

Abstract
Although much research has been conducted, the pattern of microsatellite distribution has remained ambiguous, and the development/utilization of microsatellite markers has still been limited/inefficient in Brassica, due to the lack of genome sequences. In view of this, we conducted genome-wide microsatellite characterization and marker development in three recently sequenced Brassica crops: Brassica rapa, Brassica oleracea and Brassica napus. The analysed microsatellite characteristics of these Brassica species were highly similar or almost identical, which suggests that the pattern of microsatellite distribution is likely conservative in Brassica. The genomic distribution of microsatellites was highly non-uniform and positively or negatively correlated with genes or transposable elements, respectively. Of the total of 115 869, 185 662 and 356 522 simple sequence repeat (SSR) markers developed with high frequencies (408.2, 343.8 and 356.2 per Mb or one every 2.45, 2.91 and 2.81 kb, respectively), most represented new SSR markers, the majority had determined physical positions, and a large number were genic or putative single-locus SSR markers. We also constructed a comprehensive database for the newly developed SSR markers, which was integrated with public Brassica SSR markers and annotated genome components. The genome-wide SSR markers developed in this study provide a useful tool to extend the annotated genome resources of sequenced Brassica species to genetic study/breeding in different Brassica species.
Keywords: brassica, microsatellite, distribution, marker, database
1. Introduction
Microsatellites, which are also known as simple sequence repeats (SSRs, often defined as 1–6 bp), variable numbers of tandem repeats (VNTRs) and short tandem repeats (STRs), have been found in all genomic regions of all examined organisms.1 Microsatellites have been traditionally regarded as ‘junk’ DNA and are mainly used as ‘neutral’ genetic markers.2 In recent years, microsatellites have been demonstrated to have many important biological functions (e.g. the regulation of chromatin organization, DNA metabolic processes, gene activity and RNA structure)3,4 and have therefore emerged as the third major class of genetic variations, alongside single nucleotide polymorphisms (SNPs) and copy number variations (CNVs).5 Microsatellite markers are co-dominant, multi-allelic, easily detected, hyper-variable, highly reproducible and abundant in the genome.6 Therefore, among the available genetic marker systems (e.g. RFLP, RAPD, SSR, AFLP, SRAP and SNP), the SSR marker has been the preferential choice for various applications, such as variety identification, genetic diversity evaluation, phylogenetic relationship analysis, genetic map construction, linkage/association mapping of gene/QTL, marker-assisted selection and comparative mapping.7,8
Of the 47 genera in the Brassiceae tribe within the Brassicaceae (Cruciferae) family, the genus Brassica currently comprises 38 species,9 which include economically important crops that provide many vegetables, condiments, fodders and oil products.10 The main cultivated Brassica species include three diploid species, Brassica rapa (AA, n = 10), Brassica nigra (BB, n = 8) and Brassica oleracea (CC, n = 9) and three allotetraploid species, Brassica juncea (AABB, n = 18), Brassica napus (AACC, n = 19) and Brassica carinata (BBCC, n = 17). The genetic relationship of the six widely cultivated Brassica species are described as U's triangle11 in which the three allotetraploid species originated from the chromosome doubling after the natural hybridization between the three diploid species.
Much research has been conducted to identify/characterize genomic/genic microsatellites and/or to develop markers in the Brassica species through probe (containing a repeated motif) hybridization against genomic/cDNA clones12–19 or through in silico analysis of publicly available bacterial artificial chromosome (BAC) sequences,20 BAC-end sequences (BESs),21–23 genome survey sequences (GSSs),24 whole genome shotgun sequences (WGSs),25,26 expressed sequence tag sequences27,28 and unique transcript sequences.29–32 However, the pattern of microsatellite distribution has remained ambiguous, and the development/utilization of SSR marker has still been limited/inefficient in Brassica, which is mostly due to the lack of genome sequences. First, the sequences, programmes, criteria and parameters that are used for mining microsatellites usually have differed across these previous studies, which have made it difficult to compare and integrate these results to obtain the definitive conclusions on the pattern of microsatellite distribution. Secondly, only a small part of the genomic sequences of usually one species have been analysed in each of these previous studies. Therefore, it has been impossible to obtain general conclusions on the pattern of microsatellite distribution. In addition, the total number (≈10 000) of previously developed publicly available SSR markers is still limited33 and not sufficient for many studies, which require a large number and/or high density of genetic markers, such as high-density linkage map construction, gene/QTL fine-mapping and genome-wide/regional association mapping. Thirdly, due to the lack of genome sequences, the genomic distribution of microsatellites and the physical position(s)/product(s) number of the previously developed publicly available Brassica SSR markers have been all or mostly unclear, which has hindered their exact and/or effective utilization.
Thanks to the rapid development of genome sequencing technology, the genome sequences are currently available for tens of plant species (http://www.phytozome.net), including three recently sequenced Brassica crop species, namely B. rapa,34 B. oleracea (http://www.ocri-genomics.org/bolbase/index.html) and B. napus (our unpublished data). These sequences provide a powerful tool for genome-wide microsatellite characterization and/or marker development, which has been conducted in several model and crop plants, such as Arabidopsis (http://www.arabidopsis.org/), rice,35 maize (mips.helmholtz-muenchen.de/plant/maize/), sorghum (genome.jgi-psf.org/Sorbi1/Sorbi1.home.htm), black cottonwood,36 cucumber,37 Brachypodim distachyon38 and foxtail millet39 but not Brassica. In view of this circumstance, we conducted genome-wide microsatellite characterization and marker development in the three sequenced Brassica crop species. The main objectives of this study were as follows: (i) to characterize and compare the frequency and distribution with respect to the motif length, type and repeat number of microsatellites in the assembled genomic sequences of these Brassica species; (ii) to characterize and compare the genomic distribution of microsatellites in the assembled pseudochromosomes of these Brassica species; (iii) to develop SSR markers from the assembled genomic sequences of these Brassica species and determine their copy number and positional relationship with the previously developed publicly available Brassica SSR markers and the annotated genome components; (iv) to construct a user-friendly comprehensive SSR marker database of Brassica and (v) to evaluate the newly developed genome-wide SSR markers by PCR (polymerase chain reaction) amplification in representative B. napus inbred lines.
2. Materials and methods
2.1. Sources of genome sequences
The three inbred/pure lines, namely Chiifu-401 (B. rapa), O212 (B. oleracea) and Zhongshuang11 (B. napus), were sequenced by our own and several other institutes using Illumina GA II technology, and high-quality sequence reads were assembled using stringent parameters. Finally, a total of 40 549 (283.8 Mb), 120 061 (540.0 Mb) and 5098 (1000.9 Mb) sequence scaffolds were obtained for B. rapa, B. oleracea and B. napus, respectively, which represents 58.5, 77.6 and 81.7% of the nuclear genome and covers >98% of the gene space.
2.2. Identification of microsatellites
PERL5 script MIcroSAtellite (http://pgrc.ipk-gatersleben.de/misa/)40 was used to identify and localize perfect microsatellites as well as compound microsatellites that are interrupted by a certain number of bases. The repeat unit length was defined as the default mono- to hexanucleotide because microsatellites of longer repeat units are very scarce. The minimum repeat unit was defined as 12, 6, 4, 3, 3 and 3, respectively, for the mono- to hexanucleotide. Compound microsatellites were defined as ≥2 repeats interrupted by ≤100 bp.
2.3. Development of SSR primers
Primer pairs were designed from the flanking sequences of identified microsatellites using the primer3_core program (http://www-genome.wi.mit.edu/cgi-bin/primer/primer3_ www.cgi) in batch mode. Two perl scripts, p3_in.pl and p3_out.pl, serve as interface modules for the programme-to-programme data interchange between MISA and the primer modeling software Primer3. The primer-designing parameters were 18–27 bp primer length, 57–63°C melting temperature, 30–70% GC content and 100–300 bp product size. The designed SSR primer pairs were denominated as the names of sequence scaffolds followed by a serial number of microsatellites (such as BrScaffold000001_1).
2.4. Localization/mapping of SSR markers by in silico PCR
The primer-pair sequences of previously developed publicly available Brassica SSR markers were downloaded from the brassica.info website (http://www.brassica.info/resource/markers/ssr-exchange.php) and additional files in the recent literature.20,24,26,29,30,32 To determine their physical positions and copy numbers, the previously and newly developed Brassica SSR markers were aligned to the assembled genomic sequences of the studied Brassica species. This alignment was conducted using the in silico PCR method41 with the following default parameters: 2 bp mismatch, 1 bp gap, 50 bp margin and 50–1000 bp product size.
2.5. Validation of SSR markers by PCR amplification
A total of 3974 SSR primer pairs were synthesized to test for PCR amplification in six representative B. napus cultivars/inbred lines (Tapidor, Westar, Zhongshuang11, No. 07197, No. 73290 and No. 91032), which were chosen from the core collections of a natural population and the parents of several segregating populations in our laboratory, for their large genetic distance and extreme trait(s) performance (our unpublished data).
Genomic DNA of the six accessions was isolated from young leaves. PCR was performed in 20-µl volume that contained 0.2 mM dNTP, 0.5 U of Taq DNA polymerase, 75 ng of template DNA, 0.5 µM each primer and 1× PCR buffer (10 mM Tris pH 9.0, 50 mM KCl and 1.5 mM MgCl2). DNA amplification was conducted by the ‘touchdown’ method, with the following thermal profile: initial denaturation at 94°C for 5 min; six cycles of 30 s at 94°C, 45 s at 63°C with a 1°C decrease in annealing temperature per cycle and 1 min at 72°C; 26 cycles of 30 s at 94°C, 45 s at 57°C and 1 min at 72°C and a final extension at 72°C for 10 min. The PCR products were separated on 6% denaturing polyacrylamide gels and were visualized by silver staining.
