

Abstract
Background and purpose
Voltage-activated Na+ channels contain one distinct α-subunit. In the brain NaV1.1, NaV1.2, NaV1.3 and NaV1.6 are the four most abundantly expressed α-subunits. The antiepileptic drugs (AEDs) carbamazepine, phenytoin and lamotrigine have voltage-gated Na+ channels as their primary therapeutic targets. This study provides a systematic comparison of the biophysical properties of these four α-subunits and characterizes their interaction with carbamazepine, phenytoin and lamotrigine.
Experimental approach
Na+ currents were recorded in voltage-clamp mode in HEK293 cells stably expressing one of the four α-subunits.
Key results
NaV1.2 and NaV1.3 subunits have a relatively slow recovery from inactivation, compared with the other subunits and NaV1.1 subunits generate the largest window current. Lamotrigine evokes a larger maximal shift of the steady-state inactivation relationship than carbamazepine or phenytoin. Carbamazepine shows the highest binding rate to the α-subunits. Lamotrigine binding to NaV1.1 subunits is faster than to the other α-subunits. Lamotrigine unbinding from the α-subunits is slower than that of carbamazepine and phenytoin.
Conclusions and implications
The four Na+ channel α-subunits show subtle differences in their biophysical properties, which, in combination with their (sub)cellular expression patterns in the brain, could contribute to differences in neuronal excitability. We also observed differences in the parameters that characterize AED binding to the Na+ channel subunits. Particularly, lamotrigine binding to the four α-subunits suggests a subunit-specific response. Such differences will have consequences for the clinical efficacy of AEDs. Knowledge of the biophysical and binding parameters could be employed to optimize therapeutic strategies and drug development.
Keywords: sodium channel, epilepsy, carbamazepine, phenytoin, lamotrigine, antiepileptic drug
Introduction
Voltage-gated Na+ channels are responsible for the rising phase of the action potential and play a crucial role in cellular excitability. The Na+ channel protein consists of a pore-forming α-subunit associated with auxiliary β-subunits (Catterall, 2000; Catterall et al., 2005). Expression of the α-subunit alone is sufficient for the formation of a functional Na+ channel, but β subunits (so far four types have been identified: β1 through β4) can modulate the kinetics and trafficking of the channel (Isom, 2001; Patino and Isom, 2010). Of the 10 known α-subunits, NaV1.1, NaV1.2, NaV1.3 and NaV1.6 are the four most abundantly expressed subunits in the brain (Yu and Catterall, 2003; Vacher et al., 2008; channel and receptor nomenclature follows Alexander et al., 2013).
These four brain Na+ channel α-subunits have different cellular and subcellular expression patterns which determine their functional role. The NaV1.1 subunits are primarily expressed in GABAergic interneurons in the hippocampus (Yu et al., 2006; Ogiwara et al., 2007; Lorincz and Nusser, 2010) and cortex (Ogiwara et al., 2007; Martin et al., 2010). Mutations of NaV1.1 induce an epileptic phenotype due to the decreased inhibition of GABAergic interneurons (Yu et al., 2006; Ogiwara et al., 2007; Tang et al., 2009; Martin et al., 2010). NaV1.2 is primarily expressed along axons and on nerve terminals (Gong et al., 1999; Lorincz and Nusser, 2010). This localization suggests that NaV1.2 may be involved in axonal propagation of action potentials and that it is relevant for neurotransmitter release. In rodents, NaV1.3 mRNAs have the highest levels in the embryonic and early postnatal brain, whereas NaV1.3 mRNAs (Whitaker et al., 2000) and proteins (Whitaker et al., 2001) are extensively expressed in adult human brain. The NaV1.6 subunits are highly expressed in axon initial segments and in nodes of Ranvier of axons (Debanne et al., 2011) where they have a key role in action potential initiation and propagation. NaV1.6 is also moderately expressed in the somata and the dendrites of CA1 pyramidal neurons (Lorincz and Nusser, 2010) and can play an essential role in dendritic excitability.
Voltage-gated Na+ channels are key players in cellular excitability and they are the therapeutic target of antiepileptic drugs (AEDs) like carbamazepine, phenytoin and lamotrigine. These drugs modulate voltage-gated Na+ channels in a use-and voltage-dependent manner which allows them to selectively prevent high frequency firing, with little effect on a single action potential. Carbamazepine, lamotrigine and phenytoin all have a much higher affinity for the inactivated state than for the closed and open states of the Na+ channel. Therefore, they stabilize the inactivated state, effectively blocking the Na+ conductance and delaying recovery from inactivation, which prevents synchronized high frequency firing (Ragsdale and Avoli, 1998; Rogawski and Loscher, 2004). In the majority of epileptic patients, this is an effective mechanism to reduce or even prevent epileptic seizures; however, for unknown reasons, it fails in about 30% of them (Kwan and Brodie, 2000). One possible cause, but also a solution to the problem, could lie in subtle differences in the interactions of the drugs with specific Na+ channel subtypes in combination with their regional and subcellular distribution. The interactions between the AEDs and Na+ channels or different Na+ channel α-subunits have been described in several studies (Kuo and Bean, 1994; Kuo and Lu, 1997; Kuo et al., 1997; Goldin, 2001; Catterall et al., 2005), but a systematic comparison of the interactions of carbamazepine, phenytoin and lamotrigine with the four major brain Na+ channel α-subunits NaV1.1, NaV1.2, NaV1.3 and NaV1.6 using the same expression system and identical recording conditions, is lacking.
In this study, we first compared the biophysical properties of the NaV1.1, NaV1.2, NaV1.3 and NaV1.6 pore forming α-subunits, stably expressed in HEK293 cells. We then determined their modulation by carbamazepine, phenytoin and lamotrigine, in a comparative way.
Methods
Stably transfected HEK293 cell lines
All experiments were performed in HEK293 cell lines stably expressing human NaV1.1, NaV1.2, NaV1.3 or NaV1.6 α-subunits (a kind gift of GlaxoSmithKline, Stevenage, UK) that have previously been described (Chen et al., 2000; Burbidge et al., 2002; Mantegazza et al., 2005). The cell lines were generated using the pCIN5 vector (Chen et al., 2000; Burbidge et al., 2002).
Cell culture
The HEK293 cell lines were cultured in minimum essential medium (Gibco, Life Technologies, Bleiswijk, the Netherlands), containing 10% fetal calf serum (Gibco), 1% L-glutamine (200 mM, Gibco) and 1% penicillin/streptomycin (Gibco). Cells were grown in a 95% O2/5% CO2 atmosphere at 37°C and with 95% humidity. One to two days prior to electrophysiological recordings, the cells were plated on glass coverslips.
