

Abstract
Amphibians occupy a key phylogenetic position in vertebrates and evolution of the immune system. But, the resources of its transcriptome or genome are still little now. Bombina maxima possess strong ability to survival in very harsh environment with a more mature immune system. We obtained a comprehensive transcriptome by RNA-sequencing technology. 14.3% of transcripts were identified to be skin-specific genes, most of which were not isolated from skin secretion in previous works or novel non-coding RNAs. 27.9% of transcripts were mapped into 242 predicted KEGG pathways and 6.16% of transcripts related to human disease and cancer. Of 39 448 transcripts with the coding sequence, at least 1501 transcripts (570 genes) related to the immune system process. The molecules of immune signalling pathway were almost presented, several transcripts with high expression in skin and stomach. Experiments showed that lipopolysaccharide or bacteria challenge stimulated pro-inflammatory cytokine production and activation of pro-inflammatory caspase-1. These frog's data can remarkably expand the existing genome or transcriptome resources of amphibians, especially immunity data. The entity of the data provides a valuable platform for further investigation on more detailed immune response in B. maxima and a comparative study with other amphibians.
Keywords: transcriptome, frog, immune system, tissue-specific genes, non-coding RNAs
1. Introduction
Amphibians have been used as vertebrate models for the experimentalist since the 19th century. Since 50 years or so, Xenopus laevis is the most widely used anuran amphibian research organism,1 an important model of vertebrate cell biology and development for many decades.2 However, of the thousands of species, only two closely related species (Xenopus silurana tropicalis and X. laevis) have considerable genomic resources: published genome of the Pipid, Xenopus (silurana) tropicalis, and transcript information of X. laevis.3,4 Xenopus tropicalis, of which genome analysis results show that at least 1700 predicted genes identified to be human disease genes, occupies a key phylogenetic position among amniots and teleost fish.4 But, understanding of the genome requires large quality of transcripts as reference sequences. The transcriptome is comprised of a comprehensive set of genes active in selected tissues and species. Therefore, understanding global dynamics of the transcriptome is essential for interpreting genome expanded information and unravelling phenotypic variation.5
In our previous studies, the Chinese red belly frog (Bombina maxima) has various different skin secretions defencing external stimulus, which is an endemic amphibian and lives in very harsh environments of the mountainous regions of southwestern China.6–9 This study was motivated by two reasons. First, genome-wide or transcriptome-wide approaches are now being applied to whole-genome wide duplication of Xenopodinae.10 However, the amphibian is still the lack of genomic resources like genomic sequences and transcriptome dataset, in spite of two Xenopus toads' genome or transcriptome being available. Secondly, more and more investigators found that Xenopus model offers an invaluable research tool for immunological research.11 In addition, the most ancient mechanism of immunity was considered to be direct microbicidal activity of antimicrobial peptides (AMPs) in the human immune system.12 In frogs, extremely abundant and diverse AMPs have been reported in our previous studies.13 A lot of AMPs were also rich in B. maxima.7 In recent studies, the immune system of amphibian species is nearly as complex as that of mammals.11 But, research on the immune system of anuran amphibians exclusively relies on X. laevis as a model.14 Bombina and Xenoanura diverged in the middle Jurassic and the ancestor of Bombina appeared the Eocene of Palaeogene.15 From the living environment and skin secretions of B. maxima, it could have a more mature immune system compared with Xenopus, potentially suited to be as a new model for identifying the nature of innate or adaptive immunity in vertebrates. Thus, we performed the latest paired-end RNA-sequencing (RNA-seq) techniques16 to transcriptome-wide depict its feature of the immune system. The database of the transcriptome will submit to public databases for free to use.
2. Materials and Methods
2.1. Animals and ethics
Bombina maxima, used here were obtained from the Kunming Institute of Zoology, which has been used in our previous studies. All the animal studies were reviewed and approved by the animal care and use committee of Kunming Institute of Zoology, The Chinese Academy of Sciences.
2.2. Construction of the transcriptome
Skin and blood tissues were sampled from four adult frogs with no injury. RNA extraction and library construction are described in detail in Supplementary information S1. Transcriptome-sequencing clean reads is basis for assembly and following analysis (the data are archived at the NCBI Sequence Read Archive under accession number SRA058577; http://ncbi.nlm.nih.gov/sra). Transcriptome is assembled in de novo (Supplementary information S2).
2.3. Transcriptome functional annotation
The transcript sequences were identified with the coding sequence (CDS) to search for gene ontology (GO) terms (Supplementary information S3). All translated protein sequences were identified protein domain prediction. Tissue-specific genes and comparative expression analysis are described in Supplementary information S4. Identification of novel non-coding RNAs (ncRNAs) is identified as Tan's methods.17 The detailed description is mentioned in Supplementary information S5.
2.4. Tissue expression of selected immune-related genes
We used cDNA as a template for reverse transcription PCR (RT-PCR) amplification of the coding regions of several given immune genes. Equal amounts of total RNA (1.05 μg) from each tissue were pooled for cDNA generation (Supplementary information S6). Quantitative RT-PCR (qRT-PCR) was carried out using the LightCycler® 480 (Roche, Switzerland) with SYBR premix Ex Taq II (TaKaRa, Japan). Samples were run in triplicates. Primers used in qRT-PCR are shown in Supplementary Table S1. For each primer, a melting curve of the PCR product was also performed to ensure the absence of artefacts. The comparative crossing points (ΔCP) method18 was used for the relative quantification of the respective transcript expression. Expression values were normalized using an endogenous beta-actin.
2.5. Stimulation of immune response in vitro and in vivo
In vivo immune response was stimulated by intraperitoneal injection of bacteria or lipopolysaccharide (LPS). In vitro stimulation were carried out on splenocytes from B. maxima. The detailed methods are described in Supplementary information S8.
3. Results and Discussion
3.1. Sequencing and transcriptome assembly
cDNAs prepared from skin and blood tissues of B. maxima were sequenced using Illumina HiSeq™ 2000. After sequencing, 59 820 004 and 58 492 250 raw reads was separately obtained from skin and blood tissue in B. maxima. Cleaning the low-quality reads, we acquired 55 533 592 (4 998 023 280 bp from skin tissue) and 54 967 354 (4 947 061 860 bp from blood tissue) clean reads (Table 1). Using the Trinity program to assemble clean reads to contigs in de novo, a total of 172 119 contigs were separately generated from these two tissues. After contigs assembly, a total of 88 213 putative gene objects (all unigenes) were obtained ranging from 200 to 20 882 bp and 602 bp in average length. The size of unigenes >500 bp was 27 661. The largest unigenes had 20 882 bp, and the N50 of unigenes was 868 bp (Table 1). The length of all unigene sequences was >200 bp and 41.0% (36 209) were >499 bp in length (Supplementary Fig. S1). In term of sequence completeness, we used a less stringent but broadly adopted definition, considering a sequence to be full length if it comprises at least the complete CDS.19 Of the 88 213 (41.2%) unigenes, 36 319 had CDS matches with direction possessed significant similarity (cut-off E-value of 1e−6) with best-hit blast results. Of 88 213 (3.5%) CDS sequences, 3129 were produced by ESTScan program, which cannot to be aligned to known databases. Finally, a total of 39 448 unigene sequences had the complete CDS (Supplementary Fig. S1).
