

Abstract
Combinations of homogeneous Cu salts and TEMPO have emerged as practical and efficient catalysts for the aerobic oxidation of alcohols. Several closely related catalyst systems have been reported, which differ in the identity of the solvent, the presence of 2,2′-bipyridine as a ligand, the identity of basic additives, and the oxidation state of the Cu source. These changes have a significant influence on the reaction rates, yields, and substrate scope. In this report, we probe the mechanistic basis for differences among four different Cu/TEMPO catalyst systems and elucidate the features that contribute to efficient oxidation of aliphatic alcohols.
Keywords: alcohol oxidation, aerobic, kinetics, copper, TEMPO, mechanism
Introduction
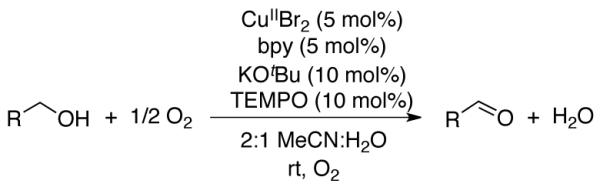
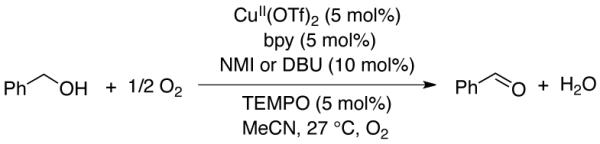
Oxidations of alcohols to aldehydes or ketones are widely used transformations in synthetic organic chemistry,1 and extensive recent efforts have focused on the development of aerobic oxidation methods.2 Homogeneous catalysts containing a copper salt in combination with TEMPO (TEMPO = 2,2,6,6-tetramethylpiperidine-N-oxyl) are among the most versatile catalysts for aerobic oxidation of primary alcohols to aldehydes, and numerous variants have been reported.3-7 The first of these was reported by Semmelhack in 1984, and consists of CuICl/TEMPO in DMF (DMF = N,N-dimethylformamide).3 In 2003, Sheldon reported a CuIIBr2/TEMPO catalyst that employed bpy as an ancillary ligand (bpy = 2,2′-bipyridine) and KOtBu as a base in an acetonitrile/water (2:1) solvent mixture.4 More recently, Koskinen reported a CuII(OTf)2/TEMPO catalyst (OTf = trifluoromethanesulfonate) that differs from the Sheldon catalyst by the replacement of KOtBu with NMI and/or DBU (NMI = N-methylimidazole, DBU = 1,8-diazabicycloundec-7-ene) and the use of non-aqueous MeCN as the solvent.5 Finally, our group recently described a CuIOTf/bpy/NMI catalyst in which the use of a CuI source improved the catalyst performance and substrate scope.6 Each of these catalyst systems, designated A, B, C, and D (Table 1), promotes aerobic oxidation of activated alcohols, such as benzyl alcohol. The latter two exhibit broad utility in the oxidation of aliphatic primary alcohols, including those with diverse functional groups.
Table 1. Representative Cu/TEMPO-based Catalyst Systems for the Aerobic Oxidation of Primary Alcohols.
| Catalyst System | [Cu] | Ligand | Base | Solvent |
|---|---|---|---|---|
|
A (Semmelhack)3 |
CuICl | – | – | DMF |
| B (Sheldon)4 | CuIIBr2 | bpy | KOtBu | 2:1 MeCN/H2O |
| C (Koskinen)5 | CuII(OTf)2 | bpy | DBU (+ NMI) |
MeCN |
| D (Stahl)6 | CuIOTf | bpy | NMI | MeCN |
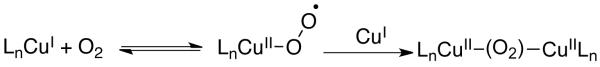
We recently investigated the mechanism of aerobic alcohol oxidation catalyzed by CuIOTf/bpy/NMI/TEMPO (catalyst D).8 Gas-uptake and in situ IR spectroscopic studies of the reaction kinetics, together with EPR and UV-visible spectroscopic analysis of the reaction mixture, provided evidence for the catalytic mechanism illustrated in Scheme 1. The reduced catalyst, consisting of LnCuI and TEMPOH, is oxidized by O2 in steps i and ii to afford LnCuII–OH and TEMPO. Oxidation of the alcohol is initiated by formation of a CuII–alkoxide (step iii), followed by TEMPO-mediated H-atom abstraction to produce the aldehyde and regenerate CuI (step iv). In order to gain a more thorough understanding of the contributions of solvent, ligand, base, and Cu oxidation state to the catalytic activity, we undertook a comparative mechanistic study of the four catalyst systems in Table 1. The results of this investigation are described below.