Whole-cell voltage-clamp recordings
Cells grown on glass coverslips were placed in a recording chamber with 0.5 mL extracellular solution containing (in mM): NaCl 140, KCl 5, CaCl2 2, MgCl2 1, HEPES 10 and glucose 11; pH was adjusted to 7.4. The patch electrodes had resistances of 2–3 MΩ and were filled with pipette solution containing (in mM): CsF 140, EGTA 10, HEPES 10, NaCl 5, MgCl2 2; the pH was adjusted to 7.3. Voltage-gated Na+ currents were recorded in whole-cell voltage-clamp mode at room temperature (20–22°C). After the whole-cell configuration was established, the cell was perfused with extracellular solution for ∼10 min allowing Na+ currents to stabilize, and then moved into either control or drug-containing extracellular solution emitted from the application pipette using the Fast-Step Perfusion system (SF-77B, Warner Instrument Corporation, Hamden, CT, USA). Voltage-step protocols were applied by a personal computer-controlled Axopatch 200A amplifier (Axon Instruments, Molecular Devices, Sunnyvale, CA, USA). The membrane capacitance was read from the amplifier dials and used to indicate membrane surface. Compensation circuitry was used to reduce the series resistance error by at least 75%. The calculated liquid junction potential was 8.5 mV but no corrections were undertaken. The holding membrane potential was set at −70 mV and currents were sampled at a frequency of 5 kHz and analysed using custom-made software. Each protocol (lasting 2–2.5 min) was performed at least twice in each extracellular solution (control or drug-containing). The control extracellular solution was applied before and after the drug-containing solution to detect possible slow rundown. Only cells that showed little current rundown over the recording time were incorporated in the analysis. Preferably, more than one concentration per cell was tested (with a maximum of three concentrations per cell). The currents were corrected offline for linear non-specific leak and residual capacitive current.
Data analysis
Data are given as the mean ± SEM. Multiple groups were compared using an (one-or two-factor) ANOVA followed by a post hoc Fisher's least significant difference (LSD) test. For comparison of the binding data, a test for homogeneity of regression coefficients was applied. Unless otherwise stated, Student's t-test was used for the direct comparison of two groups of parameters. P < 0.05 was considered to indicate a significant difference.
Materials
Carbamazepine (Sigma-Aldrich, Zwijndrecht, the Netherlands), phenytoin (Sigma) and lamotrigine (GlaxoSmithKline) were dissolved in DMSO (Sigma) to make stock solutions of 400, 100 and 333 mM respectively. They were then diluted in extracellular solutions to reach their final concentrations. DMSO concentrations in carbamazepine-, phenytoin-and lamotrigine-containing extracellular solutions were respectively 0.05, 0.2 and 0.3%; no DMSO effects on Na+ currents were observed.
Results
Biophysical properties of Na+ currents carried by NaV1.1, NaV1.2, NaV1.3 and NaV1.6 α-subunits
Voltage-dependent activation
Na+ currents were activated by a voltage-step protocol that depolarized the cell to different voltages after complete removal of inactivation at −120 mV (Figure 1Aa). The peak amplitude of the Na+ current was determined for each step and the current-voltage relationship (I-V curve) was constructed for each cell. The mean data points (I(V)) as a function of voltage (V) were fitted using a modified Goldman-Hodgkin-Katz current equation (Hille, 2001) in which a Boltzmann function is used to describe the voltage dependence of the Na+ permeability (Figure 1Ab):
Figure 1.
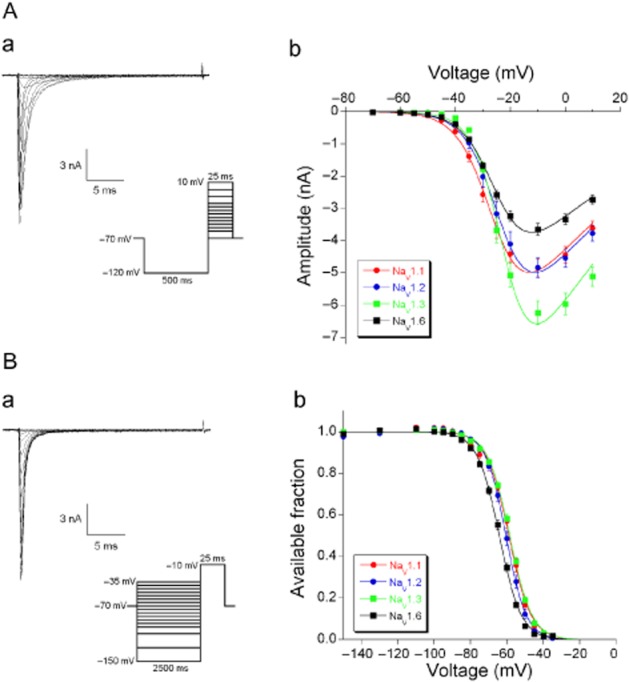
Voltage-dependent activation and steady-state inactivation of Na+ currents through the NaV1.1, NaV1.2, NaV1.3 and NaV1.6 α subunits stably expressed in HEK293 cells. (A) Voltage-dependent activation. (a) Typical traces of NaV1.3 currents. Na+ currents were activated by 25 ms depolarizing voltage steps ranging from −70 to +10 mV, following a 500 ms hyperpolarizing pre-pulse to −120 mV (protocol given as inset). (b) The mean current amplitudes for each α subunit are plotted as function of membrane voltage and fitted to the GHK equation (equation 2013). (B) Voltage-dependent steady-state inactivation. (a) Typical traces of Na+ currents. Na+ currents were activated by a depolarizing voltage step to −10 mV for 25 ms following 2500 ms hyperpolarizing pre-pulses ranging from −150 to −35 mV (protocol given as inset). (b) The mean available fraction (I/Imax) values for each α subunit are plotted as function of membrane voltage and fitted to the Boltzman function (equation 2001).
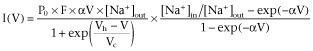 |
(1) |
Where α = F/RT with F the Faraday constant, R the gas constant and T the absolute temperature. [Na+]out and [Na+]in are the extracellular and intracellular Na+ concentrations. P0 is the maximal Na+ permeability and the voltage dependence of the conductance is described with a Boltzmann function characterized by the potential of half-maximal activation (Vh) and a slope parameter (Vc). For practical measurements, we prefer to substitute P0 F α [Na+]out = Gmax, where Gmax is the maximal conductance. The I-V curve for each cell was fitted to equation 2013 and the resulting values for Vh, Vc and Gmax for the four α-subunits are given in Table 1.
Table 1.
Activation and steady-state inactivation properties of Na+ currents carried by NaV1.1, NaV1.2, NaV1.3 and NaV1.6 α-subunits
| Activation | Inactivation | |||||
|---|---|---|---|---|---|---|
| Subtype | Vh (mV) Vc (mV) Gmax (nS) | Vh (mV) Vc (mV) | ΔVh (mV) | |||
| NaV1.1 (n = 34) | −27.1 ± 0.8aa,bb | 5.4 ± 0.2aa,bb | 74.1 ± 4.2aa,bb | −59.4 ± 0.9aa | −5.5 ± 0.3aa | 32.4 ± 1.0aa,bb,cc |
| NaV1.2 (n = 34) | −24.3 ± 1.0aa | 4.7 ± 0.2aa,cc | 76.1 ± 4.4cc,dd | −60.5 ± 0.8bb | −4.9 ± 0.1aa,bb,cc | 36.2 ± 0.9a,aa |
| NaV1.3 (n = 30) | −23.0 ± 0.7bb,cc | 4.6 ± 0.2bb,dd | 98.4 ± 5.5aa,cc,ee | −58.6 ± 0.5cc | −5.9 ± 0.1bb | 35.6 ± 0.7b,bb |
| NaV1.6 (n = 37) | −25.9 ± 0.4cc | 5.6 ± 0.1cc,dd2002 | 55.8 ± 2.7bb,dd,ee2002 | −64.3 ± 0.5aa,bb,cc | −5.7 ± 0.1cc | 38.4 ± 0.5a,b,cc |
ΔVh indicates the difference between the activation Vh and inactivation Vh values. Cell numbers are given in brackets.
P < 0.05.
P < 0.01; ANOVA, followed by Fisher's LSD post hoc test.