Table 1.
Summary of sequencing, assembly and analysis of B. maxima transcriptome
| Dataset name | All | Skin | Blood |
|---|---|---|---|
| Total no. of bases (bp) | 9 945 085 140 | 4 998 023 280 | 4 947 061 860 |
| Average read length (bp) | 90 | 90 | 90 |
| No. of reads | |||
| Raw reads | 118 312 254 | 59 820 004 | 58 492 250 |
| Clean reads | 110 500 946 | 55 533 592 | 54 967 354 |
| Q20a of clean reads | 97.75% | 97.82% | 97.67% |
| No. of contigs | |||
| Total contigs | 348 906 | 172 119 | 176 787 |
| Average contig read length | 275 | 289 | 260 |
| No. of unigenes | |||
| Total unigenes | 88 213 | 87 931 | 80 830 |
| Distinct clusters | 33 619 | 1762 | 2016 |
| Distinct singletons | 54 594 | 86 819 | 78 714 |
| N50b of unigenes | 868 | 710 | 681 |
| Average unigene read length | 602 | 498 | 481 |
| Largest unigene | 20 882 | 11 892 | 20 882 |
| No. of large unigenes >500 bp | 27 661 | 21 961 | 18 509 |
aQ20: percentage is the proportion of nucleotides with a quality value >20 in reads.
bN50: unigene length-weighted median.
3.2. Function annotation
Several complementary methods were used to annotate the assembled sequences. To begin with using BLASTX and BLASTN, the assembled sequences were searched against the Nr, Swiss-Prot protein databases and Nt nucleotides database (cut-off E-value of 1e−6). Of the 88 213 assembled sequences, 35 457 (Nr, 40.2%), 30 413 (Swiss-Prot, 34.5%) and 27 622 (Nt, 31.3%) had significant matches. The more detailed analysis of the blast top 30 hits is shown in Supplementary Fig. S2. We compared the transcriptome with CDS with that of human (Ensemble data release 69) and X. tropicalis (Xenbase, Xentr7_2). The overlap between transcripts from the transcriptome of Bombina and Xenopus is larger than the others (Fig. 1A). In total, above two times more transcripts of B. maxima have the orthologs in X. tropicalis, compared with human. However, the most of transcripts from B. maxima have no orthologs with human and X. tropicalis. Although ∼32% of transcripts from B. maxima have its orthologs in Xenopus, obviously most of the transcripts were identified to be species-specific transcripts or novel-spliced transcripts of this frog and Xenopus. To determine the expression level of the transcripts in skin and blood tissues, we mapped the raw reads to the assembled sequences. The reads per kilobase per million (RPKM) values of the transcripts are shown in Fig. 1B. It depicts that gene with high expression has distinct biases for skin, compared with the blood.
Figure 1.

Functional profiles of the transcriptome. (A) The venn diagram shows the comparison of the transcriptome between B. maxima, X. laevis and human. The data of B. maxima are composed of transcripts with CDS from the transcriptome. The transcriptome of X. laevis and human is including of all cDNA sequences. (B) All transcripts expression of skin and blood tissues in B. maxima. (C) Tissue-specific genes were identified in the transcriptome. It shows the overlap of skin- and blood-specific transcripts that passed strict filters. (D) The expression patterns of skin- and blood-specific genes under different reads per kilobase per million (RPKM) values. (E) The novel ncRNAs were revealed from RNA-seq data. A breakdown of all the transcripts into protein-coding, putative ncRNAs, and definite ncRNAs. (F) Heatmap showing distinct profiles of all definite ncRNAs was detected in the transcriptome. The RPKM values from skin and blood tissues were used for mean-centering, normalization and clustering.
The unigene sequences that had matches in Nr databases were given GO annotations with Uniprot databases. Of these, 11 905 (33.6%) were assigned to one or more 3423 GO terms. As reported in other Xenopus transcriptome studies,3 the largest proportion of transcripts fell into 2–3 broad and general categories as follows: ‘Biological Process’, ‘Molecular Function’ and ‘Cellular Component’ (Supplementary Fig. S3). These results were similarly found in Mytilus edulis,20 Hypophthalmichthys molitrix,21 Sus scrofa22. As our expectation, a great amount of transcripts were involved in localization process (2383, 20.02%) and development process (2204, 18.51%), corresponding to the results of X. tropicalis transcriptome.17 Meanwhile, annotation of the 88 213 unigene sequences using Clusters of Orthologous Groups (COG) of protein databases generated the results for 9625 putative proteins. The COG annotated putative proteins ranged functionally into at least 25 molecular families, including cell cycle control, cell membrane biogenesis, immune defence and signal transduction (Supplementary Fig. S4), in accordance with the categories of GO annotation described above.
To further analyse the tissue-specific genes, the strict filter was performed as Supplementary methods. Finally, 12 608 (14.3%) and 6771 (7.7%) unigenes were identified to be skin-specific and blood-specific genes (Fig. 1C). Compared with blood-specific genes, the skin-specific genes have a large proportion of high-expressed genes (Fig. 1D), 70 skin-specific genes whose RPKM values were >50. As our expectation, most of them were related to epithelial cell growth, wound healing and AMPs. One of high-expressed AMP—Maximin S2, isolated from skin secretion in our previous work, was identified to be as the skin-specific gene. Besides the Maximin S2, a fungicide antimicrobial peptide, Holotricin, was also a high-expressed skin-specific protein, which was not isolated from skin secretions in our previous work. In the high-expressed skin-specific genes, more and more epithelial cells' components were identified with highest RPKM values, such as WAP, Desmoglein-1. Our results responses the fact that this frog can be lived well in the harsh environment.