Scheme 1. Simplified Catalytic Cycle for CuIOTf/bpy/NMI/TEMPO Catalyzed Aerobic Alcohol Oxidation.


Results
Effect of solvent and ligand on redox potential and reactivity
Our initial mechanistic studies focused on probing mechanistic differences between CuCl/TEMPO/DMF (catalyst A) and the other three catalyst systems, which employ bpy as an ancillary ligand and acetonitrile as the solvent. Catalyst A promotes efficient oxidation of primary benzylic and allylic alcohols; however, reactions of aliphatic alcohols result in low yields of the corresponding aldehydes (~10%) unless stoichiometric quantities of Cu and TEMPO are employed.
Electrochemical data demonstrate the influence of the bpy ligand and solvent on the Cu redox properties (Figure 1). Cyclic voltammograms of TEMPO and CuCl in the presence or absence of bpy were acquired under N2 with 0.1 M LiClO4 as the electrolyte in both MeCN (solid red line) and DMF (dashed blue line). In the absence of bpy, switching from MeCN to DMF results in a significant decrease of the CuII/CuI reduction potential (ΔEmp ~0.28 V) (Figure 1a). Inclusion of the bpy ligand further lowers the CuII/CuI potential and results in a smaller solvent effect (ΔEmp ~0.10 V) (Figure 1b). The solvent has minimal effect on the potential of the TEMPO/TEMPO+ redox couple (Figure 1c).
Figure 1.

Cyclic voltammograms of CuCl (± bpy) and TEMPO in DMF (dashed blue line) and MeCN (solid red line). See Supporting Information for details.
Attempts to oxidize cyclohexylmethanol, CyCH2OH, with catalyst A led to poor conversion (Figure 2, red trace), consistent with the low reactivity of aliphatic alcohols noted in the original report.3 Addition of bpy to otherwise identical reaction conditions, however, resulted in rapid formation of the corresponding aldehyde CyCHO (Figure 2, blue trace).
Figure 2.

The oxidation of CyCH2OH by CuCl with ( ) and without (
) and without ( ) bpy in DMF. Standard conditions: 0.4 M CyCH2OH, 0.04 M TEMPO, 0.04 M CuCl, 0.04 M bpy, 600 torr O2, 27 °C.
) bpy in DMF. Standard conditions: 0.4 M CyCH2OH, 0.04 M TEMPO, 0.04 M CuCl, 0.04 M bpy, 600 torr O2, 27 °C.

Effect of water on catalyst deactivation
Catalyst B is composed of CuIIBr2 with a bpy ligand and employs a 2:1 MeCN/H2O solvent mixture (eq 1). Water is used to enhance the solubility of the KOtBu base. This system was one of the first to enable oxidation of aliphatic alcohols (such as 1-octanol);9 however, elevated temperatures and increased catalyst loading were required to achieve high yields with these substrates. In contrast, catalysts C and D mediate efficient oxidation of aliphatic alcohols at room temperature. The origin of these differences appears to be associated with the influence of water on the reactions.
 |
(1) |
Use of catalyst B in the oxidation of CyCH2OH at room temperature exhibits a slow initial reaction rate, proceeds to only low conversion (7% yield after 12 h), and forms a green precipitate. Longer reaction times did not lead to higher conversion.