Voltage-dependent steady-state inactivation
Na+ currents were activated by a voltage-step protocol where the same depolarization to −10 mV followed different pre-potential steps (Figure 1Ba). The peak amplitude of the Na+ current evoked by the standard depolarization was determined for each pre-pulse voltage. The mean data points (I(V)) as a function of pre-potential V were fitted with a Boltzmann function:
| (2) |
The available fraction is given as I(V)/Imax (Figure 1Bb). Vh is the potential of half-maximal inactivation and Vc is the slope parameter. The data points of each cell were fitted with equation 2001 and the resulting values for Vh and Vc for the four α-subunits are also given in Table 1.
Window currents
The voltage range where activation and inactivation overlap defines the so-called window current: in this range, the activated Na+ current will not completely inactivate and presents itself as a small voltage-dependent persistent current (Patlak, 1991; Johnston and Wu, 1995). Using the mean values for Vh and Vc for activation as well as inactivation (Table 1), we constructed for NaV1.1 subunits, the inactivation curve (available fraction) and the activation curve (open fraction) as a function of membrane voltage (Figure 2A). From these two curves, the window current for NaV1.1 can be constructed analytically and the procedure was repeated for the other subunits (Figure 2B). For statistical comparison of the magnitude of the window currents, we calculated the AUCs between −100 mV and 0 mV: NaV1.1: 371 ± 46 pA·mV (n = 34), NaV1.2: 91 ± 10 pA·mV (n = 34), NaV1.3: 223 ± 19 pA·mV (n = 30) and NaV1.6: 146 ± 37 pA·mV (n = 37). NaV1.1 subunits are capable of generating a larger window current than the other three subunits (P < 0.01; ANOVA, Fisher's LSD post hoc test). This is consistent with the observation that the difference between Vh for activation and inactivation is the smallest for NaV1.1 subunits (Table ). Furthermore, the NaV1.3 window current had a larger magnitude than that of the NaV1.2 (P < 0.01) and NaV1.6 (P < 0.05) α-subunits. We also determined at which membrane voltage the window currents peaked: NaV1.1: −47.6 ± 1.1 mV, NaV1.2: −44.1 ± 1.5 mV, NaV1.3: −35.0 ± 1.5 mV and NaV1.6: −47.8 ± 1.2 mV, where NaV1.3 peaked at a significantly higher value than all the others (P < 0.01; ANOVA, Fisher's LSD post hoc test). The peak of the NaV1.2 window current was at a slightly more depolarized potential than those of the NaV1.1 and NaV1.6 subunits (P < 0.05).
Figure 2.
Construction of window currents carried by the four α-subunits. (A) The mean normalized activation curve (Boltzman term of equation 2013) and the mean normalized inactivation curve (same as in Figure 1Bb) in NaV1.1-expressing cells (n = 34). (B) The window currents of the four α-subunits were constructed (as the product of the activation and inactivation functions; see A) for each cell by using the Vh and Vc parameters of the activation and inactivation curves and the Gmax value. The average window currents carried by the four α-subunits NaV1.1 (n = 34), NaV1.2 (n = 34), NaV1.3 (n = 30), NaV1.6 (n = 37) are shown.
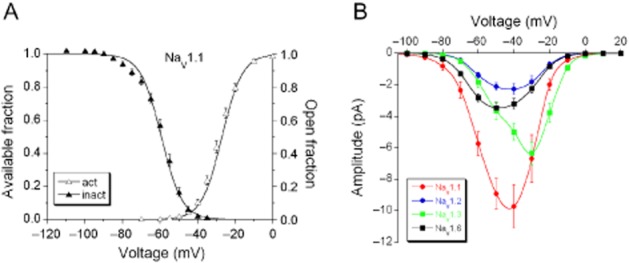
Recovery from inactivation
Na+ currents were activated by a double-pulse protocol (Figure 3A, inset). The amplitude of the Na+ current activated by the second depolarization and normalized to the first one is a function of the time interval (Δt) between them and can be fit with a single-exponential function (Figure 3Ad):
Figure 3.
Voltage-dependent recovery from inactivation of Na+ currents. (A) The time course of recovery from inactivation was determined by a double-pulse protocol (protocol given as inset). The variable pulse interval (Δt = 3.4, 6.8, 12.6, 25, 50, 100 or 200 ms, during which the current was allowed to recover) between two 25 ms depolarizing voltage steps to −10 mV, was used to determine the recovery from inactivation at the membrane voltages −80, −90 and −100 mV. Typical traces of Na+ currents are shown in (a) (Δt = 100 ms), (b) (Δt = 25 ms) and (c) (Δt = 3.4 ms). (d) The ratio of the peak amplitudes activated by the first and second pulses was plotted as a function of Δt and fitted with a mono-exponential function (equation 2001). (B) Time constant values of NaV1.1 (n = 32), NaV1.2 (n = 34), NaV1.3 (n = 32) and NaV1.6 (n = 38) currents at the three membrane voltages. The recovery from inactivation was voltage-and subunit-dependent (two-factor ANOVA, P < 0.001). For comparison between different α-subunits at the same membrane potential, *P < 0.05, **P < 0.01. For comparison of the same α-subunit at different membrane voltages, aa, bb, cc P < 0.01; Fisher's LSD post hoc test was performed for both comparisons. Because of the non-homogeneity of variance, statistical analysis was performed on the log-transformed data.
| (3) |
Where τV is the time constant that depends on the voltage during the removal of inactivation. The mean values for τV for the four α-subunits are given in Figure 3B where it can be seen that the recovery from inactivation is voltage-dependent (two-factor ANOVA, P < 0.001).
Pharmacology of NaV1.1, NaV1.2, NaV1.3 and NaV1.6 α-subunits
Frequency-dependent inhibition by carbamazepine
Carbamazepine modulates voltage-gated Na+ currents in a frequency-and use-dependent manner through its high affinity for the inactivated state (Rogawski and Loscher, 2004). The functional consequence of this phenomenon is that the current gives a strong frequency-dependent response to repetitive depolarizations, as is illustrated in Figure 4. Na+ current was activated by 3 ms depolarizations at either 10 Hz or 50 Hz depolarization steps. Even without any drug, the evoked current slowly decays due to incomplete recovery from inactivation at such frequencies (Figure 4Aa,c). In the presence of 50 μM carbamazepine that reduction is larger as binding of carbamazepine to the inactivated state will prevent a fraction of the channels from conducting upon depolarization (Figure 4Ab). The development of the carbamazepine block was isolated by subtracting the two responses; the resulting curve (Figure 4Ad) can be fit with a single-exponential function (equation 2001) to give the time constant of the process (164 ms).
Figure 4.
