

Significance
The treatment of cancer is highly reliant on drug combinations. Next-generation, targeted therapeutics are demonstrating interesting single-agent activities in clinical trials; however, the discovery of companion drugs through iterative clinical trial-and-error is not a tenable mechanism to prioritize clinically important combinations for these agents. Herein we describe the results of a large, high-throughput combination screen of the Bruton’s tyrosine kinase inhibitor ibrutinib versus a library of nearly 500 approved and investigational drugs. Multiple ibrutinib combinations were discovered through this study that can be prioritized for clinical examination.
Keywords: translational research, PCI-32765, Imbruvica
Abstract
The clinical development of drug combinations is typically achieved through trial-and-error or via insight gained through a detailed molecular understanding of dysregulated signaling pathways in a specific cancer type. Unbiased small-molecule combination (matrix) screening represents a high-throughput means to explore hundreds and even thousands of drug–drug pairs for potential investigation and translation. Here, we describe a high-throughput screening platform capable of testing compounds in pairwise matrix blocks for the rapid and systematic identification of synergistic, additive, and antagonistic drug combinations. We use this platform to define potential therapeutic combinations for the activated B-cell–like subtype (ABC) of diffuse large B-cell lymphoma (DLBCL). We identify drugs with synergy, additivity, and antagonism with the Bruton’s tyrosine kinase inhibitor ibrutinib, which targets the chronic active B-cell receptor signaling that characterizes ABC DLBCL. Ibrutinib interacted favorably with a wide range of compounds, including inhibitors of the PI3K-AKT-mammalian target of rapamycin signaling cascade, other B-cell receptor pathway inhibitors, Bcl-2 family inhibitors, and several components of chemotherapy that is the standard of care for DLBCL.
The standards of care for many diseases, including therapies for multiple types of cancer, involve drug combinations. Drug regimens can be comprised of as many as five or more agents, such as R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone), which is commonly used for the treatment of diffuse large B-cell lymphoma (DLBCL) (1). Most of these regimens were the result of protracted, empirical clinical trial-and-error. With the advent of a wide repertoire of targeted agents in oncology and other areas of medicine, unbiased preclinical methods to discover and prioritize drug combinations are urgently needed (2, 3).
Cancers use a diversity of pathological signaling and gene regulatory mechanisms to promote their survival, proliferation, and malignant phenotypes. Cancer gene sequencing across cancer cell line collections is a powerful means to identify genomic markers that may predict therapeutic response (4, 5). Targeted therapeutics have emerged as attractive strategies to block these dysregulated signaling events (6), but cancers use a variety of resistance mechanisms that must be addressed using rational drug combinations (7). The US Food and Drug Administration have indicated their willingness to consider combination of novel therapies at an earlier stage of clinical development (8), highlighting the need for innovative technologies to accelerate the discovery of novel drug combinations.
In B-cell malignancies, Bruton’s tyrosine kinase (BTK) has emerged as a particularly interesting target for therapeutic attack (9). In normal B cells, BTK activity is required to transmit signaling from the B-cell receptor (BCR) to downstream pathways, notably NF-κB. Constitutive BCR signaling has been implicated in the pathogenesis of chronic lymphocytic leukemia, mantle cell lymphoma, and the activated B-cell–like subtype (ABC) of DLBCL. Oncogenic signaling in ABC DLBCL is initiated and reinforced by activating mutations affecting BCR subunits, as well as the signaling adapters CARD11 and MYD88, triggering constitutive activation of downstream NF-κB and JAK/STAT pathways that promote cancer cell survival and proliferation (10–12). ABC DLBCL cell lines are sensitive to inhibitors of various kinases (JAK, IKKβ, BTK, IRAK4), as well as agents that target key transcription factors that modulate their oncogenic signaling pathways, suggesting drug combinations that exhibit synergistic synthetic lethality for these lymphomas (13, 14).