In order to probe the potential influence of water on Cu/TEMPO-mediated alcohol oxidation, we investigated the oxidation of PhCH2OH by catalyst D with varying amounts of water. We observe minimal rate inhibition at [H2O] ≤ 2 M (2 M = 10 equiv relative to PhCH2OH); however, higher concentrations of water inhibit the reaction (Figures 3 and S2). Catalyst B with the 2:1 MeCN/H2O solvent mixture ([H2O] = 18.5 M) exhibits a rate consistent with trend shown in Figure 3 (blue square in Figure 3). These data suggest that the water cosolvent, rather than the identity of the Cu source or base, is the origin of the decreased reaction rates with catalyst B.
Figure 3.

Rates of oxidation of PhCH2OH by CuIOTf/bpy/NMI/TEMPO with the addition of H2O. Standard conditions: 0.2 M PhCH2OH (0.5 mmol), 10 mM CuI(OTf), 10 mM bpy, 10 mM TEMPO, 20 mM NMI, 600 torr O2, 27 °C. Oxidation of PhCH2OH by CuIIBr2/bpy/KOtBu/TEMPO in MeCN/H2O ( ) included for comparison.
) included for comparison.
Copper(II) hydroxides have been proposed as products of catalyst deactivation in Cu/TEMPO catalyst systems5 and their formation is supported by the appearance of precipitates during the reaction, as noted above. After prolonged reaction times, a blue precipitate forms in reaction mixtures with the CuIOTf/bpy/NMI/TEMPO catalyst system. Dark blue crystals were isolated from a reaction mixture that was left open to air for 16 h and submitted for X-ray diffraction analysis. Structural analysis identified this species as the bis-μ-hydroxide dimer [(bpy)Cu(OH)]2(OTf)2 (Figure 4A, for details see Supporting Information).10 This CuII dimer may be used as a catalyst in the presence of added TEMPO and NMI. The oxidation of PhCH2OH exhibits an induction period, but then achieves a rate similar to that of the parent catalyst D (Figure S3), while the oxidation of CyCH2OH with this complex is somewhat slower than with catalyst D (Figure 4B).
Figure 4.

(A) X-ray structure of [(bpy)Cu(OH)]2(OTf)2, which crystallizes as a dimer of dimers (see Supporting Information for details). The triflate counterions are omitted for clarity. (B) The oxidation of CyCH2OH by CuIOTf ( ) and [(bpy)Cu(OH)]2(OTf)2. (
) and [(bpy)Cu(OH)]2(OTf)2. ( ). Data were obtained by monitoring pressure changes during catalytic turnover. Reaction conditions: 10 mM (bpy)Cu, 10 mM TEMPO, 20 mM NMI: 0.2 M CyCH2 OH, 1 atm O2, 2.5 mL MeCN, 27 °C.
). Data were obtained by monitoring pressure changes during catalytic turnover. Reaction conditions: 10 mM (bpy)Cu, 10 mM TEMPO, 20 mM NMI: 0.2 M CyCH2 OH, 1 atm O2, 2.5 mL MeCN, 27 °C.
The above data indicate that the use of water as a cosolvent leads to both decreased reaction rates and catalyst deactivation. Formation of the dimeric copper(II) hydroxide complex and potentially other Cu–hydroxide species as precipitates is promoted by the presence of large concentrations of water and makes the efficient oxidation of aliphatic alcohols difficult in the MeCN/H2O solvent mixture.
Effect of base (DBU vs. NMI) and Cu oxidation state
Catalysts C and D employ CuII(OTf)2 or CuIOTf in combination with an organic base (DBU and/or NMI) and do not employ water as a cosolvent.11 In the work described below, DBU and NMI are investigated independently to understand role of copper oxidation state and base. The kinetic dependencies of the oxidation of PhCH2OH on the major reaction components (O2, Cu/bpy, alcohol, and TEMPO) were measured with CuII(OTf)2 and either NMI or DBU as the base (eq 2). The results are compared to the previously reported kinetic data with CuIOTf and NMI. The reactions were analyzed by monitoring the change in oxygen pressure of a sealed, temperature-controlled reaction vessel using a computer interfaced gas uptake apparatus. These data are shown in Figure 5.
Figure 5.