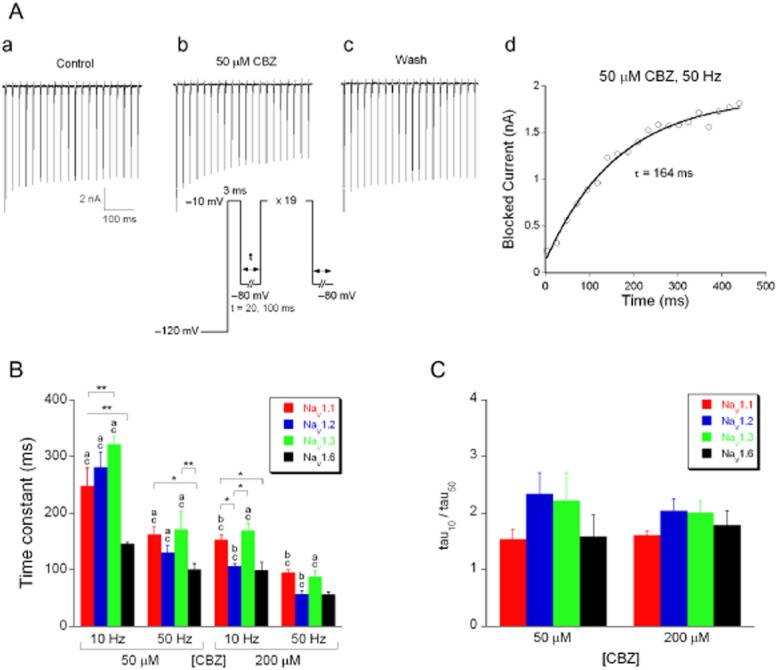
Frequency-dependent block of Na+ currents by carbamazepine. (A) Typical Na+ current traces, evoked with 20, 3 ms voltage steps to −10 mV; pulse interval at −80 mV was 20 ms) before (a), during (b) and after (c) carbamazepine (50 μM) application. Protocol given as inset: pulse interval was 20 ms (‘50 Hz’) or 100 ms (‘10 Hz’). These frequencies reflect the pulse interval frequencies and not the actual pulse frequencies (which were ∼44 Hz and ∼8 Hz respectively). (d) The blocked current amplitudes were plotted as a function of time and fitted with a mono-exponential function (equation 2001) to yield the time constant describing the development of drug block. (B) Average time constants describing the development of carbamazepine (50 and 200 μM) block of Na+ currents activated with the 10 Hz or 50 Hz stimulation protocol (n = 5–10). (C) Frequency sensitivity of carbamazepine block (ratio of tau10 and tau50 values from B) for 50 and 200 μM carbamazepine (n = 5–10). Carbamazepine showed a frequency-and concentration-dependent block (multifactor ANOVA, P < 0.001). *P < 0.05, **P < 0.01, for comparison of different α-subunits; ‘a’ P < 0.01, ‘b’ P < 0.05, for comparison of the same α-subunit at different stimulation frequencies but at the same concentration; ‘c’ P < 0.01 for comparison of the same α-subunit at different concentrations but at the same stimulation frequency (all Fisher's LSD post hoc test).
Carbamazepine block developed faster at 50 Hz than at 10 Hz and it was also faster for the higher concentration (200 μM). Although the overall trend is similar for all α-subunits, there are subtle differences in the time course of their responses (Figure 4B, multifactor ANOVA for frequency, concentration and subunit; P < 0.001). The ratio of the time constants at 10 and 50 Hz was ∼2 for 50 μM and 200 μM carbamazepine (Figure 4C), illustrating that the frequency sensitivity of the block is independent of the concentration and consistent for the four α-subunits (Figure 4C). This indicates that for these two carbamazepine concentrations, the binding rate of carbamazepine to the inactivated channels is fast enough and that carbamazepine block only depends on the fraction of inactivated channels (i.e. the substrate for this AED). In the following, we will analyse in detail the interactions between the four α-subunits and the AEDs: carbamazepine, phenytoin and lamotrigine.
AED effects on the inactivation properties of the Na+ currents
The voltage-dependent preferential binding of lamotrigine (300 μM) to the inactivated state is demonstrated using Na+ currents evoked by a depolarization to −10 mV after a pre-pulse of either −130 mV or −80 mV (Figure 5Aa). It is clear that the lamotrigine binding at −80 mV is much more effective than the one at −130 mV (Figure 5Aa).
Figure 5.

AED effects on the inactivation properties of Na+ currents. (A) (a) Typical traces of Na+ currents evoked at the testing potential of −10 mV following a 2500 ms hyperpolarizing pre-pulse at −130 mV or −80 mV in control, the presence of 300 μM lamotrigine (LTG) and wash (protocol given as inset). (b) The mean normalized steady-state inactivation curve was shifted to more hyperpolarizing direction with increasing concentrations of lamotrigine (10–1000 μM). Na+ currents were evoked and analysed in the same way as in Figure 1. (c) The absolute ΔVh values were plotted against the concentration of lamotrigine and the individual data points were fitted with a logistic function (equation 2002). (B) The average data points of the absolute ΔVh values induced by carbamazepine (CBZ; 10, 20, 50, 100 and 200 μM) (left panel), phenytoin (DPH; 10, 20, 50, 100 and 200 μM) (middle panel) and lamotrigine (10, 30, 100, 300 and 1000 μM) (right panel) were fitted with the logistic function.
The steady-state inactivation protocol (details in Figure 1B) was used to systematically investigate the concentration dependence of this phenomenon. Increasing lamotrigine concentrations (10–1000 μM) shifts the inactivation curve of the Na+ current carried by NaV1.2 to more hyperpolarizing potentials (Figure 5Ab). The major effect is on the Vh parameter of the Boltzmann fit. The shift in respect to the control situation without lamotrigine (ΔVh) was determined as a function of the applied lamotrigine concentration ([LTG]) and this relation was well fit by a first-order logistic function:
| (4) |
where ΔVhmax is the shift of Vh for saturating lamotrigine concentration and EC50 is the concentration of half-maximal ΔVh (Figure 5Ac; NaV1.2, n = 36). The analysis was repeated for all subunits and the three AEDs; the associated concentration response curves are given in Figure 5B and the data are summarized in Table 2. These results confirmed the suggestions drawn from Figure 4: the overall profile of the interaction between these drugs and the Na+ channel subunits was similar, but for this property (ΔVh) lamotrigine had a higher efficacy than carbamazepine or phenytoin (ΔVhmax is 30–43 mV and 10–20 mV, respectively) and there were subtle subunit-specific differences. The latter were evaluated using the parameters given in Table 2, but because the EC50 and the ΔVhmax parameters are dependent, direct testing might overestimate the significance. We decided only to accept the conclusions (that two subunit-specific concentration-effect relations are different) if this is also statistically supported by comparison of responses for at least one concentration. Looking at ΔVh, the NaV1.6 subunits were the most strongly modulated by carbamazepine (ΔVhmax ∼20 mV), while NaV1.3 subunits were the weakest (ΔVhmax ∼10 mV). For carbamazepine, none of the differences between EC50 values reached significance. Lamotrigine also had its strongest effect on NaV1.6 subunits (ΔVhmax ∼43 mV, ∼1.5 times larger shift of ΔVh than observed for the other subunits), while NaV1.2 subunits were most sensitive to lamotrigine (a lower EC50 value than all the others). For phenytoin, the strongest modulation is via the affinity (EC50), where NaV1.2 subunits were more sensitive to phenytoin than all other subunits.
Table 2.
AED effects on the inactivation properties of Na+ currents carried by the NaV1.1, NaV1.2, NaV1.3 and NaV1.6 α-subunits
| Phenytoin | Lamotrigine | Carbamazepine | ||||
|---|---|---|---|---|---|---|
| Subunits | EC50 (μM) | ΔVhmax (mV) | EC50 (μM) | ΔVhmax (mV) | EC50 (μM) | ΔVhmax (mV) |
| Nav1.1 | 35.4 ± 9.5 (25) | 16.6 ± 1.4 | 245.0 ± 58.7 (24) | 29.8 ± 2.4aa | 134.6 ± 41.3 (34) | 17.2 ± 2.6 |
| Nav1.2 | 16.7 ± 3.7aa,b (24) | 13.7 ± 0.9aa,b | 123.8 ± 16.8a,b (36) | 27.0 ± 1.0bb,cc | 106.0 ± 30.7 (37) | 16.8 ± 2.3 |
| Nav1.3 | 54.9 ± 6.9aa,cc (40) | 17.3 ± 0.8aa | 217.2 ± 39.8a (36) | 33.3 ± 2.0bb,dd | 49.2 ± 19.8 (17) | 9.7 ± 1.5aa |
| Nav1.6 | 31.3 ± 4.9b,cc (28) | 16.6 ± 0.8b | 230.8 ± 31.9b (36) | 43.0 ± 2.1aa,cc,dd | 116.4 ± 28.5 (18) | 20.5 ± 2.3aa |
Sample sizes are indicated in brackets.