The BTK inhibitor ibrutinib has generated particular enthusiasm because of its ability to block BCR signaling in several B-cell malignancies (9). Ibrutinib is a highly targeted agent because of its ability to form a covalent bond with a conserved cysteine residue (C481) in the active site of BTK, resulting in remarkable selectivity and pharmacodynamics. Optimal clinical development of ibrutinib will likely depend upon its pairing with other agents to increase response rates and durability, as well as to circumvent potential ibrutinib-resistance mechanisms.
Here, we describe a high-throughput combination screening platform on which a wide range of dosages for pairs of small molecules can be easily explored. We demonstrate the ability of this platform to identify agents that potentiate the toxicity of ibrutinib for cell line models of ABC DLBCL. The drug combinations discovered have both mechanistic and translational interest, including clinically actionable combinations of ibrutinib with PI3K pathway inhibitors and with chemotherapeutic agents that are part of the current standard of care for DLBCL.
Results
A High-Throughput Screening Platform for Identification of Drug–Drug Combinations.
Various screening technologies and computational methods for the discovery of combination therapies have been reported (15–18). Challenges associated with establishing a combination screening platform include the building of an appropriate library of agents, the development of a robust plating system, and the establishment of a facile data interface. In this study, we evaluated 459 agents in combination with ibrutinib within 6 × 6 dose–response (matrix) blocks. The library, termed MIPE, was composed of oncology-focused, mechanistically annotated agents that were, as much as possible, prioritized for clinical relevance (Dataset S1). Mechanistic redundancy was embraced for selected target classes (e.g., the epidermal growth factor receptor family) because each agent may possess a unique polypharmacology that could yield distinctive activities in combination studies. To achieve customizable dose–response matrix blocks at a scale capable of interrogating thousands of drug–drug combinations, we used 1,536-well plate-enabled acoustic dispensers (19). Organization of each assay plate in true physical space resulted in 35 6 × 6 matrices within one 1,536-well plate. A single plate can be created in 15 min, affording full plating of this experiment in 3.5 h. The third primary requirement for this platform was a robust method for data analysis. Data were normalized to the plate positive control (bortezomib) and negative control (DMSO), such that the DMSO response was assigned a value of 100% and the bortezomib response was considered to be complete cell death and recorded as 0%. To avoid conflicting valuations in our analyses, we bounded all data to lie between 0% and 100%. Standard Z′-factor and minimum significant ratio metrics were used to assess interplate variability within this dataset (SI Appendix, Fig. S1). Plated wells were deconvoluted into individual response matrices and their combination behavior (additivity, synergy, antagonism) was characterized using a variety of metrics based on the Loewe, Bliss, and Gaddum models (see SI Appendix for detailed description of metrics) (20–23).
Before combination analysis, we evaluated the single-agent activities of the MIPE library. The results from multiple single-agent profiles of the MIPE library suggested that a dose–response range between ∼10 nM and 2 μM would yield a range of active and inactive concentrations for the plurality of library members. Pilot combination experiments used 6 × 6 matrix block sizes, a starting concentration of 2.5 μM, and serial 1:4 dilutions of each agent. Confirmation studies with promising agents were conducted as 10 × 10 matrix blocks with customized starting concentrations and serial twofold dilutions.
Comparative Analyses Associate Viability, Induction of Apoptosis, and NF-κB Responses.
The single-agent responses of all 459 agents in MIPE were generated using several cell-based assays, including cell viability (CellTiter-Glo), apoptosis (Caspase-Glo 3/7), and an NF-κB reporter assay (summary AID 651556) (Fig. 1A). Single-agent viability was assessed in two ABC DLBCL lines (TMD8 and HBL1). The MIPE library was also assayed for single-agent viability response in a bone marrow-derived human mesenchymal stem cell (hMSC) line to help gauge each agent’s potential therapeutic index. The ability of each agent to induce caspase 3/7 activation in TMD8 cells was examined at both 8- and 16-h time points to determine which agents induced an apoptotic response in a reasonable timeframe from a therapeutic exposure perspective. Given the reliance of ABC DLBCL cells on constitutive NF-κB signaling (9–11, 24–27), we tested the MIPE agents in a TNF-α–stimulated NF-κB–based β-lactamase reporter assay using an engineered ME-180 cervical carcinoma line (28). Representative dose–response curves from these various assays are shown in Fig. 1C for the cyclin-dependent kinase (CDK) inhibitor PHA-793887 (29).