Dependence of (I) CuI(OTf)/bpy/TEMPO/NMI, (data adapted from Ref. 8). (II) CuII(OTf)2/bpy/TEMPO/DBU, and (III) CuII(OTf)2/bpy/TEMPO/NMI catalyzed alcohol oxidation on initial (A) dioxygen pressure, (B) [Cu/bpy], (C) [PhCH2OH], and (D) [TEMPO]. The curves in B are derived from a fit of the data to rate = c1[Cu]2/(c2 + c3[Cu]). The curves in C are derived from a nonlinear least-squares fit of the data to rate = c1[alcohol]/(c2 + c3[alcohol]). Standard reaction conditions: 0.2 M PhCH2OH, 10 mM [Cu], 10 mM bpy 20 mM NMI or DBU, 10 mM TEMPO, 600 torr O2, 2.5 mL MeCN, 27 °C.
The influence of the base and copper oxidation state is evident from comparison of kinetic data acquired for the three different catalytic conditions. As we reported previously, CuIOTf/bpy/NMI/TEMPO-catalyzed oxidation of PhCH2OH revealed a zero-order dependence on substrate and TEMPO, first order dependence on pO2, and a mixed first-second order dependence on CuIOTf/bpy (Figure 5, Column I).
 |
(2) |
The CuII(OTf)2/DBU conditions show kinetic dependencies that are similar to those obtained with CuIOTf/NMI, with one notable exception (Figure 5, Column II and S4-S11). CuII(OTf)2/DBU exhibits a saturation dependence on [alcohol], rather than the zero-order dependence observed with CuIOTf/NMI.
The CuII(OTf)2/NMI conditions exhibit much slower rates and very different kinetic dependencies. In this case, the oxidation of PhCH2OH is independent of the oxygen pressure, exhibits a first order dependence on [CuII(OTf)2/bpy] and [TEMPO], and a saturation dependence on [alcohol] (Figure 5, Column III).
We speculated that the change in reaction rate and kinetic dependencies associated with the use of NMI as the basic additive could be associated with the formation of the conjugate acid, NMIH+ OTf−, under the reaction conditions. (For example, NMI could serve as a Brønsted base in the formation of a CuII–alkoxide species, as shown in eq 3.) To probe this hypothesis, NMIH+ OTf− was synthesized and added at varying concentrations to catalytic reactions employing CuIOTf/bpy/NMI/TEMPO as the catalyst. The inclusion of NMIH+ OTf− led to significant inhibition of the reaction rate (Figure 6A). Addition of 1 equiv of NMIH+ OTf− shows a rate that is almost identical to that obtained with CuII(OTf)2/bpy/NMI/TEMPO (blue square, Figure 6A). In contrast, the addition of DBUH+ OTf− has no significant effect on the catalytic rate of PhCH2OH oxidation by CuIOTf/bpy/NMI/TEMPO (Figure 6B).
Figure 6.

Dependence of CuIOTf/bpy/TEMPO/NMI catalyzed benzyl alcohol oxidation on added (A) NMIH+ OTf− and (B) DBUH+ OTf−. The reaction catalyzed by CuII(OTf)2/bpy/-TEMPO/NMI ( ) is included in A for comparison. Rates were obtained by monitoring pressure changes during catalytic turnover. Standard reaction conditions: 10 mM CuI(OTf), 10 mM bpy, 20 mM NMI, 10 mM TEMPO, 0.2 M alcohol.
) is included in A for comparison. Rates were obtained by monitoring pressure changes during catalytic turnover. Standard reaction conditions: 10 mM CuI(OTf), 10 mM bpy, 20 mM NMI, 10 mM TEMPO, 0.2 M alcohol.
| (3) |
Discussion
The data presented above provide valuable insights into the roles of solvent (DMF, CH3CN, H2O), the ancillary bpy ligand, the oxidation state of the Cu source, and identity of basic additive (KOtBu, NMI, DBU). Addition of bpy to the CuCl/TEMPO catalyst system in DMF enables catalytic oxidation of aliphatic alcohols (Figure 2). Bipyridine lowers the CuII/CuI reduction potential in DMF (cf. Figures 1A and 1B), so the effect of bpy cannot be attributed to the formation of a more oxidizing catalyst capable of reacting with the more challenging aliphatic alcohol. We speculate that the bpy ligand could play at least two roles: (1) enhance the rate of CuI oxidation by O2 and (2) alter the Cu speciation to maximize the number of catalytically active Cu sites (e.g., by preventing aggregation).
