., Kholodenko B. N. (2013) Signalling by protein phosphatases and drug development. A systems-centred view. FEBS J. 280, 751–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. McConnell J. L., Wadzinski B. E. (2009) Targeting protein serine/threonine phosphatases for drug development. Mol. Pharmacol. 75, 1249–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]