Fig. 1.

(A) Heatmap representation of MIPE library activity derived from qHTS of a previously reported (28) NF-κB assay in agonist (increased transcriptional response) and antagonist (decreased transcriptional response) mode and cell viability (percent cells remaining); control viability in hMSC, TMD8, and HBL1 lines; apoptotic response in TMD8 cells at 8 and 16 h. Red represents inhibitory response; green represents activation response; color intensity represents potency (stronger intensity means lower EC50 value). (B) Expansion of these data for selected agents of interest. (C) Complete response curves from these data sets for the CDK 1–5 inhibitor PHA-793887. [○, NF-κB activation; x, NF-κB viability; □, hMSC cell viability; ■, TMD8 cell viability; ▲, TMD8 apoptotic response (8 h); △, TMD8 apoptotic response (16 h)].
The data from these assays confirmed many known pharmacologic aspects of MIPE library agents, including ibrutinib, which exhibited single-agent toxicity below 100 nM for TMD8 cells, confirming previous results (10, 12). As expected for an oncology-focused library, many MIPE agents were toxic as single agents for TMD8 cells. Several classes of agents inhibited the transcriptional induction of the NF-κB reporter, including known NF-κB modulators and inhibitors of HSP90, the proteasome, and CDKs (Fig. 1B and SI Appendix, Figs. S2–S5) (30–32). In many cases, the mechanisms behind these effects are clear: HSP90 inhibitors reduce the activity of IκB kinase (IKK), the key regulatory kinase in the NF-κB pathway; proteasome inhibitors prevent the degradation of IκBα, a negative regulator of NF-κB; and pan-CDK inhibitors affect the NF-κB reporter by targeting CDK9, an integral component of the transcriptional elongation complex pTEFb. For some agents, such as withaferin A and CDDO-Me, the mechanistic basis for NF-κB blockade is less clear (Fig. 1C and SI Appendix, Fig. S6) (33, 34).
The data from the 6 × 6 discovery screen of ibrutinib versus the MIPE library revealed many examples of synergy and additivity in ABC DLBCL viability assays (data are freely available at http://tripod.nih.gov/matrix-data/m3-btk-6x6-ls). Assignment of individual drug combinations as synergistic, additive, or antagonistic is governed by the model used (Excess HSA vs. Beta, for example). Here, we label combinations with a γ-value <0.95 as synergistic (see SI Appendix for a detailed description). Various drug classes were well represented among the combinations with the best synergistic scores, including agents targeting the PI3K pathway, as well as standard chemotherapeutic agents, as discussed below. In addition, significant interactions were identified with individual agents whose mechanism of action provides potential insight into ABC DLBCL biology. For example, strong synergy was observed with navitoclax, which targets the anti-apoptotic protein BCL2 that is highly expressed in all ABC DLBCL tumors and increased in expression by focal genomic amplifications in ∼8% of cases (35, 36).
Ibrutinib Combinations Involving PI3K Signaling Modulators.
A striking number of cooperative interactions between ibrutinib and inhibitors of the PI3K signaling pathway were observed. Previous reports demonstrated that ABC DLBCL cells have constitutive PI3K signaling that supports their viability (10, 37). In the 6 × 6 discovery screen, 12 agents reported to inhibit various PI3K isoforms interacted favorably with ibrutinib (serials 102–113), including CAL-101 (idelalisib), BKM-120, and the dual PI3K/mammalian target of rapamycin (mTOR) inhibitors BEZ-235 and GDC-0980 (38–41). The allosteric AKT inhibitor MK-2206 and the clinically approved mTORC1 inhibitor everolimus also demonstrated synergy at selected concentrations (42, 43). The chronic active BCR signaling in ABC DLBCL cells activates the kinase SYK, which is known to engage the PI3K pathway (10). Accordingly, the SYK inhibitor PRT-060318 (44) cooperated with ibrutinib in killing ABC DLBCL cells.