The reactions tolerate substantial quantities of water before experiencing inhibition (Figure 3). This observation is significant because water is a byproduct of the catalytic reaction, but stoichiometric amounts of water is well below the threshold of water inhibition (approx. 2 M). Use of water as a cosolvent, however, leads to significant inhibition of the rate. Excess water will disfavor formation of the CuII-alkoxide intermediate in favor of a CuII-hydroxide species (Scheme 2). Increased concentrations of monomeric or dimeric CuII-hydroxides can lead to catalyst deactivation via formation of off-cycle intermediates or precipitation of Cu species from the reaction mixture. For example, the isolated [bpyCu(OH)] 2+2 can enter the catalytic cycle, but it is sparingly soluble and reacts more slowly than the parent catalyst under normal reaction conditions.
Scheme 2. Formation of Bis-μ-Hydroxo CuII Dimer.
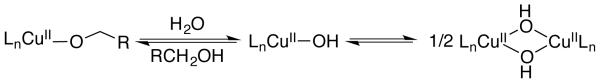
The kinetic studies of benzyl alcohol oxidation provide valuable insights into the influence of the base and Cu oxidation state effects, summarized in Scheme 3. The kinetic data for the three different catalyst systems are summarized in Table 2. The only difference among the three catalyst systems is the oxidation state of the Cu source, CuIOTf or CuII(OTf)2, and/or the organic base, NMI or DBU. Our previous mechanistic studies of Catalyst D provide a framework for understanding these results. For example, the mixed 2nd/1st-order dependence on the [Cu/bpy] and 1st-order dependence on pO2 with the first two catalyst systems is rationalized by a two-step aerobic Cu oxidation pathway that involves two metal centers (Scheme 4).
Scheme 3. Proposed Mechanism for the (bpy)CuII/TEMPO Catalyzed Aerobic Oxidation of Alcohols.

Table 2. Kinetic Dependences for Cu/bpy Systems with PhCH2OH.
| CuI/NMI | CuII/DBU | CuII/NMI | |
|---|---|---|---|
| pO2 | 1st order | 1st order | 0th order |
| [Cu] | mixed 2nd/1storder |
mixed 2nd/1st order |
1st order |
| [PhCH2OH] | 0th order | saturation | saturation |
| [TEMPO] | 0th order | 0th order | 1st order |
Scheme 4. Catalyst Oxidation Sequence Accounting for a Mixed 1st /2nd Order Cu Dependence.
When a CuII source is used, the identity of the base plays a key role in the rate of the reaction: DBU promotes fast rates, while NMI leads to slow rates. The saturation dependence on PhCH2OH concentration, observed with both CuII systems in Figure 5, suggests a substrate binding step contributes to the overall reaction rate. This step is attributed to formation of a LnCuII–alkoxide intermediate from CuII/base and PhCH2OH (eq 3).12 The strong base DBU enables formation of the CuII-alkoxide, while the weaker base NMI is less effective in this step.
Formation of a CuII–alkoxide generates an equivalent of acid, which persists throughout the reaction. In the case of CuII/NMI, the resulting acid is NMIH+ OTf−. This acid has a pKa of 14.3 in MeCN,13 which is sufficiently strong to favor protonolysis of a CuII–OH or CuII–OR group. The resulting CuII species (6) lies off the catalytic cycle and must access the active CuII–alkoxide intermediate via reaction with alcohol and the weak base, NMI. In the case of CuII/DBU, the acid is DBUH+ OTf−, which is substantially less acidic (pKa = 24 in MeCN14). These considerations account for the data shown in Figure 6, which show that NMIH+ strongly inhibits the reaction while DBUH+ lacks this inhibitory effect.