P < 0.05.
P < 0.01; Student's t-test.
AED effects on the window currents
Changes in the activation and inactivation function affect the window current. The AED effects described here mainly shift the inactivation in hyperpolarizing direction, which implies that they always reduce the amplitude of the window current, and often also change its shape as illustrated in Figure 6A for the NaV1.1 subunit and the three AEDs at relevant concentrations. We quantified the effect using the same ‘AUC’ measure (between −100 and 0 mV) as in Figure 2B. This may underestimate the consequences of strong changes in the shape of the I-V curve, but it allows the correlation of the percentage block with the drug concentration. Quantification was attained as in Figure 5B, using equation 2002 in a slightly modified form (Figure 6B). The EC50 values are given in Table 3. Not surprisingly, the effects have a tendency to follow the results obtained in Table 2, as the inactivation curve forms an essential part of the window current. The window current block by phenytoin was quite comparable to the modulation of the inactivation curve. Lamotrigine was a stronger modulator of the inactivation than phenytoin and the larger shifts of Vh induced by lamotrigine imply that complete block of the window current was attained at much lower concentrations than the maximum shift, which explains the substantially higher sensitivity (EC50) for lamotrigine.
Figure 6.

Effects of carbamazepine, phenytoin and lamotrigine on the window currents carried by NaV1.1, NaV1.2, NaV1.3 and NaV1.6 α-subunits. (A) Examples of partly blocked window current carried by NaV1.1 by 20 μM carbamazepine (CBZ; left panel; n = 7), 20 μM phenytoin (DPH; middle panel; n = 3) and 100 μM lamotrigine (LTG; right panel; n = 4). (B) Concentration-response relationships of carbamazepine (left panel), phenytoin (middle panel) and lamotrigine (right panel) for blocking the window currents carried by the four subunits. The average data points were fitted with a logistic function (equation 4).
Table 3.
AED effects on the window currents carried by the NaV1.1, NaV1.2, NaV1.3 and NaV1.6 α-subunits
| Subunits | Phenytoin EC50 (μM) | Lamotrigine EC50 (μM) | Carbamazepine EC50 (μM) |
|---|---|---|---|
| Nav1.1 | 19.9 ± 2.0a,aa* (22) | 83.4 ± 27.5* (21) | 80.2 ± 30.0 (31) |
| Nav1.2 | 10.4 ± 4.0a,bb,** (21) | 39.1 ± 16.9* (26) | 91.8 ± 12.8*,** (31) |
| Nav1.3 | 52.5 ± 6.2aa,bb,cc,* (31) | 96.4 ± 35.6 (20) | 84.7 ± 17.8* (16) |
| Nav1.6 | 20.0 ± 3.5cc (26) | 81.5 ± 36.1 (34) | 54.6 ± 23.6 (16) |
Sample sizes are indicated in brackets.
P < 0.05.
P < 0.01, comparison between effects of the same AED on different α-subunits; Student's t-test.
P < 0.05.
P < 0.01, comparison between effects of different AEDs on the same α-subunit; Student's t-test.
Binding rates of AEDs to the inactivated Na+ channel α-subunits
The pharmacological profiles reported above, all depend on the binding rates of the AEDs to the inactivated Na+ channel, which can be determined using a voltage-step protocol (Figure 7A, inset) (Kuo and Lu, 1997). The Na+ channel was exposed to a depolarizing voltage step at −40 mV of varying duration (30–2500 ms), which determined, given a specific binding rate, which fraction of the channels in the high affinity inactivated state are bound by the AED (Figure 7A). Next, this depolarization was followed by a 5 ms hyperpolarization at −120 mV, sufficient to remove inactivation from all unbound channels. Finally, the latter fraction was exposed to a 25 ms depolarization to −10 mV. Subtracting the amplitude of this current from the one evoked in the absence of the AED yielded the blocked current. This procedure also corrected for a reduction in current amplitude due to a slow inactivation process (Figure 7B). For first-order blocking kinetics, the relation between blocked current amplitude and pre-pulse duration should follow a single-exponential function (comparable to equation 2001) (Figure 7B). Then, the reciprocal time constant (i.e. rate) is a linear function of the concentration and its slope represents the binding rate constant (Figure 7C); this procedure was repeated to provide the binding rate constants of phenytoin, carbamazepine and lamotrigine for all four α-subunits (Figure 7D, Table 4). The carbamazepine binding rate constant was approximately three times larger than that of lamotrigine or phenytoin (Table 4, P < 0.01), while the latter two were not distinguishable. The NaV1.1 subunit had the largest binding rate constant with all three AEDs, although the difference with the other three subunit types only reached significance for lamotrigine.
Figure 7.

AED binding to the inactivated state of the four Na+ channel α-subunits. (A) Typical traces of Na+ currents evoked with the test potential step to −10 mV following a 30, 250 and 2500 ms pre-pulse to −40 mV in (a) the absence of and (b) the presence of 50 μM phenytoin (DPH). Pre-pulse durations (Δt) were 30, 62.5, 125, 250, 500, 1000, 1500 and 2500 ms. Before the test potential step to −10 mV the voltage was stepped back for 5 ms to −120 mV to allow the drug-free channels to recover from inactivation (protocol given as inset). (B) The blocked Na+ current amplitude was determined by subtracting the current recorded in the presence of phenytoin from the control current and was plotted against Δt. The data points were fitted with a mono-exponential function to determine the time constant (Tau) for development of block in the presence of 10, 20, 50, 100 and 200 μM phenytoin. (C) The binding rates (1/Tau, s−1) in individual NaV1.1-expressing cells are plotted against the phenytoin concentration. The slope of the linear regression gives the binding rate constant of 14.2 × 103 M−1 s−1 to NaV1.1 for phenytoin (n = 27). The binding rate constants of carbamazepine (CBZ), phenytoin and lamotrigine (LTG) for the four α-subunits are given and compared in Table 4. (D) The mean binding rates of carbamazepine (left panel), phenytoin (middle panel) and lamotrigine (right panel) to the four α-subunits, fitted with a straight line.
Table 4.
Binding rate constants of AEDs for the NaV1.1, NaV1.2, NaV1.3 and NaV1.6 α-subunits
| Binding rate constant (× 103 M−1 s−1) | |||
|---|---|---|---|
| Subunits | Phenytoin | Carbamazepine | Lamotrigine |
| NaV1.1 | 14.2 ± 2.0** (27) | 31.7 ± 2.3a (25) | 15.4 ± 0.8**;aa,bb,cc1999 (18) |
| NaV1.2 | 8.0 ± 1.2** (18) | 28.8 ± 3.6 (33) | 9.2 ± 1.0**;aa (30) |
| NaV1.3 | 10.5 ± 1.2** (29) | 26.1 ± 1.4 (14) | 9.3 ± 0.3**;bb (17) |
| NaV1.6 | 8.5 ± 1.9** (23) | 23.3 ± 0.9a (24) | 8.4 ± 0.1**;cc (30) |
Sample sizes are indicated in brackets.
P < 0.05.
P < 0.01, comparison between subunits.