2.6. Statistical analysis
The correlation analysis was performed using the SAS PROC CORR procedure incorporated into SAS version 8.0. The Excel statistical function CHISQ.TEST was used to obtain the significance level (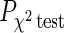 ) of the degree of fit for the practical and hypothetical distributions of microsatellites as well as genes and TEs in the assembled pseudochromosomes.
) of the degree of fit for the practical and hypothetical distributions of microsatellites as well as genes and TEs in the assembled pseudochromosomes.
3. Results
3.1. Frequency and distribution with respect to the motif length, type and number
A total of 140 998, 229 389 and 420 991 perfect mono- to hexanucleotide repeat microsatellites were identified from 283.8, 540.0 and 1000.9 Mb of assembled genomic sequences of B. rapa, B. oleracea and B. napus, respectively (Table 1), with an overall frequency of 496.8, 424.8 and 420.6 per Mb or one every 2.01, 2.35 and 2.38 kb.
Table 1.
Number, repeat number and total repeat length of the mono- to hexanucleotide repeats or motifs of microsatellites in the assembled genomic sequences of B. rapa, B. oleracea and B. napus
| Motif |
B. rapa |
B. oleracea |
B. napus |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Number (%) | Repeat number | Total length (%) | Number (%) | Repeat number | Total length (%) | Number (%) | Repeat number | Total length (%) | |
| Mono | 31 258 (22.2) | 12–307 (14.7) | 458 968 (20.1) | 55 433 (24.2) | 3–65 (15.1) | 838 104 (24.1) | 97 128 (23.1) | 12–2545 (15.2) | 147 5939 (22.9) |
| A | 29 536 (20.9) | 12–50 (14.5) | 428 733 (18.7) | 52 021 (22.7) | 3–65 (15.0) | 780 171 (22.5) | 94 281 (22.4) | 12–2545 (15.2) | 1 432 867 (22.2) |
| C | 1722 (1.2) | 12–307 (17.6) | 30 235 (1.3) | 3412 (1.5) | 3–63 (17.0) | 57 933 (1.7) | 2847 (0.7) | 12–83 (15.1) | 43 072 (0.7) |
| Di | 33 885 (24.0) | 6–3644 (11.1) | 751 910 (32.9) | 55 336 (24.1) | 6–82 (8.8) | 968 946 (27.9) | 98 816 (23.5) | 6–5556 (9.1) | 1 789 752 (27.8) |
| AT | 19 697 (14.0) | 6–419 (8.9) | 350 590 (15.3) | 33 315 (14.5) | 6–63 (8.9) | 596 070 (17.2) | 57 070 (13.6) | 6–5556 (8.7) | 996 518 (15.5) |
| AG | 11 683 (8.3) | 6–3644 (15.6) | 364 004 (15.9) | 18 593 (8.1) | 6–44 (8.7) | 322 438 (9.3) | 34 638 (8.2) | 6–1732 (9.9) | 688 124 (10.7) |
| AC | 2490 (1.8) | 6–131 (7.5) | 37 124 (1.6) | 3411 (1.5) | 6–82 (7.4) | 50 220 (1.4) | 7072 (1.7) | 6–92 (7.4) | 104 648 (1.6) |
| CG | 15 (0.0) | 6–8 (6.4) | 192 (0.0) | 17 (0.0) | 6–9 (6.4) | 218 (0.0) | 36 (0.0) | 6–8 (6.4) | 462 (0.0) |
| Tri | 32 387 (23.0) | 4–812 (4.7) | 459 039 (20.1) | 47 716 (20.8) | 4–1710 (4.7) | 670 164 (19.3) | 91 448 (21.7) | 4–1794 (4.7) | 1 277 172 (19.8) |
| AAG | 9796 (6.9) | 4–812 (4.7) | 139 239 (6.1) | 15 322 (6.7) | 4–81 (4.6) | 212 973 (6.1) | 29 395 (7.0) | 4–162 (4.6) | 408 264 (6.3) |
| AAT | 6334 (4.5) | 4–573 (4.9) | 93 240 (4.1) | 9355 (4.1) | 4–1710 (5.2) | 145 056 (4.2) | 17 722 (4.2) | 4–1794 (5.1) | 268 956 (4.2) |
| ATC | 4211 (3.0) | 4–190 (5.0) | 63 213 (2.8) | 6093 (2.7) | 4–81 (4.7) | 85 047 (2.4) | 12 314 (2.9) | 4–119 (4.6) | 170 151 (2.6) |
| AAC | 3637 (2.6) | 4–11 (4.5) | 49 146 (2.1) | 5036 (2.2) | 4–41 (4.4) | 66 705 (1.9) | 10 046 (2.4) | 4–22 (4.5) | 134 592 (2.1) |
| AGG | 3243 (2.3) | 4–179 (4.6) | 45 084 (2.0) | 5425 (2.4) | 4–13 (4.6) | 74 763 (2.2) | 8349 (2.0) | 4–14 (4.6) | 115 494 (1.8) |
| ACC | 2144 (1.5) | 4–9 (4.5) | 28 908 (1.3) | 2788 (1.2) | 4–43 (4.4) | 37 155 (1.1) | 5969 (1.4) | 4–14 (4.4) | 79 137 (1.2) |
| AGC | 1127 (0.8) | 4–11 (4.4) | 15 039 (0.7) | 1390 (0.6) | 4–22 (4.4) | 18 507 (0.5) | 2878 (0.7) | 4–18 (4.4) | 38 379 (0.6) |
| ACT | 674 (0.5) | 4–10 (4.4) | 8874 (0.4) | 818 (0.4) | 4–28 (4.5) | 10 968 (0.3) | 1754 (0.4) | 4–17 (4.4) | 23 265 (0.4) |
| CCG | 626 (0.4) | 4–8 (4.4) | 8220 (0.4) | 799 (0.3) | 4–7 (4.3) | 10 203 (0.3) | 1566 (0.4) | 4–8 (4.3) | 20 139 (0.3) |
| ACG | 595 (0.4) | 4–9 (4.5) | 8076 (0.4) | 690 (0.3) | 4–13 (4.2) | 8787 (0.3) | 1455 (0.3) | 4–10 (4.3) | 18 795 (0.3) |
| Tetra | 29 433 (20.9) | 3–264 (3.2) | 376 668 (16.5) | 48 394 (21.1) | 3–54 (3.1) | 608 452 (17.5) | 91 268 (21.7) | 3–631 (3.1) | 1 148 496 (17.8) |
| AAAT | 11 870 (8.4) | 3–8 (3.2) | 152 776 (6.7) | 18 856 (8.2) | 3–28 (3.1) | 236 560 (6.8) | 33 903 (8.1) | 3–10 (3.1) | 425 080 (6.6) |
| AAAG | 3496 (2.5) | 3–60 (3.2) | 44 608 (1.9) | 5697 (2.5) | 3–36 (3.2) | 72 224 (2.1) | 10 795 (2.6) | 3–13 (3.2) | 136 204 (2.1) |
| AAAC | 3333 (2.4) | 3–8 (3.1) | 41 960 (1.8) | 4723 (2.1) | 3–26 (3.1) | 59 076 (1.7) | 9717 (2.3) | 3–8 (3.1) | 121 768 (1.9) |
| AATT | 2534 (1.8) | 3–6 (3.1) | 31 108 (1.4) | 4088 (1.8) | 3–10 (3.1) | 51 416 (1.5) | 7863 (1.9) | 3–41 (3.1) | 98 224 (1.5) |
| AATC | 1137 (0.8) | 3–8 (3.2) | 14 368 (0.6) | 2956 (1.3) | 3–27 (3.1) | 36 908 (1.1) | 5112 (1.2) | 3–8 (3.1) | 63 884 (1.0) |
| others | 7063 (5.0) | 3–264 (3.3) | 91 848 (4.0) | 12 074 (5.3) | 3–54 (3.2) | 152 268 (4.4) | 23 878 (5.7) | 3–631 (3.2) | 303 336 (4.7) |
| Penta | 9856 (7.0) | 3–114 (3.2) | 156 510 (6.8) | 15 012 (6.5) | 3–40 (3.2) | 241 090 (6.9) | 29 058 (6.9) | 3–28 (3.1) | 457 065 (7.1) |
| AAAAT | 2758 (2.0) | 3–63 (3.1) | 42 920 (1.9) | 4051 (1.8) | 3–18 (3.4) | 67 905 (2.0) | 7617 (1.8) | 3–9 (3.1) | 118 030 (1.8) |
| AACCG | 1000 (0.7) | 3–68 (3.3) | 16 510 (0.7) | 2169 (0.9) | 3–40 (3.2) | 34 645 (1.0) | 3541 (0.8) | 3–7 (3.2) | 56 435 (0.9) |
| AAAAC | 878 (0.6) | 3–7 (3.2) | 13 905 (0.6) | 1186 (0.5) | 3–8 (3.2) | 18 715 (0.5) | 2424 (0.6) | 3–10 (3.2) | 38 280 (0.6) |
| AAAAG | 683 (0.5) | 3–7 (3.1) | 10 675 (0.5) | 1164 (0.5) | 3–14 (3.1) | 18 260 (0.5) | 2351 (0.6) | 3–7 (3.1) | 36 820 (0.6) |
| AAATT | 568 (0.4) | 3–6 (3.1) | 8790 (0.4) | 784 (0.3) | 3–5 (3.1) | 12 030 (0.3) | 1488 (0.4) | 3–7 (3.1) | 23 065 (0.4) |
| AAACC | 490 (0.3) | 3–7 (3.2) | 7850 (0.3) | 713 (0.3) | 3–6 (3.2) | 11 325 (0.3) | 1273 (0.3) | 3–9 (3.2) | 20 290 (0.3) |
| AATAT | 394 (0.3) | 3–6 (3.1) | 6085 (0.3) | 573 (0.2) | 3–6 (3.1) | 8890 (0.3) | 1083 (0.3) | 3–13 (3.1) | 16 905 (0.3) |