We propose that under the CuII/NMI conditions, an unfavorable equilibrium to form the CuII–alkoxide (i.e., eq 3) leads to a change in the turnover-limiting step. Aerobic oxidation of the CuI catalyst (step i) is no longer turnover limiting. Instead, substrate oxidation by CuII/TEMPO is rate controlling (step iv). The first order dependencies on [CuII/bpy] and [TEMPO] and the lack of a dependence on pO2 support this interpretation.
The CuI/NMI catalyst system is the most active of the three catalysts evaluated in Figure 5. This result can be rationalized by formation of a CuII–OH species in steps i and ii of the catalytic mechanism. This species can react directly with the alcohol to afford the CuII–alkoxide intermediate, without requiring involvement of the organic base. According to this proposal, the beneficial effect of NMI could arise from its role as a ligand for Cu (e.g., promoting aerobic oxidation of CuI), rather than its use as a Brønsted base. The similar efficacy of the CuI/NMI and CuII/DBU catalyst systems (i.e., C and D, Table 1) highlight the important of proper matching of the Cu oxidation state and identity of the basic additive. Perhaps most significantly, the use of CuI permits the reaction to proceed efficiently even in the absence of a strong exogenous base. This result has important synthetic implications. For example, strong bases could lead to epimerization of stereocenters adjacent to the aldehyde in the product or isomerization of (Z)-enals to the thermodynamically favored (E) product.15 The mildly basic conditions associated with the CuIOTf/NMI catalyst system avoids these complications. Electrochemical data8 show that NMI serves as a ligand for CuI and thus contributes to the aerobic oxidation step (step i, Scheme 3). In addition, NMI could hinder formation and/or facilitate dissociation of the dimer 5.
The reactivity of [(bpy)Cu(OH)]2(OTf)2 is consistent with the proposed mechanism. This CuII–hydroxide dimer is a more efficient catalyst precursor than other CuII sources because the Brønsted base (hydroxide) is present within the complex. This species enters the catalytic cycle after dissociation of the dimer into monomeric (bpy)CuII(OH)(OTf). Only 1 equiv of water is formed upon entering the catalytic cycle, and so the oxidation of aliphatic alcohols is efficient. The slower rates observed with this CuII dimer, relative to CuIOTf, probably reflects it slow dissociation into the active monomeric form and/or its poor solubility.
Summary and Conclusion
The number of Cu/TEMPO catalyst systems extend well beyond those considered in this investigation7,16 In most of these cases, the catalysts are limited in scope to the oxidation of activated alcohols (e.g., benzylic/allylic). The results of this investigation provide a framework for understanding key factors that contribute to the activity and/or limitations of these catalyst systems. Specifically, we have found that seemingly minor changes in solvent, oxidation state of the Cu source, and identity of basic additives can have a significant effect on the kinetic profile and substrate scope of the catalyst. Use of a ligand, such as bpy, can lead to catalytic activity with substrates such aliphatic alcohols that are otherwise unreactive. Water can significantly inhibit catalytic turnover when it is present in high concentration. Kinetic sutdies of several different Cu/TEMPO catalyst systems reveals that rate acceleration observed with the use of a CuI source arises from in situ formation of a hydroxide base that promotes formation of the key LnCuII–alkoxide intermediate. When CuII sources are employed, a suitably strong base must be used to deprotonate the alcohol to form the CuII-alkoxide species. The insights gained here have important implications for other emerging classes of Cu/TEMPO-mediated aerobic oxidation reactions, such as amine dehydrogenation.17
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Ilia Guzei for X-ray crystal structure determination. Financial support of this work was provided by the DOE (DE-FG02-05ER15690), the ACS GCI Pharmaceutical Roundtable and the Camille and Henry Dreyfus Postdoctoral Program in Environmental Chemistry, and NIH for a training grant (CBIT NIGMS T32 GM008505) that supported BLR. NMR spectroscopy facilities were partially supported by the NSF (CHE-9208463) and NIH (S10 RR08389).