Next, we examined the unannotated transcripts (47 582, 53.9% of all transcripts) for protein-coding transcripts potential, leaving 47 112 transcripts as potential ncRNAs. As methods of Supplementary information S5, we finally identified 1183 ncRNAs (Fig. 1E), which possess at least 70% sequence identity to known non-coding genes. These ncRNAs serve as the definite ncRNAs in B. maxima. Clustering analysis reveals that these genes can be broadly divided into three clusters (Fig. 1F). The first cluster corresponds to the transcripts whose expression levels had great variations between skin and blood tissue. The second cluster contains the genes whose high expressions were gradually increased over skin to blood. It indicates that the ncRNAs play important roles in skin and blood. The third cluster appears to contain the genes that are sharply degraded with high expression in blood, compared with skin. Not all ncRNAs derived from skin and blood tissues, we mapped the raw reads to ncRNAs transcripts and found that there were 146 (12.3%) skin-specific and 46 (3.9%) blood-specific ncRNAs (Supplementary Table S2). Collectively, our analysis has identified with confidence a total of 47 112 potential ncRNAs and 1183 definite ncRNAs. We observed that the majority of these ncRNAs appears to be lowly expressed, with 69.0% of them (or 815 transcripts) having maximum RPKM of <5. In addition, the non-coding transcripts that we found in this study appear to be performing tissue-specific function. This collection of ncRNAs provides a rich resource for future functional studies, which will greatly enhance our understanding the genetic adaption of the frog.
3.3. KEGG pathways analysis
A total of 24 579 assembled sequences were identified to occur in 242 predicted KEGG pathways. The number of sequences ranged from 2 to 2722. The top 20 pathways with the highest number of sequences are shown in Table 2, and the maximum of transcripts was found in the metabolic pathways (n = 2722). Metabolic pathways, implicated the intracellular metabolic pathways control immune tolerance and regulation of stem cell behaviour and life span,23 showed the highest number of transcripts here and corresponded to the results of GO annotation described above and published transcriptome report.21,22 As the previous report of X. tropicalis genome, 7% of protein-coding gene orthologs were identified to be human disease genes.4 We also identified 11.17% (2745) of transcripts related disease using KEGG analysis, related disease including Huntington's disease, Alzheimer's disease (n = 619), Chagas disease (American trypanosomiasis, n = 280), Parkinson's disease (191), Autoimmune thyroid disease (n = 131), Graft-versus-host disease (n = 121), Prion diseases (n = 116). Remarkably, 2693 (10.95% of 24 579 transcripts annotated by KEGG analysis) transcripts were identified to be related to cancer pathways and specified cancer (small-cell lung cancer, prostate cancer, endometrial cancer, non-small-cell lung cancer, pancreatic cancer, colorectal cancer, bladder cancer and thyroid cancer). This analysis and annotation may be useful for further study and great interest for other researchers.
Table 2.
Summary of top 20 pathways with highest gene numbers
| No. | Pathway | All genes with pathway annotation (N = 24 579), n (%) | Pathway ID |
|---|---|---|---|
| 1 | Metabolic pathways | 2722 (11.07) | ko01100 |
| 2 | Huntington's disease | 1287 (5.24) | ko05016 |
| 3 | Ubiquitin-mediated proteolysis | 1223 (4.98) | ko04120 |
| 4 | Purine metabolism | 1138 (4.63) | ko00230 |
| 5 | Pathways in cancer | 1073 (4.37) | ko05200 |
| 6 | Focal adhesion | 1014 (4.13) | ko04510 |
| 7 | Regulation of actin cytoskeleton | 946 (3.85) | ko04810 |
| 8 | Calcium signalling pathway | 821 (3.34) | ko04020 |
| 9 | Influenza A | 817 (3.32) | ko05164 |
| 10 | Lysine degradation | 794 (3.23) | ko00310 |
| 11 | Protein processing in endoplasmic reticulum | 706 (2.87) | ko04141 |
| 12 | MAPK signalling pathway | 683 (2.78) | ko04010 |
| 13 | Measles | 674 (2.74) | ko05162 |
| 14 | Endocytosis | 654 (2.66) | ko04144 |
| 15 | Alzheimer's disease | 619 (2.52) | ko05010 |
| 16 | Hepatitis C | 610 (2.48) | ko05160 |
| 17 | Basal transcription factors | 601 (2.45) | ko03022 |
| 18 | Vascular smooth muscle contraction | 598 (2.43) | ko04270 |
| 19 | Ribosome biogenesis in eukaryotes | 592 (2.41) | ko03008 |
| 20 | Tight junction | 550 (2.24) | ko04530 |
3.4. Protein domains prediction
InterProScan identified 55 758 protein domains (6912 types) among 22 241 unigene sequences. The protein domains abundantly occurred in immunoglobulin-related domains (n = 1802), Zinc finger, C2H2 (n = 864; IPR007087), followed by reverse transcriptase (n = 802; IPR000477), Zinc finger, C2H2-like (n = 748; IPR015880), Zinc finger, C2H2-type/integrase, DNA binding (n = 709; IPR013087) and Zinc finger, RING/FYVE/PHD type (n = 387; IPR013083) (Fig. 2). A total of 4416 transcripts (5.0% of the transcriptome) with protein domains including Zinc finger keyword were identified, which comprise 80 types of protein domain. Usually, the most common DNA-binding motif and transcription factors contain one or more capable of making sequence-specific recognition of DNA and also binding to RNA and protein targets.24 Comparing with other species' transcriptome or genome,25,26 Zinc finger protein domain was enriched in all scanned protein domains, such as 3.3% in Macrobrachium rosenbergii transcriptome and 2.6% in the mouse transcriptome.
Figure 2.

The distribution of protein domains predicted in B. maxima unigenes. The protein domain prediction was obtained by the InterProScan software. Remarkably, the sum of immunoglobulin-related domain is higher than others.