| Others | 3085 (2.2) | 3–114 (3.2) | 49 775 (2.2) | 4372 (1.9) | 3–16 (3.2) | 69 320 (2.0) | 9281 (2.2) | 3–28 (3.2) | 147 240 (2.3) |
| Hexa | 4179 (3.0) | 3–102 (3.4) | 85 176 (3.7) | 7498 (3.3) | 3–33 (3.3) | 146 370 (4.2) | 13 273 (3.2) | 3–1579 (3.7) | 295 236 (4.6) |
| AAAAAT | 628 (0.4) | 3–52 (3.2) | 12 000 (0.5) | 987 (0.4) | 3–22 (3.1) | 18 618 (0.5) | 1803 (0.4) | 3–35 (3.1) | 33 972 (0.5) |
| AAAATT | 267 (0.2) | 3–6 (3.0) | 4866 (0.2) | 776 (0.3) | 3–25 (3.1) | 14 376 (0.4) | 1341 (0.3) | 3–154 (3.2) | 25 614 (0.4) |
| AAAAAC | 269 (0.2) | 3–23 (3.3) | 5298 (0.2) | 395 (0.2) | 3–7 (3.2) | 7596 (0.2) | 746 (0.2) | 3–22 (3.3) | 14 562 (0.2) |
| AAAAAG | 175 (0.1) | 3–6 (3.2) | 3330 (0.1) | 357 (0.2) | 3–14 (3.3) | 6966 (0.2) | 644 (0.2) | 3–37 (3.3) | 12 612 (0.2) |
| AAATAT | 150 (0.1) | 3–52 (3.8) | 3390 (0.1) | 246 (0.1) | 3–16 (3.2) | 4704 (0.1) | 436 (0.1) | 3–29 (3.3) | 8562 (0.1) |
| Others | 2690 (1.9) | 3–102 (3.5) | 56 292 (2.5) | 4737 (2.1) | 3–33 (3.3) | 94 110 (2.7) | 8303 (2.0) | 3–1579 (4.0) | 199 914 (3.1) |
| Total | 140 998 (100) | 3–3644 (8.0) | 228 8271 (100) | 229 389 (100) | 3–1710 (7.7) | 347 3126 (100) | 420 991 (100) | 3–5556 (7.7) | 6 443 660 (100) |
In accordance with their high correlation (Supplementary Table S1), the distributions with respect to the motif length of microsatellites in the assembled genomic sequences of B. rapa, B. oleracea and B. napus were almost identical: mono-, di-, tri- and tetranucleotide repeats accounted for very similar and relatively high proportions, whereas penta- and hexanucleotide repeats were relatively uncommon (Fig. 1A).
Figure 1.

Distribution with respect to the motif length (A), type (B) and repeat number (C) of microsatellites in the assembled genomic sequences of B. rapa, B. oleracea and B. napus. The vertical axis shows the abundances (%) of microsatellites with different motif lengths, types or repeat numbers that are discriminated by the legends of different colours. For Figure 1B, because there is a limitation in the number of items (<256) in Excel, the abundances of the mono- to pentanucleotide motifs (a total of 151 types) are shown, while those for the hexanucleotide motifs (a total of 350 types) are displayed in Supplementary Table S2.
In accordance with their high correlation (Supplementary Table S1), the distributions with respect to the motif type of microsatellites in the assembled genomic sequences of B. rapa, B. oleracea and B. napus were almost identical (Fig. 1B; Supplementary Table S2). More specifically, both the dominant/major and absent/scarce mono- to hexanucleotide motifs in the assembled genomic sequences of the three Brassica species were mostly identical (Table 1; Supplementary Table S3). Interestingly, the dominant/major motifs (A, AT, AAG/AAT, AAAT, AAAAT and AAAAAT) were all A/T rich (Table 1), whereas the absent/scarce motifs were mostly C/G rich (Supplementary Table S3), which were highly consistent with the previous reports on microsatellites identified from 536 seed BACs of B. rapa,20 3500 genomic clones42 and 595 577 WGSs26 of B. oleracea and 13 794 GSSs (mainly BESs) of B. napus.24 It should be noted that the nucleotide composition characteristics of both the dominant/major and absent/scarce motifs in the assembled genomic sequences of the three Brassica species corresponded well to their much higher A/T (mean = 63.8%) than C/G (mean = 36.2%) content.
In accordance with their high correlation (Supplementary Table S1), the distributions with respect to the motif repeat number of microsatellites in the assembled genomic sequences of B. rapa, B. oleracea and B. napus were also almost identical (Fig. 1C). Obviously, the microsatellite abundances decreased significantly as the motif repeat number increased, and the rate of this change was the slowest for dinucleotide repeat, followed by mono- and trinucleotide repeats, and was faster for other long repeats (Fig. 2). As a consequence, the difference between the average and minimum motif repeat numbers was the largest for dinucleotide repeat, followed by mono- and trinucleotide repeats, and was relatively small for tetra- to hexanucleotide repeats (Table 1).
Figure 2.

Distribution with respect to the motif repeat number of the individual mono- to hexanucleotide repeat microsatellites in the assembled genomic sequences of B. rapa, B. oleracea and B. napus. The vertical axis shows the abundances of microsatellites that have different motif repeat numbers (from 3 to >20), which are discriminated by legends of different colours.
In addition, the motif repeat number of the corresponding mono- to hexanucleotide repeats or motifs of microsatellites in the assembled genomic sequences of B. rapa, B. oleracea and B. napus were highly similar (Table 1; Supplementary Table S4). As a consequence, the total repeat length (=microsatellite number × motif length × motif repeat number) proportions of the corresponding mono- to hexanucleotide repeats or motifs of microsatellites in the assembled genomic sequences of B. rapa, B. oleracea and B. napus were mostly similar (Table 1; Supplementary Table S5).
3.2. Genomic distribution
The genomic distributions of microsatellites and their relation with the annotated genome components (mainly as genes and TEs) were investigated (Fig. 3; Table 2), based on the assembled pseudochromosomes of the sequenced Brassica species (currently available for B. rapa and B. oleracea; Supplementary Table S6).
Figure 3.

Genome-wide distributions of microsatellites as well as genes and TEs in the assembled pseudochromosomes of B. rapa (A) and B. oleracea (B). The horizontal axis shows the pseudochromosomes (B. rapa: A1–A10; B. oleracea: C1–C9), which are divided into 1-Mb intervals. The left and right vertical axes show the frequencies of the microsatellites/genes and TEs, respectively. On the figure, the curves/lines of different styles represent the practical/hypothetical(average) frequencies of microsatellites, genes and TEs, respectively.
Table 2.