P < 0.01, comparison with carbamazepine (test for homogeneity of regression coefficients).
Unbinding rates of AEDs from the Na+ channel α-subunits
The affinity of an AED is a combination of the binding rate of the drug and the rate at which the drug dissociates from its binding site. The latter was determined with a voltage-step protocol that resembled the one used in Figure 7: a depolarization from −120 mV to −40 mV lasted 2 s and allowed complete binding of the AED to the inactivated channels (Figure 8A, inset). In the following hyperpolarization to −120 mV, the unbinding of the drug is rate limiting as it is much slower than the removal of voltage-dependent inactivation. Varying the duration of the hyperpolarization (5–500 ms), testing the result with a standard depolarization to −10 mV and subtracting the current evoked in the absence of the drug, allowed us to determine the single exponential that relates blocked current to pulse duration (Figure 8Ac). The reciprocal of the time constant (i.e. the off rate) is independent of drug concentration and defines the subunit-specific unbinding rate for carbamazepine, lamotrigine and phenytoin (at −120 mV, Figure 8B). The off rates were determined for each drug using two concentrations which always gave the same result (phenytoin: 20 and 100 μM, carbamazepine: 20 and 100 μM, lamotrigine: 30 and 300 μM). The off rate of lamotrigine was much lower than that of phenytoin and carbamazepine (Figure 8B; P < 0.01). The off rates for all AEDs were always the lowest for the NaV1.6 α-subunit. For lamotrigine, the off rate for NaV1.3 was also lower than that for the NaV1.1 and NaV1.2 α-subunits.
Figure 8.
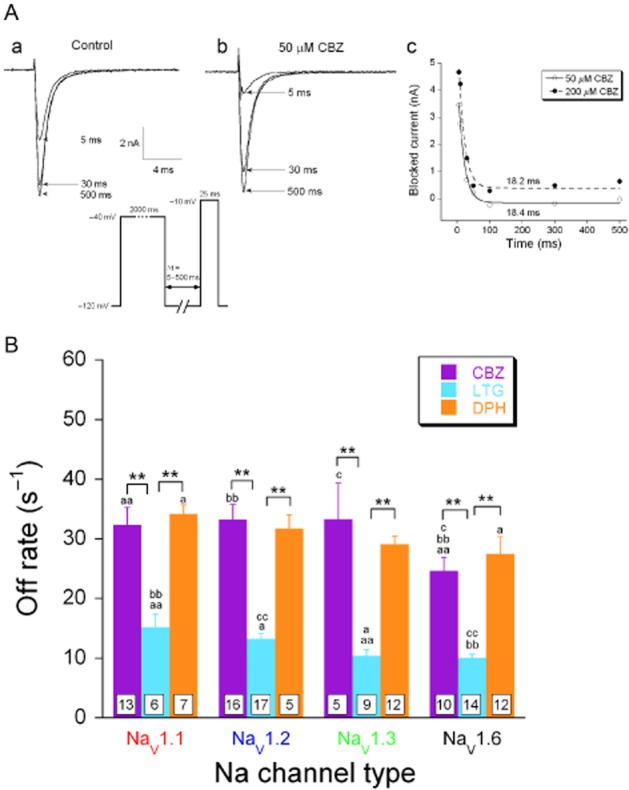
Unbinding rates of AEDs from the inactivated NaV1.1, NaV1.2, NaV1.3 and NaV1.6. (A) Typical traces of NaV1.3 currents evoked with a testing step potential to −10 mV, following 5-, 30-and 500-ms pre-pulses to −120 mV in the absence (a) and presence (b) of 50 μM carbamazepine (CBZ). The membrane was held at −40 mV for 2000 ms to permit drug binding to the inactivated channels, followed by a step to a recovery potential at −120 mV with a variable time duration Δt to facilitate channels to recover from inactivation and to allow drug dissociation (protocol shown as inset). The blocked current was determined by subtracting the current recorded in the presence of carbamazepine from the control current (measured in the absence of carbamazepine) and plotted against the time duration of the recovery Δt (c). The data points were fitted with a mono-exponential equation to determine the time constant (Tau) of carbamazepine unbinding, showing that the unbinding rate is concentration-independent (tau ∼18 ms for 50 and 200 μM carbamazepine). (B) Carbamazepine, lamotrigine (LTG) and phenytoin (DPH) unbinding rates from all four α-subunits. These off rate values were determined by calculating the reciprocal of the time constants. For all four α-subunit subtypes, lamotrigine showed a slower off rate than carbamazepine and phenytoin (multifactor ANOVA followed by Fisher's LSD post hoc test). *P < 0.05, **P < 0.01, comparison between different drugs dissociating from the same α-subunit. a P < 0.05; aa, bb and cc, P < 0.01; comparison between different α-subunits from which the same drug dissociates. Because of the non-homogeneity of variance, the statistical analysis was performed on the log-transformed data.
Discussion
In the present study, we performed a detailed comparison of the biophysical and pharmacological properties of human NaV1.1, NaV1.2, NaV1.3 and NaV1.6 α-subunits stably expressed in HEK293 cells. It is unclear whether differences in biophysical properties that have been reported for these subunits (Goldin, 2001; Catterall et al., 2005) were tangible differences or whether they should be attributed to differences in cell host systems (e.g. CHO cells vs. HEK cells), transfection methods (transient vs. stable) and/or recording solution compositions. Our study represents a systematic comparison of the four major brain Na+ channel α-subunits using the same expression system and identical recording conditions. We compared the biophysical properties of the four α-subunits and observed subtle differences (Table 1). The study focuses on the properties of the α-subunits, where the main interaction with the three AEDs we used, takes place. When extrapolating our conclusions to more complete (neuronal) systems, potential modulatory actions of β-subunits of these voltage-gated Na+ channels should also be taken into consideration.
The recovery from inactivation for the NaV1.1 and NaV1.6 subunits were relatively fast. Fast recovery from inactivation for the NaV1.1 subunit could facilitate fast action potential spiking observed in NaV1.1-expressing interneurons (Galarreta and Hestrin, 2002; Ogiwara et al., 2007). NaV1.6 subunits are present at high densities in axon initial segments and nodes of Ranvier (Boiko et al., 2001; Ogiwara et al., 2007; Lorincz and Nusser, 2008) and fast recovery from inactivation could also facilitate action potential initiation and propagation along axons. The NaV1.2 and NaV1.3 subunits displayed a slow recovery from inactivation, suggesting a low excitability at (subcellular) sites where these subunits are present. Together with the knowledge of the precise distribution patterns of the four Na+ channel α subunits, our data can provide more insight into neural computation and signal processing of single neurons. Moreover, subtle differences in the interactions between the AEDs and the subunits could lead to functional differences of their antiepileptic profile. In addition, a shift in subunit expression during epilepsy (Aronica et al., 2001; Qiao et al., 2013) could change the relative efficacy of the AEDs.
A smaller difference between Vh for activation and Vh for inactivation results in a larger window current (Patlak, 1991; Johnston and Wu, 1995; Ketelaars et al., 2001). The window current for the NaV1.1 subunit was indeed larger than those of the other three subunits. This may indicate that the cellular or subcellular locations where the NaV1.1 subunit is densely expressed would be more excitable. Recent studies showed that the NaV1.1 subunit is primarily expressed in GABAergic interneurons and largely coexpressed in parvalbumin-and KV3.1b-positive interneurons (Yu et al., 2006; Ogiwara et al., 2007; Martin et al., 2010; Lorincz and Nusser, 2010). The larger window current for the NaV1.1 subunit could facilitate fast-spiking of these interneurons. The NaV1.3 subunit also generates a considerable window current although not as large as that of the NaV1.1 subunits. The peak of the NaV1.3 subunit window current was at a relatively depolarized membrane potential (∼ −35 mV). In addition to this window current, the NaV1.3 subunit also carries a persistent Na+ current, which peaks at an even more depolarized membrane potential (Sun et al., 2007). The presence of a window current and a persistent Na+ current in cells that express the NaV1.3 subunit may have profound consequences for neuronal excitability.