Among the immunoglobulin-related domains, a total of 653 immunoglobulin (Ig)-like-fold domains (IPR013783) and 318 immunoglobulin-like domains (IPR007110) were predicted (Fig. 2). Ig-like-fold domains are involved in a variety of functions playing essential roles in the vertebrate immune response and many other processes, involved with protein–protein interactions with their β-sheets. Ig-like-related domains were also found in other published species' transcriptome. However, its proportion in M. rosenbergii (0.9% of Ig-like-fold domains in 8411 detected contigs) was much less than that in B. maxima (2.9% of 22 241 input sequences). Moreover, the immunoglobulin provides further links between vertebrate immune systems. Interestingly, we found a high number of repeating units ending with Trp-Asp (WD)-containing domains (4.6% of the CDS), including WD40/YVTN repeat-like-containing domains (IPR015943; n = 423), WD40-repeat-like-containing domains (IPR011046; n = 303), WD40-repeat subgroup (IPR019781; n = 300), WD40-repeat domains (IPR001680; n = 281), WD40-repeat-containing domains (IPR017986; n = 267) and WD40-repeat 2 (IPR019782; n = 254) domains. In contrast to published domain prediction data, 2.1% of the transcripts with WD40-repeat domains in the M. rosenbergii transcriptome. These domains are involved in functions ranging from signal transduction transcription regulation to cell cycle control and apoptosis.27 In Xenopus, the WD40-repeat domains were present in wdr36, traf51 and pik3r4 related to transcriptional regulation and epigenetic regulation in the latest report.17
3.5. Genes related to the innate immune system
Innate immune system (IIS) is the ancient defence system and first defence line of multicellular organisms against various infectious agents. We identified 1501transcripts (570 genes) involved in the immune system process. Of these, a total of 32 transcripts and at least 11 different Toll-like receptors (TLRs) were identified in B. maxima, including TLR1, TLR2, TLR3, TLR4, TLR6, TLR7, TLR8 and Toll/interleukin-1 receptor (Fig. 3 and Supplementary Table S3). The TLRs signalling pathway is primarily dependent on TLR2 and TLR4. In humans, TLR2 binds LPS in the presence of LPS-binding protein (LBP) and CD14, and then induces nuclear factor (NF)-κB activation. In B. maxima, lipoprotein and peptidoglycan can induce the TLR signalling pathway in B. maxima, indicated by the predicted pathway map (Fig. 4A). The expression of several key molecules activated by the signalling pathway was validated in B. maxima (Fig. 4B and Supplementary Table S4). We detected the expression of TLR2 and TLR4 using qRT-PCR, and then found that TLR2 and TLR4 show a peak expression in spleen, kidney, stomach and liver (Fig. 4D). In digital expression genes analysis (Fig. 4C), the clustered expression of genes in skin and blood was divided into major three clusters. The first cluster contains genes whose expression in skin and blood keep in the medium level, comparing with others. Most of them are downstream genes of the signalling pathway. The second cluster corresponds to genes whose expression in skin was higher than that in blood except for its first branch. The third cluster contains genes with highest expression in the TLR pathway of B. maxima.
Figure 3.

The putative genes related to the immune system in B. maxima. Bars with dots represent the number of transcripts with the annotated functions; diamond bars represent the number of different genes (transcripts <10 are not shown).
Figure 4.

The signalling pathway activated by TLRs in B. maxima. (A) The putative TLRs pathway of B. maxima predicted by KEGG pathway annotation (KO04620). Red coloured genes on the map were definitely identified in the transcriptome. In MyD88-dependent pathway, it leads to the activation of IRAK. In MyD88-independent pathway, TRIF and Rac1 are the main adaptor protein and interact with TRAF3 or PI3K to activate IRF or AKT. Finally, NF-κB is activated. All the members shown in this figure have homologous proteins in other species. (B) The key molecules of TLR pathway were amplified by reverse transcription PCR (RT-PCR) using RNA from skin and blood tissues. Primers were designed according to the transcripts. (C) Clustered expression profile for 56 genes mapped to the KEGG pathway is organized by different tissues. The RPKM values were normalized, with each row representing a different gene. (D) Expression detection of eight key components of the TLR signalling pathway was measured by quantitative RT-PCR (qRT-PCR). Results are the mean crossing points (ΔCP) value ± SEM (n = 3 technical triplicates).
CD14 transcripts of B. maxima were not mapped to the pathway determined by KEGG annotation. The similarity of CD14 in B. maxima was closely identical to that of Taeniopygia guttata and also to TLR2 type 2 of Amazona albifrons. In addition to previous studies, fish, of which TLR4 is not response to LPS, are resistant to endotoxic shock.28 In frog, the TLR4 is respond to LPS as a receptor in urinary bladder epithelial cells.29 As the pathway prediction, it showed that TLR4 might be failing to respond to LPS through LBP binding to CD14 induced the pathway. Hence, the fact that TLR4 of B. maxima can recognize the LPS to induce the pathway remains to be elucidated. Then, in addition to in silico analysis, we detected the response to LPS or bacteria both in vitro and in vivo. Interleukin (IL)-1β and IL-8 are pro-inflammatory cytokines that play pivotal role in the innate immunity. In response to LPS stimulation, it can be activated in dependent of TLRs activation.30 We detected the IL-1β and IL8 secretion by B. maxima splenocytes after LPS or bacteria stimulation. The results from RT-PCR demonstrated that the mRNA level elevated than that of unstimulated (Fig. 5A). LPS and bacteria stimulation upregulated the intracellular level of pro-IL-1β, but LPS only slightly enhanced the secretion of mature IL-1β. It seems that bacteria strongly enhanced the expression and secretion of IL-1β (Fig. 5B). Western blotting of IL-8 revealed that LPS and bacteria enhanced the secretion of IL-8 by splenocytes (Fig. 5B). The secretion of IL-1β and IL-8 upon LPS and bacteria stimulation were also tested in vivo. The results demonstrated that after intraperitoneal administration of either LPS or bacteria, IL-1β and -8 could be found in the peritoneum exudate fluids (Fig. 5D). LPS and bacteria induced the change of peritoneum cell proportion, and also the upregulated proportion of cells was identified to be monocyte-like cells (Fig. 5E).
Figure 5.
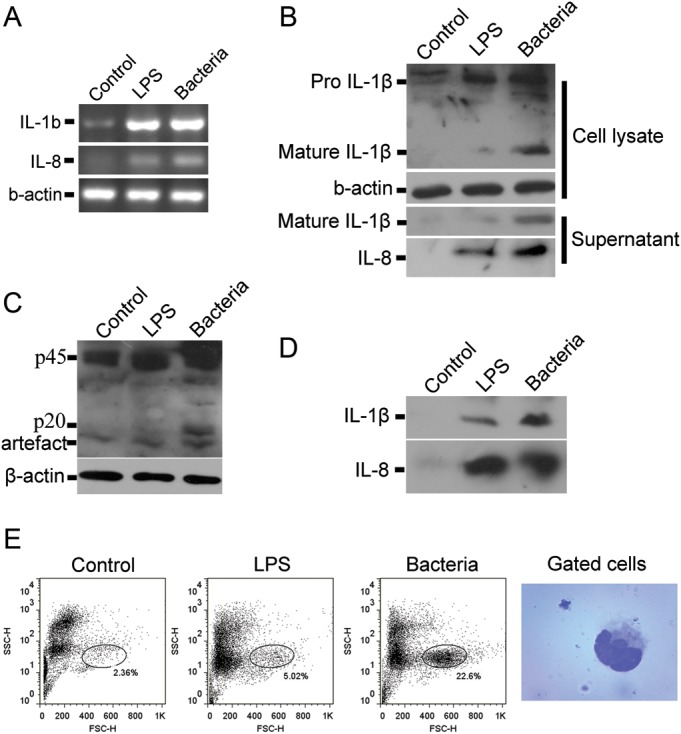
Lipopolysaccharide (LPS) and bacteria induced pro-inflammatory caspase activation and pro-inflammatory cytokine secretion in vitro and in vivo. (A) Splenocytes were isolated from B. maxima spleen and stimulated with LPS (8 μg/ml) or bacteria (3 × 108cfu/ml) for 2 h. The cells were collected for RT-PCR of IL-1βand IL-8. Either LPS or bacteria can upregulate the mRNA level of IL-1β and IL-8. (B) Splenocytes were incubated with LPS or bacteria. The cells and the supernatants were collected for western blotting of IL-1β and IL-8. (C) Splenocytes stimulated with LPS or bacteria were lysed for the detection of caspase-1 activation by western blotting. The band of ∼20 kDa represents the presence of activated caspase-1. (D) Bombina maxima weighted 25 ± 5 g were injected intraperitoneally with LPS (50 mg/kg in 0.1 ml PBS) or bacteria (3 × 108cfu in 0.1 ml PBS, Staphylococcus aureus, ATCC25923). Two hours after injection, peritoneum exudates were harvested, IL-1β and IL-8 were detected by western blot. (E) Two hours after injection, peritoneal cells were harvested and analysed by flow cytometry. The mostly changed subset of cells was stained and photographed.