χ/italic>2 test between the practical and hypothetical/average distribution of microsatellites and their correlation with genes and TEs, for all pseudochromosomes of B. rapa and B. oleracea
| Species | Chromosome | Microsatellites |
Genes |
TEs |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Frequency | 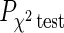 |
rgene | rTE | Frequency | 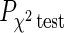 |
Frequency | 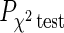 |
||
| A01 | 488 | 1.9E−42 | 0.76 | −0.69 | 146 | 8.7E−60 | 841 | 0.0E+00 | |
| A02 | 522 | 1.6E−16 | 0.78 | −0.65 | 150 | 8.3E−41 | 869 | 4.0E−220 | |
| A03 | 539 | 1.2E−13 | 0.46 | −0.29 | 179 | 8.9E−15 | 725 | 9.9E−163 | |
| A04 | 515 | 6.5E−14 | 0.53 | −0.49 | 149 | 1.2E−17 | 887 | 1.9E−134 | |
| A05 | 500 | 9.8E−37 | 0.77 | −0.69 | 153 | 1.8E−63 | 828 | 2.9E−249 | |
| B. rapa | A06 | 509 | 6.1E−49 | 0.81 | −0.58 | 153 | 7.6E−45 | 828 | 7.6E−122 |
| A07 | 524 | 5.2E−32 | 0.79 | −0.65 | 155 | 2.3E−38 | 810 | 1.6E−173 | |
| A08 | 502 | 1.8E−34 | 0.74 | −0.66 | 150 | 1.8E−56 | 867 | 5.0E−206 | |
| A09 | 505 | 4.3E−58 | 0.78 | −0.63 | 150 | 9.2E−77 | 884 | 3.1E−255 | |
| A10 | 522 | 6.1E−20 | 0.75 | −0.57 | 166 | 6.5E−36 | 767 | 3.0E−210 | |
| Total | 512 | 2.4E−287 | 0.75 | −0.61 | 155 | 0.0E+00 | 831 | 0.0E+00 | |
| C01 | 442 | 1.5E−81 | 0.90 | −0.82 | 93 | 1.7E−112 | 1570 | 0.0E+00 | |
| C02 | 429 | 8.2E−108 | 0.81 | −0.74 | 78 | 2.4E−103 | 1673 | 6.2E−248 | |
| C03 | 459 | 1.4E−190 | 0.85 | −0.71 | 96 | 1.0E−174 | 1606 | 0.0E+00 | |
| C04 | 446 | 6.2E−59 | 0.86 | −0.62 | 95 | 1.8E−48 | 1590 | 2.2E−258 | |
| B. oleracea | C05 | 439 | 1.6E−93 | 0.94 | −0.84 | 97 | 4.2E−100 | 1535 | 8.2E−288 |
| C06 | 430 | 2.3E−139 | 0.90 | −0.79 | 89 | 4.0E−150 | 1606 | 0.0E+00 | |
| C07 | 435 | 4.7E−83 | 0.88 | −0.74 | 86 | 6.6E−92 | 1630 | 3.5E−240 | |
| C08 | 448 | 4.0E−163 | 0.87 | −0.72 | 99 | 2.1E−173 | 1566 | 0.0E+00 | |
| C09 | 445 | 3.8E−124 | 0.86 | −0.73 | 96 | 3.3E−127 | 1566 | 0.0E+00 | |
| Total | 442 | 0.0E+00 | 0.87 | −0.73 | 92 | 0.0E+00 | 1596 | 0.0E+00 | |
For both B. rapa and B. oleracea, the frequency of microsatellites was high at/near both ends but low in/near the middle of all the pseudochromosomes (Fig. 3), which likely corresponded to the peri- telomere and centromere, respectively.43 The frequencies of microsatellites for the different pseudochromosomes of B. rapa or B. oleracea were generally comparable, which was in accordance with the similar frequencies of genes/TEs for these chromosomes (Fig. 3; Table 2). Interestingly, the homoeologous chromosomes A3 and C3 both exhibited the highest microsatellite frequency among all the pseudochromosomes of B. rapa or B. oleracea, respectively, which was in accordance with their highest gene frequency among these chromosomes (Fig. 3; Table 2). In accordance with the high significance of the P-values of the χ2 test between the practical and hypothetical/average frequencies of microsatellites in the 1-Mb genomic intervals (Table 2), the physical distribution of microsatellites on all the pseudochromosomes of both B. rapa and B. oleracea were highly non-uniform (Fig. 3), which suggests the non-random occurrence of microsatellites. In accordance with the usually higher P-values of the χ2 test between the practical and hypothetical/average frequencies of microsatellites for the 9 pseudochromosomes of B. oleracea than for the 10 pseudochromosomes of B. rapa (Table 2), the distribution of microsatellites was more uneven in B. oleracea than in B. rapa (Fig. 3), which was likely attributable to the more concentrated distribution of genes/TEs in B. rapa than in B. oleracea. For both B. rapa and B. oleracea, the frequencies of microsatellites in the 1-Mb genomic intervals studied were significantly positively or negatively correlated with those of genes (total r = 0.75 and 0.87) or TEs (total r = −0.61 and −0.73), respectively (Table 2), which was accordant with one of the interesting findings in this study, that the genomic distribution of microsatellites was generally in accordance with that of genes but opposite to that of TEs (Fig. 3). These results were in agreement with the previous findings, which showed that microsatellites are preferentially associated with non-repetitive DNA/gene sequences in the plant genome.5,44 The high agreement of microsatellites and genes strongly suggests the putative role of microsatellites in regulating gene function3–5 and the use of SSR markers for tagging/cloning genes.
In conclusion, the genomic distributions of microsatellites in the assembled pseudochromosomes of B. rapa and B. oleracea were generally similar.
3.3. Development and database of genome-wide SSR markers
A total of 115 869 (92.1%), 185 662 (91.4%) and 356 522 (95.0%) primer pairs were successfully designed from the flanking sequences of 125 856, 203 161 and 375 214 mono- to hexanucleotide and compound microsatellites identified from the assembled genomic sequences of B. rapa, B. oleracea and B. napus, respectively (Table 3). The primer pairs could not be designed for the remaining microsatellites, mostly due to the constraint of obtaining sufficient flanking sequences from either side of the identified microsatellites. Similar observations have also been observed in other genome-wide microsatellite marker development studies in plants, such as rice,35 black cottonwood,36 cucumber,37 Brachypodim distachyon38 and foxtail millet.39 The frequencies of newly developed genome-wide SSR markers of B. rapa, B. oleracea and B. napus were 408.2, 343.8 and 356.2 per Mb or one every 2.45, 2.91 and 2.81 kb, respectively. Most of the genome-wide SSR markers of B. rapa (91.9%) and B. oleracea (75.4%) were developed from the mapped sequence scaffolds and thus have determined physical positions. The physical positions of the newly developed genome-wide SSR markers of B. napus will be determined soon because the anchoring of its sequence scaffolds will be completed after several months (our unpublished data).
Table 3.
Number (%) of newly developed genome-wide SSR markers that generated certain numbers (from 0 to >3) of in silico PCR products in the assembled genomic sequences of B. rapa, B. oleracea and B. napus, respectively
| Markers from | In silico PCR in | Zero | One | Two | Three | >Three | Total |
|---|---|---|---|---|---|---|---|
| B. rapa | 47 (0.0) | 92 517 (79.8) | 10 977 (9.5) | 3309 (2.9) | 9019 (7.8) | ||
| B. rapa | B. oleracea | 59 201 (51.1) | 40 493 (34.9) | 6370 (5.5) | 2100 (1.8) | 7705 (6.6) | 115 869 (100) |
| B. napus | 12 765 (11.0) | 36 030 (31.1) | 38 162 (32.9) | 10 666 (9.2) | 18 246 (15.7) | ||
| B. rapa | 110 784 (59.7) | 48 423 (26.1) | 8486 (4.6) | 3349 (1.8) | 14 620 (7.9) | ||
| B. oleracea | B. oleracea | 20 (0.0) | 121 169 (65.3) | 18 299 (9.9) | 7140 (3.8) | 39 034 (21.0) | 185 662 (100) |
| B. napus | 13 192 (7.1) | 54 749 (29.5) | 55 160 (29.7) | 14 156 (7.6) | 48 405 (26.1) | ||
| B. rapa | 157 473 (44.2) | 138 202 (38.8) | 20 604 (5.8) | 7758 (2.2) | 32 485 (9.1) | ||
| B. napus | B. oleracea | 106 706 (29.9) | 153 690 (43.1) | 24 601 (6.9) | 10 264 (2.9) | 61 261 (17.2) | 356 522 (100) |
| B. napus | 0 (0.0) | 93 084 (26.1) | 110 106 (30.9) | 44 138 (12.4) | 109 194 (30.6) |
Because of the polyploidy nature of Brassica,45 SSR markers usually amplify multiple fragments from homologous DNA sequences, which could complicate or cause errors in the genotype scoring. Therefore, all of the newly developed genome-wide SSR markers were subjected to in silico PCR analysis in the assembled genomic sequences of B. rapa, B. oleracea and B. napus, and the numbers of in silico PCR product(s) were recorded and summarized (Table 3). For the 115 869 SSR markers developed from B. rapa, 47 (0.0%), 92 517 (79.8%), 10 977 (9.5%), 3309 (2.9%) and 9019 (7.8%) markers generated 0, 1, 2, 3 and >3 in silico PCR product(s), respectively, from the assembled genomic sequences of B. rapa; 59 201 (51.1%), 40 493 (34.9%), 6370 (5.5%), 2100 (1.8%) and 7705 (6.6%) markers generated 0, 1, 2, 3 and >3 in silico PCR product(s), respectively, from the assembled genomic sequences of B. oleracea and 12 765 (11.0%), 36 030 (31.1%), 38 162 (32.9%), 10 666 (9.2%) and 18 246 (15.7%) markers generated 0, 1, 2, 3 and >3 in silico PCR product(s), respectively, from the assembled genomic sequences of B. napus. For the 185 662 SSR markers developed from B. oleracea, 20 (0.0%), 121 169 (65.3%), 18 299 (9.9%), 7140 (3.8%) and 39 034 (21.0%) markers generated 0, 1, 2, 3 and >3 in silico PCR product(s), respectively, from the assembled genomic sequences of B. oleracea; 110 784 (59.7%), 48 423 (26.1%), 8486 (4.6%), 3349 (1.8%) and 14 620 (7.9%) markers generated 0, 1, 2, 3 and >3 in silico PCR product(s), respectively, from the assembled genomic sequences of B. rapa and 13 192 (7.1%), 54 749 (29.5%), 55 160 (29.7%), 14 156 (7.6%) and 48 405 (26.1%) markers generated 0, 1, 2, 3 and >3 in silico PCR product(s), respectively, from the assembled genomic sequences of B. napus. For the 356 522 SSR markers developed from B. napus, 0 (0.0%), 93 084 (26.1%), 110 106 (30.9%), 44 138 (12.4%) and 109 194 (30.6%) markers generated 0, 1, 2, 3 and >3 in silico PCR product(s), respectively, from the assembled genomic sequences of B. napus; 157 473 (44.2%), 138 202 (38.8%), 20 604 (5.8%), 7758 (2.2%) and 32 485 (9.1%) markers generated 0, 1, 2, 3 and >3 in silico PCR product(s), respectively, from the assembled genomic sequences of B. rapa and 106 706 (29.9%), 153 690 (43.1%), 24 601 (6.9%), 10 264 (2.9%) and 61 261 (17.2%) markers generated 0, 1, 2, 3 and >3 in silico PCR product(s), respectively, from the assembled genomic sequences of B. oleracea. Interestingly, the SSR markers that generated tens to thousands of in silico PCR products were mostly associated with the annotated TEs, especially the retrotransposons.