Combinations of ibrutinib with various PI3K pathway inhibitors (MK-2206, CAL-101, BKM-120, BEZ-235, GDC-0941, GDC-0980, everolimus, PRT-060318) were confirmed to have synergistic/additive effects on ABC DLBCL viability in 10 × 10 matrix studies (http://tripod.nih.gov/matrix-data/btk-10x10-ctg-48hr/) (Fig. 2A and SI Appendix, Fig. S7). Representative results are shown for BKM-120, a pan-class I PI3K inhibitor that is currently in phase II clinical trials (Fig. 2 A–D). One method of data visualization is a heat map indicating the degree of toxicity for each of the combinations in the 10 × 10 matrix (Fig. 2A). An isobologram representation shows that ibrutinib and BKM-120 cooperate in killing TMD8 cells in certain dose ranges of each drug (Fig. 2B and SI Appendix, Fig. S16).
Fig. 2.
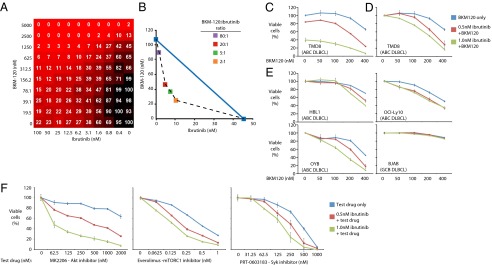
The combination responses for ibrutinib and BKM-120 as judged by: (A) 10 × 10 matrix block experiment informing on TMD8 viability as judged by CellTiter Glo in a 1,536-well plate; (B) an isobologram analysis (SI Appendix). (C and D) Single-agent and combination responses reported by an MTS viability assay in TMD8 cells. See text for details. (E) Single-agent and combination MTS viability assays conducted in the indicated cell lines, plotted as in D. (F) single-agent and combination responses informing on TMD8 viability for combinations of ibrutinib plus specific inhibitors of the PI3K-AKT-mTOR signaling cascade including MK-2206, everolimus, and PRT-0603183, as judged by MTS response in 96-well plates.
We conducted confirmatory MTS viability assays investigating the combination of ibrutinib with BKM-120 in several ABC DLBCL lines. Two methods were used to depict the cooperative effects of drug combinations. Data were normalized either to cells growing in the absence of either drug (Fig. 2C) or to the signal obtained with the specified concentration of ibrutinib alone (Fig. 2D). Left-curve shifts in the former method indicate superadditive effects of the combination. The BKM-120–ibrutinib combination produced cooperative toxicity in the TMD8, OCI-Ly10, and OYB ABC DLBCL lines, but had less activity in the HBL1 ABC DLBCL line and no combination activity in the GCB DLBCL line BJAB (Fig. 2 D and E). This viability assay also confirmed good responses for the combinations of ibrutinib with MK-2206, everolimus, and PRT-060318 in most ABC DLBCL lines, although HBL1 had less combination activity for MK-2206 and everolimus (Fig. 2F and SI Appendix, Fig. S7). Importantly, the favorable interactions of ibrutinib with BKM-120, MK-2206, and everolimus were noted at concentrations that can be achieved by each agent in a clinical setting.
Ibrutinib Combinations with Bcl-2 Family Inhibitors.