Footnotes
ASSOCIATED CONTENT
Supporting Information Available: Experimental details, kinetic time course traces, CIF for [(bpy)Cu(OH)]2(OTf)2. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Tojo G, Fernández M. In: Oxidation of Alcohols to Aldehydes and Ketones. Tojo G, editor. Springer; New York: 2010. [Google Scholar]
- (2).(a) Arends IWCE, Sheldon RA. In: Modern Oxidation Methods. Bäckvall J-E, editor. Wiley-VCH Verlag Gmb & Co.; Weinheim: 2004. pp. 83–118. [Google Scholar]; (b) Sheldon RA, Arends IWCE, ten Brink G-J, Dijksman A. Acc. Chem. Res. 2002;35:774–781. doi: 10.1021/ar010075n. [DOI] [PubMed] [Google Scholar]; (c) Zhan BZ, Thompson A. Tetrahedron. 2004;60:2917–2935. [Google Scholar]; (d) Mallat T, Baiker A. Chem. Rev. 2004;104:3037–3058. doi: 10.1021/cr0200116. [DOI] [PubMed] [Google Scholar]; (e) Stahl SS. Angew. Chem., Int. Ed. 2004;43:3400–3420. doi: 10.1002/anie.200300630. [DOI] [PubMed] [Google Scholar]; (f) Markó IE, Giles PR, Tsukazaki M, Chellé-Regnaut I, Gautier A, Dumeunier R, Philippart F, Doda K, Mutonkole J-L, Brown SM, Urch CJ. Adv. Inorg. Chem. 2004;56:211–240. [Google Scholar]; (g) Schultz MJ, Sigman MS. Tetrahedron. 2006;62:8227–8241. [Google Scholar]; (h) Sigman MS, Jensen DR. Acc. Chem. Res. 2006;39:221–229. doi: 10.1021/ar040243m. [DOI] [PubMed] [Google Scholar]; (i) Karimi B, Zamani A. J Iran Chem Soc. 2008;5:S1–S20. [Google Scholar]; (j) Matsumoto T, Ueno M, Wang N, Kobayashi S. Chem-Asian J. 2008;3:196–214. doi: 10.1002/asia.200700359. [DOI] [PubMed] [Google Scholar]; (k) Parmeggiani C, Cardona F. Green Chem. 2012;14:547–564. [Google Scholar]
- (3).Semmelhack MF, Schmid CR, Cortés DA, Chou CS. J. Am. Chem. Soc. 1984;106:3374–3376. [Google Scholar]
- (4).(a) Gamez P, Arends IWCE, Reedijk J, Sheldon RA. Chem. Commun. 2003:2414–2415. doi: 10.1039/b308668b. [DOI] [PubMed] [Google Scholar]; (b) Gamez P, Arends IWCE, Sheldon RA, Reedijk J. Adv. Synth. Catal. 2004;346:805–811. [Google Scholar]
- (5).Kumpulainen ETT, Koskinen AMP. Chem. Eur. J. 2009;15:10901–10911. doi: 10.1002/chem.200901245. [DOI] [PubMed] [Google Scholar]
- (6).Hoover JM, Stahl SS. J. Am. Chem. Soc. 2011;133:16901–16910. doi: 10.1021/ja206230h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hoover JM, Steves JE, Stahl SS. Nat. Protoc. 2012;7:1161–1166. doi: 10.1038/nprot.2012.057. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hoover JM, Stahl SS. Org. Synth. 2013;90:2357–2367. [Google Scholar]
- (7).(a) Ragagnin G, Betzemeier B, Quici S, Knochel P. Tetrahedron. 2002;58:3985–3991. [Google Scholar]; (b) Contel M, Izuel C, Laguna M, Villuendas PR, Alonso PJ, Fish RH. Chem. Eur. J. 2003;9:4168–4178. doi: 10.1002/chem.200304771. [DOI] [PubMed] [Google Scholar]; (c) Contel M, Villuendas PR, Fernandez-Gallardo J, Alonso PJ, Vincent J-M, Fish RH. Inorg. Chem. 2005;44:9771–9778. doi: 10.1021/ic051220m. [DOI] [PubMed] [Google Scholar]; (d) Geisslmeir D, Jary WG, Falk H. Monatsh. Chem. 2005;136:1591–1599. [Google Scholar]; (e) Jiang N, Ragauskas AJ. J. Org. Chem. 2006;71:7087–7090. doi: 10.1021/jo060837y. [DOI] [PubMed] [Google Scholar]; (f) Mannam S, Alamsetti SK, Sekar G. Adv. Synth. Catal. 2007;349:2253–2258. [Google Scholar]; (g) Figiel PJ, Sibaouih A, Ahmad JU, Nieger M, Räisänen MT, Leskelä M, Repo T. Adv. Synth. Catal. 2009;351:2625–2632. [Google Scholar]
- (8).Hoover JM, Ryland BL, Stahl SS. J. Am. Chem. Soc. 2013;135:2357–2367. doi: 10.1021/ja3117203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).For an earlier system that catalyzes oxidation of aliphatic alcohols see Tretyakov VP, Chudaev VV, Zimtseva GP. Ukr. Khim. Zh. 1985;51:942–946.