The ligation of TLRs signalling occurs primarily through MyD88 (MyD88-dependent pathway) or other adapter proteins (TRAM, TRIF and MyD88-independent pathway). It leads to the induction of antimicrobial genes, cytokines and chemokines and maturation of dendritic cells (Fig. 4A). In TLR4 signal transduction, Mal (MyD88) is a necessary adaptor.31 We surely found MyD88 transcripts and that had nearly 2-fold expression in skin than in blood, containing Toll/interleukin-1 receptor domain. But, the RPKM value of MyD88 in skin was a little great than in blood. In addition, MyD88 is required for Xenopus axis formation as binding protein with Toll/IL-1 receptors.32 In the MyD88-independent pathway, the putative transcripts of components were identified such as TRAF3, TBK1 and several IRFs (Fig. 4A and Supplementary Table S3).
The complement system is a key subsystem in innate immunity, which is composed of many serum proteins associated with many important activities.33 A total of 157 transcripts were identified including C2, C3, C3d, C4a, C5, C6, factor B, factor D and factor I, etc. The most of key complement components were also detected with high expression in skin, stomach and lung tissues of B. maxima (Supplementary Fig. S5 and Table S3). The complement components usually share the short consensus domains, such as C9 with perforin-like domain. We analysed the domains contained in complement components of B. maxima. Most of the complement components shared CCP domain and Tryp SPc (trypsin-like serine protease) domain (Supplementary Table S5). But, the factor P (properdin factor) and factor B have thrombospondin_1 domain. These domains may play a important role in complement initiation and amplification. The regulator of complement activation (RCA) is essential for excessive C3 activation that often drives the consumption of the complement pathway and damages self tissue.34 We identified two RCA-coding proteins in B. maxima, ARC1 and ARC3, which are closely identical to the ARCs of Xenopus (Supplementary Fig. S5D and E). In addition to the lectin pathway, we also identified that a number of 22 transcripts highly expressed in B. Maxima, including Fcn1 (M-ficolin), Fcn2 (L-ficolin) and Fcn3 (H-ficolin). In our results, it shows a peak expression in spleen, kidney and stomach (Supplementary Fig. S5C).
The cytokine network is an important homeostatic system with potent activities in immune surveillance, growth, developmental and repair processes.35 The important role of cytokines is controlling immune responses.36 We identified 42 interleukin/interleukin receptor transcripts in the B. maxima dataset. We did not found IL-2 transcripts in B. maxima the same as in Xenopus that involved in the bactericidal activity in the adaptive immune system (AIS).37 However, IL-34 and IL-7 receptors were identified in B. Maxima, with no orthologs of Xenopus (Supplementary Table S6). Another ancient and conserved cytokine is the macrophage migration inhibitory factor (MIF) in the immune system. We identified one full-length MIF transcripts (345 bp), which is closely identical to the MIF of Xenopus, mouse and human (Supplementary Table S6). MIF is conserved across taxa from invertebrate to vertebrates,38 emerging as an important mediator of IIS.
In human, TLRs engage in Type I interferon (IFN) (IFN-α/β) production. In our results, the full-length Type I IFNs were not identified in the transcriptome. But, we found short-length (<200 bp) small contigs with the medium identity (40–60%) to Xenopus Type I IFNs, which have low consensus overlaps for further assembly to long transcripts. In interferon receptor pathways, Interferon-α/β receptor (IFNAR) expressed eight transcripts in the transcriptome and mapped to the pathway (Fig. 4A and Supplementary Table S3). It shows high similarity to IFNAR of chicken (Supplementary Fig. S6A). The Type I IFNs all bind to IFNAR, which is composed of two chains: IFNAR1 and IFNAR2. The intracellular domain of IFNAR1 associates with Tyk2, whereas IFNAR2 associates with JAK1 and JAK2. Type I IFNs can induce the evolutionary pathway, JAK/STAT pathway. As expected, 12 transcripts with a high identity to JAK2 and STATs were found in B. maxima (Supplementary Table S3). These cytokines also regulate the expression of over 1200 genes, the products of which function as mediators of host immune responses,39 such as four families of IFN-inducible GTPases including dynamin-like GTPases, Mx proteins, p47 GTPases and very large inducible GTPase-1 (VLIG-1).40 These four families of IFN-inducible GTPases were all expressed in the B. maxima transcriptome (Supplementary Table S7). There were also great amounts of VLIG-1 from two major groups expressed in this frog (Supplementary Fig. S6B).
NOD-like receptors (NLRs) and RIG-like receptors (RLRs) act as pathogen sensors for the recognition of intracellular bacteria and viruses. Twenty-nine transcripts of NLRs were present in B. maxima, including of 10 members such as NOD1, NLRC3, NLRC5, NAIP. Another intracellular sensor is retinoic acid-inducible gene I (RIG-I) receptors. Two transcripts of RIG-I were identified in this transcriptome, with high similarity to Xenopus, then to mouse and human (Supplementary Table S6). Caspase-1 is considered as a typical member of pro-inflammatory caspases, including caspase-1, caspase-4, caspase-5 and caspase-12 in human and caspase-1, caspase-11 and caspase-12 in mouse.41 Caspase-1 is activated upon the assembly of inflammasome, which is assembled by the activation of NOD-like receptors including NLRP3, NLRP1 and NLRC4. In our present study, we tested whether the pro-inflammatory caspase, caspase-1, could be activated. The pro-enzyme form of caspase-1 is an ∼45 kDa protein and cleaved into a p10 and p20 subunit after activation. The results showed that, after bacteria stimulation, the appearance of p20 was determined by western blotting using a rabbit polyclonal antibody against caspase-1 p20. Although it seems that LPS was also able to induce the activation of caspase-1, the ability is much weaker than bacteria (Fig. 5C). As the downstream executor of inflammasome, caspase-1 activation implied the presence of the NLR–inflammasome–caspase-1 pathway in this frog.