Our screen revealed strong cooperation between ibrutinib and the Bcl-2 family inhibitor, navitoclax (ABT-263), in killing ABC DLBCL cells. Bcl-2 family members regulate apoptosis and are frequently deregulated in lymphoma. In particular, BCL2 is the target of translocations and amplifications in many B-cell lymphomas. Navitoclax specifically targets the prosurvival Bcl-2 family members Bcl-2, Bcl-xL, and Bcl-w (45). Results from the 6 × 6 discovery screen revealed excellent combination activity of ibrutinib plus navitoclax (Fig. 3A). This synergy was confirmed by MTS viability assays in the ABC DLBCL cell lines TMD8 and HBL1 (Fig. 3B). Although navitoclax had promising preclinical results in solid tumors and hematological malignancies, a high incidence of thrombocytopenia (a result of inhibition of Bcl-xL in platelets) has limited its clinical promise (46). ABT-199 was developed to specifically target Bcl-2 but not Bcl-xL (46) and is currently in phase Ia/II clinical trials in chronic lymphocytic leukemia. We observed greater synergy between ibrutinib and ABT-199 than with navitoclax in all ABC DLBCL cell lines tested (Fig. 3C).
Fig. 3.
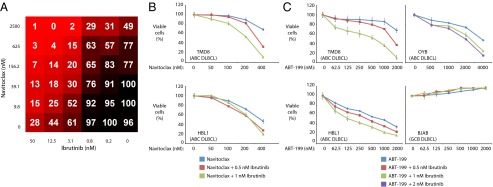
Viability of lymphoma cells treated with ibrutinib plus Bcl-2 family inhibitors. The combination responses for ibrutinib and navitoclax as judged by: (A) 6 × 6 matrix block evaluation of ibrutinib plus navitoclax in TMD8 cells; (B) MTS assay in 96-well plates of ibrutinib plus navitoclax in the indicated lines, plotted as in Fig. 2D; (C) MTS assay in 96-well plates of ibrutinib plus ABT-199 in the indicated lines, plotted as in Fig. 2D.
Ibrutinib Combinations with Common Chemotherapeutic Agents.
We observed significant enhancement of ibrutinib activity in combination with selected agents that are included in current therapeutic regimens for DLBCL. The combination regimen R-CHOP is the current standard of care for DLBCL, achieving a 43.5% 10-year overall survival benefit (1). The dose-adjusted regimen EPOCH-R (etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, rituximab) also has significant activity in DLBCL (47). The combination of ibrutinib with the topoisomerase II inhibitor doxorubicin was among the most impressive in the 6 × 6 and 10 × 10 matrix data (Fig. 4 A and B, and SI Appendix, Fig. S16). In addition, ibrutinib also acted synergistically/additively with other clinically available cytotoxic agents, including the DNA replication inhibitor gemcitabine and the microtubule depolymerization inhibitors docetaxel and plinabulin. Confirmatory viability assays revealed good combination responses in the ABC DLBCL line TMD8 with ibrutinib plus doxorubicin, etoposide, and dexamethasone (Fig. 4C and SI Appendix, Fig. S8). Ibrutinib did not influence the toxicity of these chemotherapy agents for GCB DLBCL cell lines. Taken together, these results suggest that inclusion of ibrutinib in the R-CHOP or EPOCH-R regimens could prove beneficial.
Fig. 4.
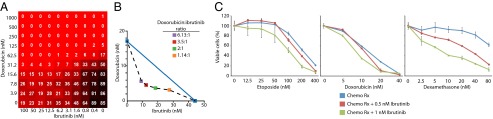
Viability of lymphoma cells treated with ibrutinib plus common chemotherapeutics. The combination responses for ibrutinib and doxorubicin as judged by: (A) 10 × 10 matrix block evaluation of ibrutinib plus doxorubicin in TMD8 cells, with viability judged by CellTiter Glo in a 1,536-well plate; (B) an isobologram analysis (see SI Appendix); (C) MTS assay in 96-well plates of ibrutinib plus the indicated chemotherapeutic agents in TMD8 cells, plotted as in Fig. 2D.
