
Figure 1.
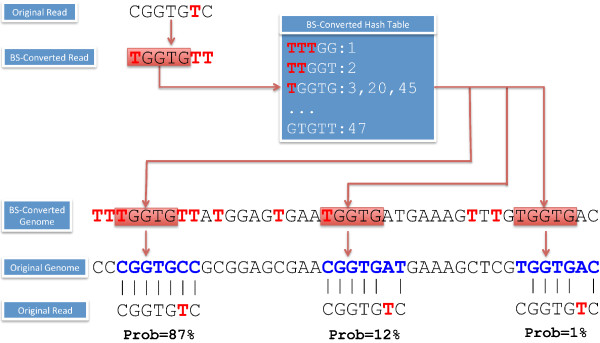
GNUMAP-bs workflow for mapping high-throughput bisulfite reads. A flow-chart of the GNUMAP-bs algorithm is shown. For the reference genome, all Cs are converted to Ts and then each k-mer in the genome is hashed, producing a list of positions in the genome to which the k-mer sequence is mapped. Given a completely BS-converted read (e.g. TGGTGTT) where the corresponding original bisulfite read (BSR) is known (e.g. CGGTGTC), a query k-mer (e.g. TGGTG) is searched against the BS-converted hash table. Locations with a k-mer match to both the genome and the read are aligned using the original read and original genome and the GNUMAP-bs probabilistic Needleman–Wunsch (N-W) algorithm. If the alignment score passes a quality threshold, the location is considered a match and recorded on the genome for future output. The posterior probability is computed for each mapped location using Equation (1). Finally, all the mapped BSRs are used to quantify the fraction of methylation at each CG location across the reference genome.