3.6. Genes related to the adaptive immune system
Currently available data indicate that the AIS suddenly emerged on vertebrate with jaws like a ‘big-bang’ event.42,43 So, it is fascinating to many scientist and investigators for its origin and evolution. The highly complex of AIS is based on intact B-cell receptor (BCR)–T-cell receptor (TCR)–major histocompatibility complex (MHC)-based AIS.43 We identified the transcripts of BCR, TCR and MHC in B. maxima (Fig. 3 and Supplementary Table S3). Moreover, we amplified some key components of the AIS and sequencing the amplified results (Fig. 6A). All of validated molecules were high expressed in B. Maxima, excluding IgM compared with the others (Fig. 6B and Supplementary Table S4). From the components of AIS, the immune system of B. maxima is nearly parallel to the mammals'. BCR is composed of two parts, ligand-binding moiety and signal transduction moiety. We identified that IgM, Ig CH II/IgY, IgG and many segments of heavy chain and light chains were definitely presented in the B. maxima transcriptome (Supplementary Table S3). IgM, generally evolutionarily conserved, is most ancient antibody class and secreted monomer.44 IgY is first found in amphibians, the function of which is equal to IgG and IgE.45 Shorter forms of IgY are believed to neutralize pathogen without activation of inflammatory response.43 In addition, we identified the IgG-gamma heavy chain in B. maxima that is searched with no result in Xenopus (Supplementary Table S6). IgG is involved in high-affinity memory responses. In summary, frog is the most primitive species, whose IgY and IgG play a important role in immune response, with a classical memory response that is based on class switching after antigen stimulation. This result showed that IgY has already taken rearrangement and is essential for immune response in B. maxima. CD79A was also identified in the transcriptome and had two transcripts. Therefore, the component of BCRs is complete in the AIS of B. maxima. TCR is responsible for recognizing antigens bound to MHC molecules. Beta-chain TCR and gamma-chain TCR were identified, but alpha-chain and delta-chain had no transcripts expressed in skin and blood tissues. TCR is a heterodimer, consisting of α/β-chains and γ/δ-chains. A translocon organization of both α- and β-TCR genes is found in all animals, and the δ- and α-TCR gene with close linkage.43 Hence, the α- and δ-chain of TCR probably existed in B. maxima.
Figure 6.

Genes related to the adaptive immune system. (A) Several selected molecules of the AIS in B. maxima were amplified by RT-PCR using RNA from skin and blood tissues, primers designed according to the assembled sequences of the transcriptome. These molecules were identified by searching against known database and domain prediction. Each two transcripts from 18 MHC I and 11 MHC II were detected by RT-PCR. (B) Five key immune components were detected its expression in seven different tissues using qRT-PCR. Results are the mean ΔCP value ± SEM (n = 3 technical triplicates). (C) Eighteen MHC Class I amino sequences were from B. maxima, and MHC I of Gallus gallus, MHC Ia and Ib of Homo sapiens, MHC Ia and Ib of Mus musculus and MHC I of X. laevis. The bootstrap values next to the nodes represent the percentage of 1000 replicate trees supporting the corresponding clade. (D) Eleven MHC Class II amino sequences were from B. maxima, and other MHC II proteins from above species.
The MHC is defined by MHC Class I and Class II; the MHC Class I/II molecules are closely involved in antigen presentation and usually has multiple copies in the vertebrates.46 We identified 18 transcripts of MHC Class I and 11 transcripts of MHC Class II expressed in B. maxima. As the previous methods,47 we conservatively estimated that the minimum number of putative expressed MHC Class I and Class II was three and two loci indicated by transcripts clustering into three and two major groups without a reference genome (Fig. 6C and D). Multiple loci for MHC Class II have a profound impact on peptide binding and anti-infections.48 With multiple loci for MHC Class II, the frog has an ability to battle constantly intracellular infection, such as virus. Most Xenopus species have a single-expressed MHC Class I gene (Class Ia), which encodes receptor molecules to present intracellular pathogen and is closely linked to other genes assisting in antigen processing.49 MHC Class Ib gene belongs to a separate MHC lineage. The expressed multiple loci of MHC Class I/II is considered to be the crucial role acted by one or two rounds of whole-genome duplication (WGD) at stage of fish or amphibians.50 In fact, X. laevis expresses non-classical MHC Ib genes with similarity in sequence and structure to classical genes.51 From the constructed a phylogenetic tree (Fig. 6C), the MHC Class Ib have rapid rate of evolution compared with classical MHC Class Ia of B. maxima and X. laevis. The multiple sequence alignment of the selected MHC Class I sequences (Supplementary Fig. S7A) also shows that the rate of residue substitution to MHC Class Ia lower than that of MHC Class Ib from this frog and other vertebrates. The MHC Class Ia of X. laevis shows high identity to that of B. maxima, except in very few sites. And the conserved sites of MHC Class I exist great variances in this frog (marked with red box). Counterpart to MHC Class II (Supplementary Fig. S7B, marked with blue box), it shows that the conserved domain of four transcripts in B. maxima is high similarity to the human MHC Class II, instead of Xenopus, which is correspond to the phylogenetic tree analysis (Fig. 6D). The phylogenetic analysis revealed that these sequences clustered into distinct two clades. One of these clades is grouped into MHC Class Ib genes from higher vertebrates. From bootstrap values, MHC Class Ib was indeed a more rapid evolutional rate than MHC Class Ia. As to a single classical Class I locus expressed in X. tropicalis and X. laevis, the degree to which B. maxima is a good potential model for the anuran MHC.
In summary, we have developed comprehensive transcriptome for B. maxima. Although there is extensive genome or transcriptome achieved in Xenopus, comparative analysis showed that difference exists between these two species' transcriptome. The data obtained is rich in gene transcripts related to disease and disease pathway, especially to immunity. Based on pathway prediction, we uncover the fact that TLRs can recognize the lipoproteins, peptidoglycan, LPS or bacteria in this frog. Furthermore, our results showed that NLR–inflammasome–caspase-1 pathway is present in this frog, which is not clear in other frogs including of Xenopus. Besides that active IIS, this frog can live well in harsh environment owing to its nearly parallel AIS to the mammal. In addition, tissue-specific genes and ncRNAs analysis initially reveal the genetic adaption of this frog.