Discussion
Therapeutic options for cancers rely heavily on drug combinations, but the strategies to define effective combinations need to evolve. Empirical clinical trials are not optimal because only a limited number of drug combinations can be realistically evaluated. In the current report, we present experimental and technical details of an unbiased matrix screening platform that promises to accelerate our search for effective drug combinations. A key feature of this approach is the dense dose titration that is afforded by the high-throughput screening platform, which enables the discovery of dose ranges in which drugs synergize. Application of this platform to the BTK inhibitor ibrutinib demonstrated impressive combination activity with a variety of mechanistically distinct drug classes against cell line models of ABC DLBCL. Because ibrutinib is yielding impressive response rates in a variety of B-cell malignancies, including ABC DLBCL, several of the drug combinations described here should be tested in clinical trials (48–50). Current clinical trials of ibrutinib in DLBCL and other B-cell malignancies have shown this agent to be very well tolerated, with toxicity occurring in a small fraction of patients and generally limited to low-grade (≤2) diarrhea, fatigue, and upper respiratory tract infections (49, 50). This safety profile, combined with the extraordinary pharmacodynamic profile of ibrutinib (full target occupancy maintained for 24 h after single-dose administration), allows considerable flexibility in conducting combination drug trials.
Striking combination responses with ibrutinib were observed with multiple inhibitors of the PI3K pathway, including agents targeting the PI3K catalytic subunit, AKT, and the mTORC1 complex. PI3K pathway synergism/additivity is consistent with our current mechanistic understanding of pathological signaling in ABC DLBCL. BTK is a key signaling intermediate between the BCR and IKK, thereby engaging the potent anti-apoptotic NF-κB pathway (13). The PI3K pathway is a parallel prosurvival mechanism, potentially explaining the synergy between ibrutinib and PI3K pathway inhibitors that we observed. Indeed, previous work showed that the mTORC1 inhibitor rapamycin cooperated with an inhibitor of IKKβ in killing ABC DLBCL cells (10). In a subset of ABC DLBCL lines, it has been suggested that the PI3K pathway controls NF-κB, potentially through the action of PDK1 (37). The proposed cross-talk between the PI3K pathway and the NF-κB pathway was not affected by an AKT inhibitor (37). In contrast, we observed cooperativity between ibrutinib and inhibitors of AKT and mTORC1, suggesting that the PI3K pathway promotes the survival of ABC DLBCL cells by mechanisms beyond regulating NF-κB.
The present study demonstrates a favorable interaction between ibrutinib and the BCL2 family antagonists navitoclax and ABT-199, which have yielded single-agent activity in B-cell malignancies (51). The exact mechanism behind this combination activity is unclear. One possibility is that BCL2 family members function in a salvage pathway to keep ABC DLBCL cell lines alive once ibrutinib treatment diminishes their constitutive anti-apoptotic NF-κB activity. Although BCL2 translocations are rare or absent in ABC DLBCL, copy number amplification of the BCL2 locus is more common in ABC DLBCL (36) and high expression of BCL2 is a hallmark of this DLBCL subtype (35). The fact that ABT-199 had more combination activity with ibrutinib than navitoclax suggests that BCL2 plays a more central prosurvival function in ABC DLBCL than other BCL2 family members. Our data support future clinical investigations of the ibrutinib–ABT-199 combination in ABC DLBCL, especially given the favorable safety profiles of both drugs.
Also promising are the combinations of ibrutinib with cytotoxic chemotherapeutic agents, including several components of the R-CHOP and EPOCH-R regimens that are currently used to treat DLBCL. Previous work demonstrated that DNA damage activates NF-κB (52) and that inhibition of NF-κB sensitizes cancer cells to undergo apoptosis when treated with DNA-damaging chemotherapeutic agents. Hence, the ability of ibrutinib to inhibit NF-κB in ABC DLBCL provides a mechanistic explanation for its cooperativity with chemotherapeutic agents. These results support the initiation of trials in ABC DLBCL comparing R-CHOP with and without ibrutinib.