Although the AED binding sites are located on the α-subunits of the Na+ channels (Rogawski and Loscher, 2004), β-subunits can still modulate AED efficacy (Lucas et al., 2005; Uebachs et al., 2010). Our cell system is devoid of β-subunits and it should be kept in mind that in neurons (with β-subunits present) AED efficacy may be slightly different. The three AEDs block voltage-gated Na+ channels in a use-or frequency-dependent manner. High frequency activity pushes many Na+ channels in the inactivated state and because these AEDs have a much higher affinity for the inactivated state than for the closed and open states, the channels will not conduct current (Macdonald and Kelly, 1995; Rogawski and Loscher, 2004). This was illustrated by the observation that carbamazepine blocked Na+ currents faster with higher frequency depolarization steps (50 Hz vs. 10 Hz) in a concentration-dependent manner. The increase in the degree of block from 10 to 50 Hz was not influenced by the concentration of carbamazepine, suggesting that within the concentration range used (50 and 200 μM), carbamazepine binding rate is fast enough to effectively block Na+ currents with either protocol (50 Hz vs. 10 Hz). Therefore, the development of carbamazepine block depends solely on the availability of inactivated channels. AEDs are able to concentration-dependently shift the steady-state inactivation curves of the Na+ currents to more hyperpolarizing potentials. When comparing the effects of the three AEDs, we observed that the maximal shift of Vh (ΔVhmax) for steady-state inactivation evoked by lamotrigine was twice as large as that of carbamazepine and phenytoin (∼30 mV vs. ∼15 mV). When comparing AED affinities for the four subunits, subtle differences were observed. The efficacy of carbamazepine and phenytoin in blocking the window current was in the same concentration range as the effects on the steady-state inactivation. However, the EC50 values for lamotrigine were smaller than those obtained for shifting the steady-state inactivation function (Tables 2 and 3). This could result from the relatively slow unbinding of lamotrigine (as compared with carbamazepine and phenytoin), which would induce a more efficient block of the window current.
Possibly, the differences in binding rates to the α-subunits underlie the frequency sensitivity of AED block. We found that carbamazepine had a (much) higher binding rate to the inactivated Na+ channel than phenytoin and lamotrigine. The magnitude of these rates is in the range previously described for native voltage-gated Na+ channels (Kuo and Bean, 1994; Kuo et al., 1997; Kuo and Lu, 1997). The different binding rate constants of these three AEDs could explain their differences in efficacy to control epileptic discharges. With the slowest binding rate of the three AEDs, phenytoin would require a prolonged depolarization for at least a few hundred milliseconds in clinical situations to exert its antiepileptic action. In contrast to phenytoin, carbamazepine has the fastest binding rate, which may make it more effective than phenytoin against ictal discharges with relatively short depolarizations. The patients who respond well to carbamazepine, but not to phenytoin might have bursts of discharges with shorter depolarization phases. Another parameter that determines AED efficacy is the dissociation rate. The low off rate of lamotrigine explains why lamotrigine is able to induce a larger shift in the steady-state inactivation function than carbamazepine or phenytoin. Because lamotrigine occupies its binding site longer, this AED evokes a larger shift of Vh and a stronger block. This slow dissocation also explains the efficacy of lamotrigine in blocking the window current.
The differences in Na+ channel make-up as observed in various brain regions (Clare et al., 2000; Trimmer and Rhodes, 2004) could result in a region-specific AED sensitivity. The actual neuronal situation is much more complex than our expression system for α-subunits and we should therefore extrapolate our conclusions to the physiology and pharmacology of intact neuronal systems with great caution.
The use-dependent block of the tested AEDs points to a preferential inhibition of high frequency discharges and this, in turn, suggests that AEDs would most effectively inhibit fast-spiking interneurons. We found that lamotrigine has a higher binding rate to the NaV1.1 subunit (than to the other three subtypes), the predominant Na+ channel subtype in certain subpopulations of inhibitory interneurons (Yu et al., 2006; Ogiwara et al., 2007; Lorincz and Nusser, 2008). However, the net outcome of an AED in a local network that contains different types of neurons (with various firing patterns), which express different subunits, might be hard to predict.
Our study has uncovered the subtle differences in biophysical properties of four brain Na+ channel α-subunits and how they interact with the most common AEDs. These differences are not huge, but they might be relevant to understand the net effect of AEDs on a local circuit that contains several classes of neurons that express various Na+ channel α-subunits. For the same reason, development of new AEDs and screening for their antiepileptic effects should take these properties into account.
Acknowledgments
This work was supported by the Dutch National Epilepsy Foundation ‘the Power of the Small’ (06-13, WW) and grant 114000091 from the ‘Platform alternatief voor dierproeven’ of the Dutch organization for fundamental research.
Glossary
- AED
antiepileptic drug
Conflict of interest
None declared.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, Spedding M, Peters JA, Harmar AJ, Collaborators CGTP. The Concise Guide to PHARMACOLOGY 2013/14: Overview. Br J Pharmacol. 2013;170:1449–1867. [Google Scholar]
- Aronica E, Yankaya B, Troost D, van Vliet EA, Lopes da Silva FH, Gorter JA. Induction of neonatal sodium channel II and III alpha-isoform mRNAs in neurons and microglia after status epilepticus in the rat hippocampus. Eur J Neurosci. 2001;13:1261–1266. doi: 10.1046/j.0953-816x.2001.01502.x. [DOI] [PubMed] [Google Scholar]
- Boiko T, Rasband MN, Levinson SR, Caldwell JH, Mandel G, Trimmer JS, et al. Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron. 2001;30:91–104. doi: 10.1016/s0896-6273(01)00265-3. [DOI] [PubMed] [Google Scholar]
- Burbidge SA, Dale TJ, Powell AJ, Whitaker WR, Xie XM, Romanos MA, et al. Molecular cloning, distribution and functional analysis of the NA(V)1.6. Voltage-gated sodium channel from human brain. Brain Res Mol Brain Res. 2002;103:80–90. doi: 10.1016/s0169-328x(02)00188-2. [DOI] [PubMed] [Google Scholar]
- Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev. 2005;57:397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- Chen YH, Dale TJ, Romanos MA, Whitaker WR, Xie XM, Clare JJ. Cloning, distribution and functional analysis of the type III sodium channel from human brain. Eur J Neurosci. 2000;12:4281–4289. [PubMed] [Google Scholar]
- Clare JJ, Tate SN, Nobbs M, Romanos MA. Voltage-gated sodium channels as therapeutic targets. Drug Discov Today. 2000;5:506–520. doi: 10.1016/s1359-6446(00)01570-1. [DOI] [PubMed] [Google Scholar]