Authors' Contributions
Y.Z., F.Z. and W.L. conceived the study. Manuscript wrote by F.Z. Sequence assemble, bioinformatics analysis and expression validation were performed by F.Z. Samples and RNA extraction were carried out by Y.Y. and G.W. Figure 5 was performed by C.Y., X.W. and Y.X. Y.X. gives great help to manuscript revision. All authors read and approved the final version of manuscript.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
This work was supported by grants from the National Basic Research Program of China (973 Program, 2010CB529800), the National Natural Science Foundation of China (NSFC-Yunnan joint funding U1132601, 31071926 and 31270835) and the Key Research Program of the Chinese Academy of Sciences (KJZD-EW-L03).
Supplementary Material
Footnotes
Edited by Dr Shoji Tsuji
References
- 1.Pollet N., Mazabraud A. Insights from Xenopus genomes. Genome Dyn. 2006;2:138–53. doi: 10.1159/000095101. [DOI] [PubMed] [Google Scholar]
- 2.Yergeau D.A., Mead P.E. Manipulating the Xenopus genome with transposable elements. Genome Biol. 2007;8(Suppl 1):S11. doi: 10.1186/gb-2007-8-s1-s11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morin R.D., Chang E., Petrescu A., et al. Sequencing and analysis of 10,967 full-length cDNA clones from Xenopus laevis and Xenopus tropicalis reveals post-tetraploidization transcriptome remodeling. Genome Res. 2006;16:796–803. doi: 10.1101/gr.4871006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hellsten U., Harland R.M., Gilchrist M.J., et al. The genome of the Western clawed frog Xenopus tropicalis. Science. 2010;328:633–36. doi: 10.1126/science.1183670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rockman M.V., Kruglyak L. Genetics of global gene expression. Nat. Rev. Genet. 2006;7:862–72. doi: 10.1038/nrg1964. [DOI] [PubMed] [Google Scholar]
- 6.Zhang J., Zhang Y., Wan S.G., Wei S.S., Lee W.H. Bm-TFF2, a trefoil factor protein with platelet activation activity from frog Bombina maxima skin secretions. Biochem. Biophys. Res. Commun. 2005;330:1027–33. doi: 10.1016/j.bbrc.2005.03.077. [DOI] [PubMed] [Google Scholar]
- 7.Lee W.H., Li Y., Lai R., Li S., Zhang Y., Wang W. Variety of antimicrobial peptides in the Bombina maxima toad and evidence of their rapid diversification. Eur. J. Immunol. 2005;35:1220–9. doi: 10.1002/eji.200425615. [DOI] [PubMed] [Google Scholar]
- 8.Xiang Y., Gao Q., Su W., et al. Establishment, characterization and immortalization of a fibroblast cell line from the Chinese red belly toad Bombina maxima skin. Cytotechnology. 2012;64:95–105. doi: 10.1007/s10616-011-9399-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu S.B., He Y.Y., Zhang Y., et al. A novel non-lens betagamma-crystallin and trefoil factor complex from amphibian skin and its functional implications. PLoS One. 2008;3:e1770. doi: 10.1371/journal.pone.0001770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu Y., Tao X., Shen B., et al. Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature. 2000;408:111–5. doi: 10.1038/35040600. [DOI] [PubMed] [Google Scholar]
- 11.Robert J., Cohen N. The genus Xenopus as a multispecies model for evolutionary and comparative immunobiology of the 21st century. Dev. Comp. Immunol. 2011;35:916–23. doi: 10.1016/j.dci.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wiesner J., Vilcinskas A. Antimicrobial peptides: the ancient arm of the human immune system. Virulence. 2010;1:440–64. doi: 10.4161/viru.1.5.12983. [DOI] [PubMed] [Google Scholar]
- 13.Yang X., Lee W.H., Zhang Y. Extremely abundant antimicrobial peptides existed in the skins of nine kinds of Chinese odorous frogs. J. Proteome Res. 2012;11:306–19. doi: 10.1021/pr200782u. [DOI] [PubMed] [Google Scholar]
- 14.Du Pasquier L., Schwager J., Flajnik M.F. The immune system of Xenopus. Annu. Rev. Immunol. 1989;7:251–75. doi: 10.1146/annurev.iy.07.040189.001343. [DOI] [PubMed] [Google Scholar]
- 15.Blackburn D.C., Bickford D.P., Diesmos A.C., Iskandar D.T., Brown R.M. An ancient origin for the enigmatic flat-headed frogs (Bombinatoridae: Barbourula) from the islands of Southeast Asia. PloS One. 2010;5:e12090. doi: 10.1371/journal.pone.0012090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shendure J., Ji H. Next-generation DNA sequencing. Nat. Biotechnol. 2008;26:1135–45. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 17.Tan M.H., Au K.F., Yablonovitch A.L., et al. RNA sequencing reveals a diverse and dynamic repertoire of the Xenopus tropicalis transcriptome over development. Genome Res. 2013;23:201–16. doi: 10.1101/gr.141424.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coppe A., Pujolar J.M., Maes G.E., et al. Sequencing, de novo annotation and analysis of the first Anguilla anguilla transcriptome: EeelBase opens new perspectives for the study of the critically endangered European eel. BMC Genomics. 2010;11:635. doi: 10.1186/1471-2164-11-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma K., Qiu G., Feng J., Li J. Transcriptome analysis of the oriental river prawn, Macrobrachium nipponense using 454 pyrosequencing for discovery of genes and markers. PloS One. 2012;7:e39727. doi: 10.1371/journal.pone.0039727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fu B., He S. Transcriptome analysis of silver carp (Hypophthalmichthys molitrix) by paired-end RNA sequencing. DNA Res. 2012;19:131–42. doi: 10.1093/dnares/dsr046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nie Q., Fang M., Jia X., et al. Analysis of muscle and ovary transcriptome of Sus scrofa: assembly, annotation and marker discovery. DNA Res. 2011;18:343–51. doi: 10.1093/dnares/dsr021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Procaccini C., Galgani M., De Rosa V., Matarese G. Intracellular metabolic pathways control immune tolerance. Trends Immunol. 2012;33:1–7. doi: 10.1016/j.it.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 24.Font J., Mackay J.P. Beyond DNA: zinc finger domains as RNA-binding modules. Methods Mol. Biol. 2010;649:479–91. doi: 10.1007/978-1-60761-753-2_29. [DOI] [PubMed] [Google Scholar]