We also determined the relationship between the physical positions of the newly developed genome-wide SSR markers and the previously developed publicly available Brassica SSR markers as well as the annotated genome components (mainly as genes and TEs) (Supplementary Table S7). Of the 115 869 SSR markers developed from B. rapa, 5991 (5.2%), 22 596 (19.5%) and 32 648 (28.2%) were involved in public Brassica SSR markers, genes and TEs, respectively. Of the 185 662 SSR markers developed from B. oleracea, 12 322 (6.6%), 33 228 (17.9%) and 73 487 (39.6%) were involved in public Brassica SSR markers, genes and TEs, respectively. Of the 356 522 SSR markers developed from B. napus, 23 928 (6.7%), 58 952 (16.5%) and 161 090 (45.2%) were involved in public Brassica SSR markers, genes and TEs, respectively. Interestingly, the TE-associated SSR markers were rarely involved in the annotated genes and mostly generated tens to thousands of in silico PCR products.
To facilitate the access and effective utilization of the Brassica SSR markers, we constructed an integrative database (http://oilcrops.info/SSRdb), which has search tools to obtain much useful information for the newly developed genome-wide SSR markers from the sequenced Brassica species and the previously developed publicly available Brassica SSR markers (Fig. 4). For the previously developed publicly available Brassica SSR markers, this information includes the primer-pair sequences, microsatellite repeat, source, reference and number of in silico PCR product(s) in the assembled genomic sequences of the sequenced Brassica species (currently only for B. rapa, B. oleracea and B. napus). For the newly developed genome-wide SSR markers from the sequenced Brassica species, this information includes the following: (i) the sequence, type, length and physical position of microsatellite repeat; (ii) the serial number, sequences, annealing temperatures, lengths and expected product size of primer pair; (iii) the number of in silico PCR product(s) in the assembled genomic sequences of the sequenced Brassica species (currently only for B. rapa, B. oleracea and B. napus) and (iv) the positional relationship with the previously developed publicly available Brassica SSR markers and the annotated genome components (mainly genes and TEs). In addition, this database also provides useful analysis tools (such as BLAST, e-PCR, Primer3 and ExtractSeq) and web links to other databases (e.g. http://brassicadb.org/brad/) and websites (e.g. http://www.brassica.info/) related to Brassica research. More importantly, this SSR marker database for Brassica will update as the number of sequenced Brassica species increases.
Figure 4.

Brassica SSR marker database homepage (A) and search results pages (B–D). This database provides three search approaches: by the name of the previously developed publicly available Brassica SSR markers (e.g. BN12A), by the serial number of the SSR markers (e.g. BnScaffold000001_12) and by the start to end positions on the chromosome/scaffold (e.g. BnScaffold000001:20–20 000 bp). This database also provides many useful tools, such as e-PCR, BLAST and primer3. Figure B–D show the results of examples of the three search approaches that are provided in the homepage.
3.4. Experimental evaluation of the newly developed genome-wide SSR markers
A total of 3974 newly developed SSR markers from B. rapa and B. oleracea were tested for amplification in the six representative B. napus cultivars/inbred lines (Table 4). Of these, 3880 SSR markers (97.6%) successfully amplified at least one clear fragment, while the remaining 94 (2.4%) failed to amplify, which could be due to the differences between the genome sequences of B. napus and its two progenitors, B. rapa and B. oleracea.46,47 The amplification rate (97.6%) of the tested SSR markers in the six B. napus cultivars/inbred lines was slightly or much higher than the corresponding rates (94.3/82.9, 89.2 and 77.4%, respectively) for the previously developed SSR markers from GSSs (mainly BESs)/unique transcripts of B. napus,24,30 BACs of B. rapa20 and WGSs of B. oleracea,26 which suggests that there is a high quality in the SSR markers that were developed from the assembled genomic sequences. The amplification rate of the tested SSR markers showed small variations for different motif lengths, motif repeat numbers and repeat lengths (i.e. motif length × motif repeat number), which was consistent with the previous reports in Brassica20,24,26 and rice.35 For the majority of the tested SSR markers, the numbers of fragment(s) amplified from the six representative B. napus cultivars/inbred lines were equal or very close to those of in silico PCR product(s) in the assembled genomic sequences of B. napus (Supplementary Table S8). In particular, most (1602 of 1813; 88.4%) of the tested SSR markers that generated one in silico PCR product in the assembled genomic sequences of B. napus also amplified only a single clear fragment from the six representative B. napus cultivars/inbred lines. A considerable proportion (1099 of 3880; 28.3%) of the successfully amplified SSR markers also produced weak fragment(s), which could correspond to non-specific amplification(s) from homologous DNA sequences.
Table 4.
Amplification and polymorphism rate of the tested SSR markers and their association with the number of amplified fragment(s), the motif length, the motif repeat number and the repeat length
| Total markers |
Amplified markers |
Polymorphic markers |
|||||
|---|---|---|---|---|---|---|---|
| Number | % | Number | % | Number | % | ||
| 0 | 94 | 2.4 | / | / | / | / | |
| 1 | 1859 | 46.8 | / | / | 1255 | 67.5 | |
| Fragment (s) number | 2 | 1541 | 38.8 | / | / | 1124 | 72.9 |
| 3 | 352 | 8.9 | / | / | 276 | 78.4 | |
| >3 | 128 | 3.2 | / | / | 110 | 85.9 | |
| Mono | 35 | 0.9 | 34 | 97.1 | 26 | 76.5 | |
| Di | 3156 | 79.4 | 3077 | 97.5 | 2195 | 71.3 | |
| Tri | 594 | 14.9 | 584 | 98.3 | 397 | 68.0 | |
| Motif length | Tetra | 74 | 1.9 | 73 | 98.6 | 41 | 56.2 |
| Penta | 21 | 0.5 | 20 | 95.2 | 15 | 75.0 | |
| Hexa | 12 | 0.3 | 12 | 100.0 | 11 | 91.7 | |
| Compound | 82 | 2.1 | 80 | 97.6 | 80 | 100.0 | |
| 5 | 496 | 12.5 | 488 | 98.4 | 316 | 64.8 | |
| 6 | 98 | 2.5 | 96 | 98.0 | 67 | 69.8 | |
| 7 | 59 | 1.5 | 57 | 96.6 | 41 | 71.9 | |
| 10 | 791 | 19.9 | 765 | 96.7 | 539 | 70.5 | |
| 11 | 528 | 13.3 | 513 | 97.2 | 347 | 67.6 | |
| Motif repeat number | 12 | 388 | 9.8 | 375 | 96.6 | 252 | 67.2 |
| 13 | 576 | 14.5 | 567 | 98.4 | 386 | 68.1 | |
| 14 | 295 | 7.4 | 288 | 97.6 | 209 | 72.6 | |
| 15 | 98 | 2.5 | 95 | 96.9 | 76 | 80.0 | |
| 16 | 140 | 3.5 | 139 | 99.3 | 114 | 82.0 | |
| 17 | 105 | 2.6 | 105 | 100.0 | 88 | 83.8 | |
| Others | 400 | 10.1 | 392 | 98.0 | 330 | 84.2 | |
| 15 | 412 | 10.4 | 405 | 98.3 | 266 | 65.7 | |
| 18 | 82 | 2.1 | 80 | 97.6 | 55 | 68.8 | |
| 20 | 840 | 21.1 | 814 | 96.9 | 561 | 68.9 | |
| 21 | 58 | 1.5 | 56 | 96.6 | 40 | 71.4 | |
| 22 | 526 | 13.2 | 511 | 97.1 | 344 | 67.3 | |
| Repeat length | 24 | 418 | 10.5 | 405 | 96.9 | 275 | 67.9 |
| 26 | 577 | 14.5 | 568 | 98.4 | 387 | 68.1 | |
| 28 | 303 | 7.6 | 295 | 97.4 | 213 | 72.2 | |
| 30 | 122 | 3.1 | 119 | 97.5 | 96 | 80.7 | |
| 32 | 133 | 3.3 | 132 | 99.2 | 107 | 81.1 | |
| 34 | 97 | 2.4 | 97 | 100.0 | 80 | 82.5 | |
| Others | 406 | 10.2 | 398 | 98.0 | 341 | 85.7 | |
| Total | 3974 | 100 | 3880 | 97.6 | 2765 | 71.3 | |
The majority (2765 of 3880; 71.3%) of the successfully amplified SSR markers was polymorphic across the six representative B. napus cultivars/inbred lines (Table 4). The polymorphism rate of the tested SSR markers was almost equal to or slightly higher than the corresponding rates (73.4/65.9, 57.9, 69.5%) for the previously developed SSR markers from GSSs (mainly BESs)/unique transcripts of B. napus,24,30 BACs of B. rapa20 and WGSs of B. oleracea.26 Obviously, the polymorphism rate of the tested SSR markers increased (from 67.5% to 85.9%) as the number of amplified bands increased (from 1 to >3). The polymorphism rate of the tested SSR markers decreased slightly from the mono- to tetranucleotide repeats, while it increased quickly from the penta- to hexanucleotide repeats. This inconsistency of the relationship between the SSR marker polymorphism level and the motif length was also observed frequently in the previous SSR marker evaluation experiments, such as in the tests of the 627 and 1000 SSR markers from the GSSs (mainly BESs) and unique transcripts, respectively, of B. napus,24,30 the 890 SSR markers from the BACs of B. rapa,20 the 1398 SSR markers from the WGSs of B. oleracea26 and the 1009 SSR markers from the assembled genomic sequences of cucumber.37 This type of inconsistency could be attributable to the observation that only a small number of SSR markers of the specific (usually long) motif length(s) have been used to investigate this relationship in all of the above-mentioned studies (e.g. only 21 and 12 penta- and hexanucleotide repeat SSR markers were tested in the current investigation), which worthwhile to develop more SSR markers with long motifs to further investigate the relationship between the SSR marker polymorphism level and the motif length. The polymorphism rate of the tested SSR markers was highly positively correlated with both the motif repeat number and the repeat length (r = 0.74 and 0.86, respectively), which was basically consistent with the previous reports in Brassica24,30 and other plant species, including cucumber37 and carrot.48 Both correlation coefficients in the current investigation were much higher than or equal to the corresponding values (0.21 and 0.41; 0.74 and _) that were estimated with the 627 SSR markers from the GSSs (mainly BESs) of B. napus24 or the 1009 SSR markers from the assembled genomic sequences of cucumber,37 respectively. Strikingly, the tested SSR markers that were designed from compound repeats were almost all (80 of 82; 97.6%) polymorphic across the six representative B. napus cultivars/inbred lines (Supplementary Table S8).