- (10).A crystal structure of [Cu(bpy)(OH)]2(OTf)2 has been previously reported, although the packing of the dimers within the unit cell is significantly different. Castro I, Faus J, Julve M, Bois C, Real JA, Lloret F. J. Chem. Soc. Dalton Trans. 1992:47–52. The crystals in the present work consist of dimers of [Cu(bpy)(OH)] 2+2 dimers in which long axial Cu–O bonds (2.4Å) connect adjacent dimers (Figure S2).
- (11).With CuII(OTf)2, use of both DBU and NMI was shown to afford higher yields with aliphatic alcohols (see ref. 5). For example, applying the mixed DBU/NMI catalyst system to the oxidation of CyCH2OH provided higher yields of the corresponding aldehyde than a reaction with DBU alone (94% vs. 85% 1H NMR spectroscopy). The initial rates for the two systems were similar suggesting that employing NMI in addition to DBU helps mitigate the catalyst deactivation observed when DBU is the only base present.
- (12).The data cannot exclude equilibrium formation of a LnCuI–alkoxide species.
- (13).Pawlak Z. J. of Mol. Struct. 1986;143:369–374. [Google Scholar]
- (14).(a) Lemaire CF, Aerts JJ, Voccia S, Libert LC, Mercier F, Goblet D, Penevaux AR, Luxen AJ. Angew. Chem. Int. Ed. 2010;49:3161–3164. doi: 10.1002/anie.200906341. [DOI] [PubMed] [Google Scholar]; (b) Kaljurand I, Kütt A, Sooväli L, Rodima T, Mäemets V, Leito I, Koppel IA. J. Org. Chem. 2005;70:1019–1028. doi: 10.1021/jo048252w. [DOI] [PubMed] [Google Scholar]
- (15).Koenning D, Hiller W, Christmann M. Org. Lett. 2012;14:5258–5261. doi: 10.1021/ol302420k. [DOI] [PubMed] [Google Scholar]
- (16).For additional catalyst systems, see ref. 8 (cf. refs. 9 and 10 in this publication).
- (17).See, for example: Sonobe T, Oisaki K, Kanai M. Chem. Sci. 2012;3:3249–3255. Han B, Yang X-L, Wang C, Bai Y-W, Pan T-C, Chen X, Yu W. J. Org. Chem. 2012;77:1136–1142. doi: 10.1021/jo2020399. Hu Z, Kerton FM. Org. Biomol. Chem. 2012;10:1618–1624. doi: 10.1039/c2ob06670j. Tao C, Liu F, Zhu Y, Liu W, Cao Z. Org. Biomol. Chem. 2013;11:3349–3354. doi: 10.1039/c3ob00002h. Yin W, Wang C, Huang Y. Org. Lett. 2013;15:1850–1853. doi: 10.1021/ol400459y. Dornan LM, Cao Q, Flanagan JCA, Cook MJ, Crawford MJ, Muldoon MJ. Chem. Commun. 2013;49:6030–6032. doi: 10.1039/c3cc42231c. Kim J, Stahl SS. ACS Catal. 2013;3:1652–1656. doi: 10.1021/cs400360e.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.