- Debanne D, Campanac E, Bialowas A, Carlier E, Alcaraz G. Axon physiology. Physiol Rev. 2011;91:555–602. doi: 10.1152/physrev.00048.2009. [DOI] [PubMed] [Google Scholar]
- Galarreta M, Hestrin S. Electrical and chemical synapses among parvalbumin fast-spiking GABAergic interneurons in adult mouse neocortex. Proc Natl Acad Sci U S A. 2002;99:12438–12443. doi: 10.1073/pnas.192159599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin AL. Resurgence of sodium channel research. Annu Rev Physiol. 2001;63:871–894. doi: 10.1146/annurev.physiol.63.1.871. [DOI] [PubMed] [Google Scholar]
- Gong B, Rhodes KJ, Bekele-Arcuri Z, Trimmer JS. Type I and type II Na(+) channel alpha-subunit polypeptides exhibit distinct spatial and temporal patterning, and association with auxiliary subunits in rat brain. J Comp Neurol. 1999;412:342–352. [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membrane. 3rd edn. Sinauer, Sunderland, MA; 2001. [Google Scholar]
- Isom LL. Sodium channel beta subunits: anything but auxiliary. Neuroscientist. 2001;7:42–54. doi: 10.1177/107385840100700108. [DOI] [PubMed] [Google Scholar]
- Johnston D, Wu SMS. Foundations of Cellular Neurophysiology. Cambridge: MIT Press; 1995. [Google Scholar]
- Ketelaars SO, Gorter JA, van Vliet EA, Lopes da Silva FH, Wadman WJ. Sodium currents in isolated rat CA1 pyramidal and dentate granule neurones in the post-status epilepticus model of epilepsy. Neuroscience. 2001;105:109–120. doi: 10.1016/s0306-4522(01)00176-2. [DOI] [PubMed] [Google Scholar]
- Kuo CC, Bean BP. Slow binding of phenytoin to inactivated sodium channels in rat hippocampal neurons. Mol Pharmacol. 1994;46:716–725. [PubMed] [Google Scholar]
- Kuo CC, Lu L. Characterization of lamotrigine inhibition of Na+ channels in rat hippocampal neurones. Br J Pharmacol. 1997;121:1231–1238. doi: 10.1038/sj.bjp.0701221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CC, Chen RS, Lu L, Chen RC. Carbamazepine inhibition of neuronal Na+ currents: quantitative distinction from phenytoin and possible therapeutic implications. Mol Pharmacol. 1997;51:1077–1083. doi: 10.1124/mol.51.6.1077. [DOI] [PubMed] [Google Scholar]
- Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342:314–319. doi: 10.1056/NEJM200002033420503. [DOI] [PubMed] [Google Scholar]
- Lorincz A, Nusser Z. Cell-type-dependent molecular composition of the axon initial segment. J Neurosci. 2008;28:14329–14340. doi: 10.1523/JNEUROSCI.4833-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz A, Nusser Z. Molecular identity of dendritic voltage-gated sodium channels. Science. 2010;328:906–909. doi: 10.1126/science.1187958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas PT, Meadows LS, Nicholls J, Ragsdale DS. An epilepsy mutation in the beta1 subunit of the voltage-gated sodium channel results in reduced channel sensitivity to phenytoin. Epilepsy Res. 2005;64:77–84. doi: 10.1016/j.eplepsyres.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Kelly KM. Antiepileptic drug mechanisms of action. Epilepsia. 1995;36(Suppl. 2):S2–S12. doi: 10.1111/j.1528-1157.1995.tb05996.x. [DOI] [PubMed] [Google Scholar]
- Mantegazza M, Yu FH, Powell AJ, Clare JJ, Catterall WA, Scheuer T. Molecular determinants for modulation of persistent sodium current by G-protein betagamma subunits. J Neurosci. 2005;25:3341–3349. doi: 10.1523/JNEUROSCI.0104-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MS, Dutt K, Papale LA, Dube CM, Dutton SB, de Haan G, et al. Altered function of the SCN1A voltage-gated sodium channel leads to gamma-aminobutyric acid-ergic (GABAergic) interneuron abnormalities. J Biol Chem. 2010;285:9823–9834. doi: 10.1074/jbc.M109.078568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, et al. Na(v)1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27:5903–5914. doi: 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patino GA, Isom LL. Electrophysiology and beyond: multiple roles of Na+ channel beta subunits in development and disease. Neurosci Lett. 2010;486:53–59. doi: 10.1016/j.neulet.2010.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patlak J. Molecular kinetics of voltage-dependent Na+ channels. Physiol Rev. 1991;71:1047–1080. doi: 10.1152/physrev.1991.71.4.1047. [DOI] [PubMed] [Google Scholar]
- Qiao X, Werkman TR, Gorter JA, Wadman WJ, van Vliet EA. Expression of sodium channel alpha subunits 1.1, 1.2 and 1.6 in rat hippocampus after kainic acid-induced epilepsy. Epilepsy Res. 2013;106:17–28. doi: 10.1016/j.eplepsyres.2013.06.006. [DOI] [PubMed] [Google Scholar]
- Ragsdale DS, Avoli M. Sodium channels as molecular targets for antiepileptic drugs. Brain Res Brain Res Rev. 1998;26:16–28. doi: 10.1016/s0165-0173(97)00054-4. [DOI] [PubMed] [Google Scholar]
- Rogawski MA, Loscher W. The neurobiology of antiepileptic drugs. Nat Rev Neurosci. 2004;5:553–564. doi: 10.1038/nrn1430. [DOI] [PubMed] [Google Scholar]
- Sun GC, Werkman TR, Battefeld A, Clare JJ, Wadman WJ. Carbamazepine and topiramate modulation of transient and persistent sodium currents studied in HEK293 cells expressing the Na(v)1.3 alpha-subunit. Epilepsia. 2007;48:774–782. doi: 10.1111/j.1528-1167.2007.01001.x. [DOI] [PubMed] [Google Scholar]
- Tang B, Dutt K, Papale L, Rusconi R, Shankar A, Hunter J, et al. A BAC transgenic mouse model reveals neuron subtype-specific effects of a Generalized Epilepsy with Febrile Seizures Plus (GEFS+) mutation. Neurobiol Dis. 2009;35:91–102. doi: 10.1016/j.nbd.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer JS, Rhodes KJ. Localization of voltage-gated ion channels in mammalian brain. Annu Rev Physiol. 2004;66:477–519. doi: 10.1146/annurev.physiol.66.032102.113328. [DOI] [PubMed] [Google Scholar]
- Uebachs M, Opitz T, Royeck M, Dickhof G, Horstmann MT, Isom LL, et al. Efficacy loss of the anticonvulsant carbamazepine in mice lacking sodium channel beta subunits via paradoxical effects on persistent sodium currents. J Neurosci. 2010;30:8489–8501. doi: 10.1523/JNEUROSCI.1534-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacher H, Mohapatra DP, Trimmer JS. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol Rev. 2008;88:1407–1447. doi: 10.1152/physrev.00002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker WR, Clare JJ, Powell AJ, Chen YH, Faull RL, Emson PC. Distribution of voltage-gated sodium channel alpha-subunit and beta-subunit mRNAs in human hippocampal formation, cortex, and cerebellum. J Comp Neurol. 2000;422:123–139. doi: 10.1002/(sici)1096-9861(20000619)422:1<123::aid-cne8>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Whitaker WR, Faull RL, Waldvogel HJ, Plumpton CJ, Emson PC, Clare JJ. Comparative distribution of voltage-gated sodium channel proteins in human brain. Brain Res Mol Brain Res. 2001;88:37–53. doi: 10.1016/s0169-328x(00)00289-8. [DOI] [PubMed] [Google Scholar]
- Yu FH, Catterall WA. Overview of the voltage-gated sodium channel family. Genome Biol. 2003;4:207. doi: 10.1186/gb-2003-4-3-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–1149. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]