Because the 1055 and 2919 tested SSR markers were developed from the sequence scaffolds of B. rapa and B. oleracea, respectively, they were thus designated as ‘BrSF’ and ‘BoSF’. To facilitate the effective utilization of these tested newly developed BrSF and BoSF SSR markers, the following useful information was provided (Supplementary Table S8): (i) the type, length, position and sequence of the microsatellite repeat; (ii) the name, sequences, annealing temperatures and expected product size of the primer pair; (iii) the number of in silico PCR product(s) in the assembled genomic sequences of the sequenced Brassica species (currently for B. rapa, B. oleracea and B. napus) and (iv) the polymorphism survey and number of fragment(s) amplified in six representative B. napus cultivars/inbred lines.
4. Discussion
4.1. The pattern of microsatellite distribution is likely conservative in Brassica
In the current study, almost all of the important characteristics of microsatellite distribution in the assembled genomic sequences of the three recently sequenced Brassica crop species have been analysed and compared. To the best of our knowledge, this study is the first report on the genome-wide analysis and comparison of the pattern of microsatellite distribution across the different species within the same genus in plants.
First, the frequencies of microsatellites in the assembled genomic sequences of B. rapa (496.8 per Mb), B. oleracea (424.8 per Mb) and B. napus (420.6 per Mb) were similar, and all were higher than almost all of the previous estimations.20,21,24,26,42 The slightly higher frequency of microsatellites in B. rapa than in both B. oleracea and B. napus is likely attributable to the more concentrated distribution and lower content of TEs in the assembled genomic sequences of B. rapa than in B. oleracea and B. napus (Fig. 3) because the frequencies (285.5, 272.0 and 285.4 per Mb) of microsatellites in the coding DNA sequences of the three species are almost equal.49 Secondly, in accordance with the high correlation between these variables (Supplementary Table S1), the distributions with respect to the motif length, type and repeat number of microsatellites in the assembled genomic sequences of the three Brassica species were almost identical (Fig. 1; Supplementary Table S2). More specifically, both the dominant/major and absent/scarce mono- to hexanucleotide motifs in the assembled genomic sequences of the three Brassica species were mostly identical (Table 1; Supplementary Table S3). Interestingly, the dominant/major motifs were all A/T rich, while the absent/scarce motifs were mostly C/G rich, which corresponded well to the much higher A/T than C/G content in the analysed sequences. Thirdly, the repeat numbers of the corresponding repeats or motifs for the three Brassica species were mostly similar (Table 1; Supplementary Table S4). Fourthly, the total repeat length (=microsatellite number × motif length × motif repeat number) proportions of the corresponding repeats or motifs of microsatellites in the assembled genomic sequences of the three Brassica species were also mostly similar (Table 1; Supplementary Table S5). In addition, the genomic distributions of microsatellites in the assembled pseudochromosomes of B. rapa and B. oleracea were generally similar (Fig. 3).
In conclusion, almost all of the analysed important characteristics of microsatellite distribution in the assembled genomic sequences of the three sequenced Brassica crop species were highly similar or almost identical, which suggests that the pattern of microsatellite distribution is likely conservative in Brassica. This circumstance is understandable because B. napus (AACC, 2n = 38) originated from the chromosome doubling after the very recent (≈0.01 MYA) natural hybridization between B. rapa (AA, 2n = 20) and B. oleracea (CC, 2n = 18),11 which diverged from a common ancestor only ≈5 MYA.50
4.2. Usefulness of the newly developed genome-wide Brassica SSR markers
In the current study, a total of 115 869, 185 662 and 356 522 SSR markers were successfully developed from the assembled genomic sequences of B. rapa, B. oleracea and B. napus, respectively (Table 3), with the frequencies of 408.2, 343.8 and 356.2 per Mb or one every 2.45, 2.91 and 2.81 kb. To the best of our knowledge, this study is the first report on genome-wide SSR marker development in Brassica. Only a small proportion of the newly developed genome-wide SSR markers (5.2, 6.6 and 6.7% for B. rapa, B. oleracea and B. napus, respectively) were involved in the previously developed publicly available Brassica SSR markers (Supplementary Table S7), which suggests that most of the newly developed genome-wide SSR markers should represent the new SSR markers. The huge-number and high-frequency genome-wide SSR markers developed from the sequenced Brassica species in this study could be useful for many studies that require large-number and/or high-density molecular markers, such as high-density linkage map construction, gene/QTL fine mapping and genome-wide/regional association mapping.
The acute physical positions of the majority of the newly developed genome-wide SSR markers of the sequenced Brassica species have been determined (http://oilcrops.info/SSRdb) based on the mapped sequence scaffolds (Supplementary Table S6) from which they are designed. In fact, the physical positions of most of the previously developed publicly available Brassica SSR markers have also been determined by in silico mapping against the pseudochromosomes of these sequenced Brassica species (http://oilcrops.info/SSRdb). The high-density SSR marker-based physical maps constructed in this study could be useful for the rapid selection of genome-wide SSR markers that are well distributed over these chromosomes for various genotyping applications.
Because of the polyploidy nature of Brassica,45 the developed SSR markers usually amplify multiple fragments from the homologous DNA sequences, as revealed in the current (Supplementary Table S8) and previous12–14,22,24,26,27,29,30,42,51 studies in Brassica. This could complicate or cause errors in the genotype scoring due to the reciprocal overlapping and uncertain allelism of these fragments.33 However, only a small proportion of the previously developed publicly available Brassica SSR markers have been alleged to be single locus.33. Therefore, there is an urgent need to develop more single-locus SSR markers to facilitate their application in Brassica. Previously, the single-locus SSR markers were developed by practical PCR amplification in a panel of inbred lines,33 which was time consuming, labour intensive, high cost and, thus, inefficient. In the current study, through the highly efficient in silico PCR analysis, a large number of newly developed genome-wide SSR markers (92 517, 121 169 and 93 084 for B. rapa, B. oleracea and B. napus, respectively) were found to generate one in silico PCR product in the assembled genomic sequences of the three sequenced Brassica species (Table 3). In addition, thousands of previously developed publicly available Brassica SSR markers were also found to generate one in silico PCR product in the assembled genomic sequences of these Brassica species (http://oilcrops.info/SSRdb). More importantly, most (88.4%) of the tested SSR markers, that generated one in silico PCR product in the assembled genomic sequences of B. napus, also amplified a single clear fragment in the six representative B. napus cultivars/inbred lines (Supplementary Table S8). These results suggest that SSR markers that generate one in silico PCR product should be the putative single-locus markers and could be especially useful. Interestingly, the proportion (27.9%) of the newly developed genome-wide Brassica SSR markers (Table 3), which generated one in silico PCR product in the assembled genomic sequences of B. napus, was close to the corresponding proportion (33.8%) of the previously developed 9858 SSR marker from the GSSs/unique transcripts of B. napus, the BACs of B. rapa and the GSSs of B. oleracea,33 which amplified a single clear fragment in six B. napus inbred lines.
Also known as ‘functional’ markers,52 genic SSR markers are developed from genes and have a high transferability across related species.52 Although several studies have been conducted to develop genic SSR markers from the ESTs/unique transcripts of B. rapa,29,31,32 B. oleracea31 and B. napus,30–32 the total number (<5000) of publicly available genic SSR markers has remained limited in Brassica (http://oilcrops.info/SSRdb). In the current study, a large number of newly developed genome-wide SSR markers (32 648, 33 228 and 58 952 for B. rapa, B. oleracea and B. napus, respectively) were involved in the annotated genes (Supplementary Table S7) and thus belonged to the genic SSR markers. Of these, only a small proportion (7.2, 6.1, 6.7% for B. rapa, B. oleracea and B. napus, respectively) was involved in the previously developed publicly available Brassica SSR markers (http://oilcrops.info/SSRdb). This finding suggests that most of these newly developed Brassica genic SSR markers could represent the new ‘functional’ markers, which should be highly useful in evolutionary studies,29 comparative mapping,32 candidate gene association mapping53 and molecular breeding.