- 25.Jung H., Lyons R.E., Dinh H., Hurwood D.A., McWilliam S., Mather P.B. Transcriptomics of a giant freshwater prawn (Macrobrachium rosenbergii): de novo assembly, annotation and marker discovery. PLoS One. 2011;6:e27938. doi: 10.1371/journal.pone.0027938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ravasi T., Huber T., Zavolan M., et al. Systematic characterization of the zinc-finger-containing proteins in the mouse transcriptome. Genome Res. 2003;13:1430–42. doi: 10.1101/gr.949803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohta Y., Flajnik M. IgD, like IgM, is a primordial immunoglobulin class perpetuated in most jawed vertebrates. Proc. Natl Acad. Sci. USA. 2006;103:10723–28. doi: 10.1073/pnas.0601407103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iliev D.B., Roach J.C., Mackenzie S., Planas J.V., Goetz F.W. Endotoxin recognition: in fish or not in fish? FEBS Lett. 2005;579:6519–28. doi: 10.1016/j.febslet.2005.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nikolaeva S., Bachteeva V., Fock E., et al. Frog urinary bladder epithelial cells express TLR4 and respond to bacterial LPS by increase of iNOS expression and l-arginine uptake. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012;303:R1042–52. doi: 10.1152/ajpregu.00045.2012. [DOI] [PubMed] [Google Scholar]
- 30.Chow J.C., Young D.W., Golenbock D.T., Christ W.J., Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J. Biol. Chem. 1999;274:10689–92. doi: 10.1074/jbc.274.16.10689. [DOI] [PubMed] [Google Scholar]
- 31.Fitzgerald K.A., Palsson-McDermott E.M., Bowie A.G., et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature. 2001;413:78–83. doi: 10.1038/35092578. [DOI] [PubMed] [Google Scholar]
- 32.Prothmann C., Armstrong N.J., Rupp R.A. The Toll/IL-1 receptor binding protein MyD88 is required for Xenopus axis formation. Mech. Develop. 2000;97:85–92. doi: 10.1016/s0925-4773(00)00447-0. [DOI] [PubMed] [Google Scholar]
- 33.Li D., Roberts R. WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cell. Mol. Life Sci. 2001;58:2085–97. doi: 10.1007/PL00000838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oshiumi H., Suzuki Y., Matsumoto M., Seya T. Regulator of complement activation (RCA) gene cluster in Xenopus tropicalis. Immunogenetics. 2009;61:371–84. doi: 10.1007/s00251-009-0368-9. [DOI] [PubMed] [Google Scholar]
- 35.Del Rio-Tsonis K., Tsonis P.A., Zarkadis I.K., Tsagas A.G., Lambris J.D. Expression of the third component of complement, C3, in regenerating limb blastema cells of urodeles. J. Immunol. 1998;161:6819–24. [PubMed] [Google Scholar]
- 36.Huising M.O., Stet R.J., Savelkoul H.F., Verburg-van Kemenade B.M. The molecular evolution of the interleukin-1 family of cytokines; IL-18 in teleost fish. Dev. Comp. Immunol. 2004;28:395–413. doi: 10.1016/j.dci.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 37.Wargnier A., Lagrange P.H. [Bactericidal activity of cells of the immune system] Pathol. Biol. 1993;41:887–96. [PubMed] [Google Scholar]
- 38.Calandra T., Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat. Rev. Immunol. 2003;3:791–800. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taylor G.A., Feng C.G., Sher A. p47 GTPases: regulators of immunity to intracellular pathogens. Nat. Rev. Immunol. 2004;4:100–9. doi: 10.1038/nri1270. [DOI] [PubMed] [Google Scholar]
- 40.Klamp T., Boehm U., Schenk D., Pfeffer K., Howard J.C. A giant GTPase, very large inducible GTPase-1, is inducible by IFNs. J. Immunol. 2003;171:1255–65. doi: 10.4049/jimmunol.171.3.1255. [DOI] [PubMed] [Google Scholar]
- 41.Martinon F., Mayor A., Tschopp J. The inflammasomes: guardians of the body. Annu. Rev. Immunol. 2009;27:229–65. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 42.Schluter S.F., Bernstein R.M., Bernstein H., Marchalonis J.J. ‘Big Bang’ emergence of the combinatorial immune system. Dev. Comp. Immunol. 1999;23:107–11. doi: 10.1016/s0145-305x(99)00002-6. [DOI] [PubMed] [Google Scholar]
- 43.Flajnik M.F., Kasahara M. Origin and evolution of the adaptive immune system: genetic events and selective pressures. Nat. Rev. Genet. 2010;11:47–59. doi: 10.1038/nrg2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flajnik M.F. Comparative analyses of immunoglobulin genes: surprises and portents. Nat. Rev. Immunol. 2002;2:688–98. doi: 10.1038/nri889. [DOI] [PubMed] [Google Scholar]
- 45.Warr G.W., Magor K.E., Higgins D.A. IgY: clues to the origins of modern antibodies. Immunol. Today. 1995;16:392–8. doi: 10.1016/0167-5699(95)80008-5. [DOI] [PubMed] [Google Scholar]
- 46.Kasahara M. Genome dynamics of the major histocompatibility complex: insights from genome paralogy. Immunogenetics. 1999;50:134–45. doi: 10.1007/s002510050589. [DOI] [PubMed] [Google Scholar]
- 47.Kiemnec-Tyburczy K.M., Richmond J.Q., Savage A.E., Lips K.R., Zamudio K.R. Genetic diversity of MHC class I loci in six non-model frogs is shaped by positive selection and gene duplication. Heredity (Edinb) 2012;109:146–55. doi: 10.1038/hdy.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McConnell T.J., Godwin U.B., Cuthbertson B.J. Expressed major histocompatibility complex class II loci in fishes. Immunol Rev. 1998;166:294–300. doi: 10.1111/j.1600-065x.1998.tb01270.x. [DOI] [PubMed] [Google Scholar]
- 49.Flajnik M.F., Canel C., Kramer J., Kasahara M. Evolution of the major histocompatibility complex: molecular cloning of major histocompatibility complex class I from the amphibian Xenopus. Proc. Natl Acad. Sci. USA. 1991;88:537–41. doi: 10.1073/pnas.88.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Panopoulou G., Poustka A.J. Timing and mechanism of ancient vertebrate genome duplications—the adventure of a hypothesis. Trends Genet. 2005;21:559–67. doi: 10.1016/j.tig.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 51.Flajnik M.F., Kasahara M., Shum B.P., Salter-Cid L., Taylor E., Du Pasquier L. A novel type of class I gene organization in vertebrates: a large family of non-MHC-linked class I genes is expressed at the RNA level in the amphibian Xenopus. EMBO J. 1993;12:4385–96. doi: 10.1002/j.1460-2075.1993.tb06123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.