For the high transferability of SSR markers across the cultivated and wild Brassica species,27,33,54,55 the developed genome-wide SSR markers from B. rapa (AA, 2n = 20), B. oleracea (CC, 2n = 18) and B. napus (AACC, 2n = 38) should also be useful for B. nigra (BB, 2n = 16), B. juncea (AABB, 2n = 36), B. carinata (BBCC, 2n = 34) and other Brassica species. In addition, according to the previous marker transferability research,15,16,22,25 a considerable proportion of the newly developed genome-wide Brassica SSR markers (especially the genic SSR markers) should also be useful for the species that belong to other genera and tribes within the Brassicaceae family.
More importantly, we also constructed an integrative SSR marker database for Brassica (http://oilcrops.info/SSRdb), which not only provides useful information on the newly developed genome-wide SSR markers from the sequenced Brassica species (currently only for B. rapa, B. oleracea and B. napus) but is also integrated with the previously developed publicly available Brassica SSR markers and the annotated genome components (mainly as genes and TEs). To the best of our knowledge, this is the first comprehensive SSR marker database for Brassica until now, and it should be a significant contribution to the Brassica research community.
4.3. Implications for SSR marker development
The numbers of clear fragment(s) amplified in the six representative B. napus cultivars/inbred lines for the 3974 tested SSR markers were usually equal or close to the numbers of in silico PCR product(s) in the assembled genomic sequences of B. napus (Supplementary Table S8). This finding suggests that the number of products amplified by SSR markers can be relatively accurately estimated by in silico PCR, which was in accordance with the previous reports in plants such as rice56 and Brachypodium.38 Therefore, the target microsatellite should be subjected to BLAST/in silico PCR analysis to estimate its copy number before SSR marker development, especially for the polyploidy species. In addition, most (88.4%) of the tested SSR markers that generate one in silico PCR product were also confirmed by practical PCR analysis (Supplementary Table S8). Therefore, the in silico identified single/low copy microsatellites should be preferential for marker development.
Replication slippage and recombination are currently two major mechanisms that are responsible for microsatellite expansion or contraction.2,3,5,57 Because of the small numbers of the tested SSR markers of specific motif length(s), the relationship between the SSR marker polymorphism level and the motif length was usually inconsistent in both the current (Table 4) and previous20,24,26,30,37 studies. However, the general trend was similar: the SSR marker polymorphism level tended to decrease as the motif length increased. This relationship is understandable because shorter motifs allow more possible replication slippage events per unit length of DNA.58,59 In addition, the SSR marker polymorphism level was positively correlated with both the motif repeat number and the repeat length in both the current (r = 0.74 and 0.86, respectively) and previous37,47,48,60 studies. More importantly, the tested compound SSR markers were almost all (97.6%) polymorphic. These relationships are also understandable because more motifs, larger motif repeat number and longer repeat length give more opportunity for replication slippage.2 Therefore, microsatellites with a shorter motif length, larger motif repeat number, longer repeat length and especially the compound repeat should be preferential for marker development.
It should be noted that a considerable proportion (Supplementary Table S7) of the newly developed genome-wide SSR markers from the sequenced Brassica species were involved in the so-called ‘mobile DNA sequences’ TEs61 and should thus be unstable. In addition, the SSR markers that are associated with TEs (especially retrotransposons) mostly generated tens to thousands of in silico PCR products (http://oilcrops.info/SSRdb). Therefore, caution should be observed with respect to marker development based on microsatellites that are associated with TEs (especially retrotransposons).
Supplementary Data
Supplementary Data are available at www.dnaresearch.oxfordjournals.org.
Funding
This work was supported by the National Science and Technology Supporting Program (2010BAD01B02), the National Rapeseed Industry Technology System (CARS-13) and the Hubei Agricultural Science and Technology Innovation Center of China.
Supplementary Material
Footnotes
Edited by Dr Satoshi Tabata
References
- 1.Tautz D., Renz M. Simple sequences are ubiquitous repetitive components of eukaryotic genomes. Nucleic Acids Res. 1984;12:4127–38. doi: 10.1093/nar/12.10.4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ellegren H. Microsatellites: simple sequences with complex evolution. Nat. Rev. Genet. 2004;5:435–45. doi: 10.1038/nrg1348. [DOI] [PubMed] [Google Scholar]
- 3.Li Y.C., Korol A.B., Fahima T., Beiles A., Nevo E. Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review. Mol. Ecol. 2002;11:2453–65. doi: 10.1046/j.1365-294x.2002.01643.x. [DOI] [PubMed] [Google Scholar]
- 4.Li Y.C., Korol A.B., Fahima T., Nevo E. Microsatellites within genes: structure, function, and evolution. Mol. Biol. Evol. 2004;21:991–1007. doi: 10.1093/molbev/msh073. [DOI] [PubMed] [Google Scholar]
- 5.Gemayel R., Vinces M.D., Legendre M., Verstrepen K.J. Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu. Rev. Genet. 2010;44:445–77. doi: 10.1146/annurev-genet-072610-155046. [DOI] [PubMed] [Google Scholar]
- 6.Morgante M., Olivieri A.M. PCR-amplified microsatellites as markers in plant genetics. Plant J. 1993;3:175–82. [PubMed] [Google Scholar]
- 7.Gupta P.K., Varshney R.K. The development and use of microsatellite markers for genetic analysis and plant breeding with emphasis on bread wheat. Euphytica. 2000;113:163–85. [Google Scholar]
- 8.Beckmann J.S., Soller M. Toward a unified approach to genetic mapping of eukaryotes based on sequence tagged microsatellite sites. Biotechnology. 1990;8:930–2. doi: 10.1038/nbt1090-930. [DOI] [PubMed] [Google Scholar]
- 9.Cheng F., Mandakova T., Wu J., Xie Q., Lysak M.A., Wang X. Deciphering the diploid ancestral genome of the Mesohexaploid Brassica rapa. Plant Cell. 2013;25:1541–54. doi: 10.1105/tpc.113.110486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Labana K.S., Gupta M.L. Breeding Oilseed Brassicas. Berlin, Germany: Springer-Verlag; 1993. [Google Scholar]
- 11.U, N. Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Japan J. Bot. 1935;7:389–452. [Google Scholar]
- 12.Lowe A.J., Moule C., Trick M., Edwards K.J. Efficient large-scale development of microsatellites for marker and mapping applications in Brassica crop species. Theor. Appl. Genet. 2004;108:1103–12. doi: 10.1007/s00122-003-1522-7. [DOI] [PubMed] [Google Scholar]
- 13.Piquemal J., Cinquin E., Couton F., et al. Construction of an oilseed rape (Brassica napus L.) genetic map with SSR markers. Theor. Appl. Genet. 2005;111:1514–23. doi: 10.1007/s00122-005-0080-6. [DOI] [PubMed] [Google Scholar]
- 14.Plieske J., Struss D. Microsatellite markers for genome analysis in Brassica. I. Development in Brassica napus and abundance in Brassicaceae species. Theor. Appl. Genet. 2001;102:689–94. [Google Scholar]
- 15.Suwabe K., Iketani H., Nunome T., Kage T., Hirai M. Isolation and characterization of microsatellites in Brassica rapa L. Theor. Appl. Genet. 2002;104:1092–8. doi: 10.1007/s00122-002-0875-7. [DOI] [PubMed] [Google Scholar]
- 16.Suwabe K., Iketani H., Nunome T., Ohyama A., Hirai M., Fukuoka H. Characteristics of microsatellites in Brassica rapa genome and their potential utilization for comparative genomics in Cruciferae. Breeding Sci. 2004;54:85–90. [Google Scholar]
- 17.Kresovich S., Szewcmcfadden A.K., Bliek S.M., Mcferson J.R. Abundance and characterization of simple-sequence repeats (SSRs) isolated from a size-fractionated genomic library of Brassica napus L (rapeseed) Theor. Appl. Genet. 1995;91:206–11. doi: 10.1007/BF00220879. [DOI] [PubMed] [Google Scholar]
- 18.SzewcMcFadden A.K., Kresovich S., Bliek S.M., Mitchell S.E., McFerson J.R. Identification of polymorphic, conserved simple sequence repeats (SSRs) in cultivated Brassica species. Theor. Appl. Genet. 1996;93:534–8. doi: 10.1007/BF00417944. [DOI] [PubMed] [Google Scholar]
- 19.Uzunova M.I., Ecke W. Abundance, polymorphism and genetic mapping of microsatellites in oilseed rape (Brassica napus L.) Plant Breeding. 1999;118:323–6. [Google Scholar]
- 20.Xu J., Qian X., Wang X., et al. Construction of an integrated genetic linkage map for the A genome of Brassica napus using SSR markers derived from sequenced BACs in B. rapa. BMC Genomics. 2010;11:594. doi: 10.1186/1471-2164-11-594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hong C.P., Piao Z.Y., Kang T.W., et al. Genomic distribution of simple sequence repeats in Brassica rapa. Mol. Cells. 2007;23:349–56. [PubMed] [Google Scholar]
- 22.Ling A.E., Kaur J., Burgess B., et al. Characterization of simple sequence repeat markers derived in silico from Brassica rapa bacterial artical chromosome sequences and their application in Brassica napus. Mol. Ecol. Notes. 2007;7:273–7. [Google Scholar]
- 23.Choi S.R., Teakle G.R., Plaha P., et al. The reference genetic linkage map for the multinational Brassica rapa genome sequencing project. Theor. Appl. Genet. 2007;115:777–92. doi: 10.1007/s00122-007-0608-z. [DOI] [PubMed] [Google Scholar]
- 24.Cheng X., Xu J., Xia S., et al. Development and genetic mapping of microsatellite markers from genome survey sequences in Brassica napus. Theor. Appl. Genet. 2009;118:1121–31. doi: 10.1007/s00122-009-0967-8. [DOI] [PubMed] [Google Scholar]
- 25.Burgess B., Mountford H., Hopkins C.J., et al. Identification and characterization of simple sequence repeat (SSR) markers derived in silico from Brassica oleracea genome shotgun sequences. Mol. Ecol. Notes. 2006;6:1191–4. [Google Scholar]
- 26.Li H.T., Chen X., Yang Y., et al. Development and genetic mapping of microsatellite markers from whole genome shotgun sequences in Brassica oleracea. Mol. Breeding. 2011;28:585–96. [Google Scholar]
- 27.Batley J., Hopkins C.J., Cogan N.O.I., et al. Identification and characterization of simple sequence repeat markers from Brassica napus expressed sequences. Mol. Ecol. Notes. 2007;7:886–9. [Google Scholar]
- 28.Hopkins C.J., Cogan N.O.I., Hand M., et al. Sixteen new simple sequence repeat markers from Brassica juncea expressed sequences and their cross-species amplication. Mol. Ecol. Notes. 2007;7:697–700. [Google Scholar]
- 29.Ramchiary N., Nguyen V.D., Li X., et al. Genic microsatellite markers in Brassica rapa: development, characterization, mapping, and their utility in other cultivated and wild Brassica relatives. DNA Res. 2011;18:305–20. doi: 10.1093/dnares/dsr017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang F., Wang X.F., Chen X., et al. Abundance, marker development and genetic mapping of microsatellites from unigenes in Brassica napus. Mol. Breeding. 2012;30:731–44. [Google Scholar]
- 31.An Z., Gao C., Li J., Fu D., Tang Z., Ortegon O. Large-scale development of functional markers in Brassica species. Genome. 2011;54:763–70. doi: 10.1139/g11-042. [DOI] [PubMed] [Google Scholar]
- 32.Parida S.K., Yadava D.K., Mohapatra T. Microsatellites in Brassica unigenes: relative abundance, marker design, and use in comparative physical mapping and genome analysis. Genome. 2010;53:55–67. doi: 10.1139/g09-084. [DOI] [PubMed] [Google Scholar]
- 33.Li H., Younas M., Wang X., et al. Development of a core set of single-locus SSR markers for allotetraploid rapeseed (Brassica napus L.) Theor. Appl. Genet. 2013;126:937–47. doi: 10.1007/s00122-012-2027-z. [DOI] [PubMed] [Google Scholar]
- 34.Wang X., Wang H., Wang J., et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 2011;43:1035–9. doi: 10.1038/ng.919. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Z., Deng Y., Tan J., Hu S., Yu J., Xue Q. A genome-wide microsatellite polymorphism database for the indica and japonica rice. DNA Res. 2007;14:37–45. doi: 10.1093/dnares/dsm005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin T.M., Zhang X.Y., Gunter L.E., et al. Microsatellite primer resource for Populus developed from the mapped sequence scaffolds of the Nisqually-1 genome. New Phytol. 2009;181:498–503. doi: 10.1111/j.1469-8137.2008.02663.x. [DOI] [PubMed] [Google Scholar]
- 37.Cavagnaro P.F., Senalik D.A., Yang L., et al. Genome-wide characterization of simple sequence repeats in cucumber (Cucumis sativus L.) BMC Genomics. 2010;11:569. doi: 10.1186/1471-2164-11-569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sonah H., Deshmukh R.K., Sharma A., et al. Genome-wide distribution and organization of microsatellites in plants: an insight into marker development in Brachypodium. PLoS One. 2011;6:e21298. doi: 10.1371/journal.pone.0021298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pandey G., Misra G., Kumari K., et al. Genome-wide development and use of microsatellite markers for large-scale genotyping applications in foxtail millet [Setaria italica (L.)] DNA Res. 2013;20:197–207. doi: 10.1093/dnares/dst002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thiel T., Michalek W., Varshney R.K., Graner A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.) Theor. Appl. Genet. 2003;106:411–22. doi: 10.1007/s00122-002-1031-0. [DOI] [PubMed] [Google Scholar]
- 41.Rotmistrovsky K., Jang W., Schuler G.D. A web server for performing electronic PCR. Nucleic Acids Res. 2004;32:W108–112. doi: 10.1093/nar/gkh450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iniguez-Luy F.L., Voort A.V., Osborn T.C. Development of a set of public SSR markers derived from genomic sequence of a rapid cycling Brassica oleracea L. genotype. Theor. Appl. Genet. 2008;117:977–85. doi: 10.1007/s00122-008-0837-9. [DOI] [PubMed] [Google Scholar]
- 43.Ott A., Trautschold B., Sandhu D. Using microsatellites to understand the physical distribution of recombination on soybean chromosomes. PLoS One. 2011;6:e22306. doi: 10.1371/journal.pone.0022306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morgante M., Hanafey M., Powell W. Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat. Genet. 2002;30:194–200. doi: 10.1038/ng822. [DOI] [PubMed] [Google Scholar]
- 45.Lysak M.A., Koch M.A., Pecinka A., Schubert I. Chromosome triplication found across the tribe Brassiceae. Genome Res. 2005;15:516–25. doi: 10.1101/gr.3531105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mun J.H., Kwon S.J., Yang T.J., et al. Genome-wide comparative analysis of the Brassica rapa gene space reveals genome shrinkage and differential loss of duplicated genes after whole genome triplication. Genome Biol. 2009;10:R111. doi: 10.1186/gb-2009-10-10-r111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheung F., Trick M., Drou N., et al. Comparative analysis between homoeologous genome segments of Brassica napus and its progenitor species reveals extensive sequence-level divergence. Plant Cell. 2009;21:1912–28. doi: 10.1105/tpc.108.060376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cavagnaro P.F., Chung S.M., Manin S., et al. Microsatellite isolation and marker development in carrot—genomic distribution, linkage mapping, genetic diversity analysis and marker transferability across Apiaceae. BMC Genomics. 2011;12:386. doi: 10.1186/1471-2164-12-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shi J., Huang S., Fu D., et al. Evolutionary dynamics of microsatellite distribution in plants: insight from the comparison of sequenced brassica, Arabidopsis and other angiosperm species. PLoS One. 2013;8:e59988. doi: 10.1371/journal.pone.0059988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang Y.W., Lai K.N., Tai P.Y., Li W.H. Rates of nucleotide substitution in angiosperm mitochondrial DNA sequences and dates of divergence between Brassica and other angiosperm lineages. J. Mol. Evol. 1999;48:597–604. doi: 10.1007/pl00006502. [DOI] [PubMed] [Google Scholar]
- 51.Saal B., Plieske J., Hu J., Quiros C.F., Struss D. Microsatellite markers for genome analysis in Brassica. II. Assignment of rapeseed microsatellites to the A and C genomes and genetic mapping in Brassica oleracea L. Theor. Appl. Genet. 2001;102:695–9. [Google Scholar]
- 52.Varshney R.K., Graner A., Sorrells M.E. Genic microsatellite markers in plants: features and applications. Trends Biotechnol. 2005;23:48–55. doi: 10.1016/j.tibtech.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 53.Du Q., Gong C., Pan W., Zhang D. Development and application of microsatellites in candidate genes related to wood properties in the Chinese white poplar (Populus tomentosa Carr.) DNA Res. 2013;20:31–44. doi: 10.1093/dnares/dss031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ge Y., Ramchiary N., Wang T., et al. Development and linkage mapping of unigene-derived microsatellite markers in Brassica rapa L. Breeding Sci. 2011;61:160–7. [Google Scholar]
- 55.Marquez-Lema A., Velasco L., Perez-Vich B. Transferability, amplification quality, and genome specificity of microsatellites in Brassica carinata and related species. J. Appl. Genet. 2010;51:123–31. doi: 10.1007/BF03195720. [DOI] [PubMed] [Google Scholar]
- 56.McCouch S.R., Teytelman L., Xu Y., et al. Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.) DNA Res. 2002;9:199–207. doi: 10.1093/dnares/9.6.199. [DOI] [PubMed] [Google Scholar]
- 57.Oliveira E.J., Padua J.G., Zucchi M.I., Vencovsky R., Vieira M.L.C. Origin, evolution and genome distribution of microsatellites. Genet. Mol. Biol. 2006;29:294–307. [Google Scholar]
- 58.Katti M.V., Ranjekar P.K., Gupta V.S. Differential distribution of simple sequence repeats in eukaryotic genome sequences. Mol. Biol. Evol. 2001;18:1161–7. doi: 10.1093/oxfordjournals.molbev.a003903. [DOI] [PubMed] [Google Scholar]
- 59.Schug M.D., Hutter C.M., Wetterstrand K.A., Gaudette M.S., Mackay T.F., Aquadro C.F. The mutation rates of di-, tri- and tetranucleotide repeats in Drosophila melanogaster. Mol. Biol. Evol. 1998;15:1751–60. doi: 10.1093/oxfordjournals.molbev.a025901. [DOI] [PubMed] [Google Scholar]
- 60.Cloutier S., Miranda E., Ward K., et al. Simple sequence repeat marker development from bacterial artificial chromosome end sequences and expressed sequence tags of flax (Linum usitatissimum L.) Theor. Appl. Genet. 2012;125:685–94. doi: 10.1007/s00122-012-1860-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McClintock B. Chromosome organization and genic expression. Cold Spring Harb. Symp. Quant. Biol. 1951;16:13–47. doi: 10.1101/sqb.1951.016.01.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.