Ibrutinib also combines well with other agents with distinct mechanisms of action. For example, lenalidomide potentiates the toxic effects of ibrutinib for ABC DLBCL cells by simultaneously inhibiting CARD11-dependent NF-κB signaling and inducing the secretion of type I interferon, which is itself cytotoxic (13). Given the synergy between ibrutinib and multiple agents uncovered thus far, it may eventually be possible to combine three or more agents to overcome the aggressive nature of ABC DLBCL. Given the large number of potential combinations, extensive preclinical development of mechanism-based combinations will be needed to identify those with the greatest efficacy and potential safety. Based on the test case of ibrutinib, we are excited by the prospect of matrix drug screening to uncover actionable drug combinations in cancer and for other diseases. The high-throughput nature of the platform allows a scale and reproducibility of experimentation that cannot be achieved by conventional methods. We hope that the methods and software that we provide in this report will enable researchers to identify drug combinations for a variety of human cancer subtypes.
Methods
HTS Viability and Apoptosis Assays.
For each cell line tested, a total of 1,000 cells per well in 5 μL of media were dispensed using a Multidrop Combi dispenser (Thermo Fisher Scientific) and a small cassette into barcoded 1,536 solid-bottom white Greiner Bio-one tissue culture-treated plates (catalog #789173-F). Standard RPMI -1640 supplemented with 5% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin, and 2 mM glutamine was used (Gibco). For the generation of standard 11-point dose–response curves, the cells were plated, followed by the immediate pintool addition of 23 nL of control compound (bortezomib) and library compounds using a Kalypsys pintool. For preplated matrix plates, the cells were added directly to the plates immediately after compounds were acoustically dispensed using an ATS-100 (EDC Biosystems). The plates were then covered with stainless steel cell culture Kalypsys lids and incubated at 37 °C with 5% CO2 under 95% humidity. For cell proliferation assays, the cells were incubated for 48 h and then 3 μL of CellTiter Glo luminescent cell viability assay reagent (Promega) was added using a Bioraptor Flying Reagent Dispenser (Aurora Discovery-BD). The plates were then incubated for 15 min at room temperature. The signal was measured using a 10-s exposure with a ViewLux (Perkin-Elmer) with a luminescent filter. For apoptosis assays, the cells were incubated at either 8 or 16 h and then 3 μL of Caspase Glo 3/7 luminescent apoptosis assay reagent (Promega) was added using the same method described above. Relative luminescence units (RLU) for each well were normalized to the median RLUs from the DMSO control wells as 100% viability or 0% caspase activation, and median RLUs from the bortezomib control wells as 0% viability or 100% caspase activation.
NF-κB Assay.
CellSensor NF-κB-bla ME180 cell line (NF-κB-bla cells) were obtained from Invitrogen. Cells were cultured in High Glucose GlutaMAX DMEM supplemented with 10% dialyzed FBS, 0.1 mM nonessential amino acids, 25 mM Hepes (pH 7.3), 100 U/mL penicillin and 100 μg/mL streptomycin, and 5 μg/mL of blasticidin. The cultures were maintained in a 37 °C incubator with 5% CO2 and under at 95% humidity. The NF-κB-bla cells were suspended in OPTI-MEM medium supplemented with 0.5% dialyzed FBS, 0.1 mM nonessential amino acids, 25 mM sodium pyruvate, 100 U/mL penicillin, and 100 μg/mL streptomycin. Compounds (20 nL, final concentration range from 2.32 nM to 91.6 µM agonist mode; 1.94 nM to 76.4 µM antagonist mode; 2.90 nM to 114 µM viability mode) and positive control titration (MG132, final concentration range 15.2 nM to 33.2 µM) were transferred to a 1,536-well black wall/clear-bottom plates (Greiner Bio-one) using the ATS Acoustic Liquid Dispenser. Cells were then dispensed at 2,000 cells per 5 μL per well using a Thermo Scientific Multidrop Combi. Cells were incubated (37 °C) 15 min, followed by addition of either 1 µL assay buffer with or without TNF-α (final concentration 1 ng/mL). The plates were incubated for 5 h at 37 °C, then 1 µL of LiveBLAzer B/G FRET substrate (Invitrogen) detection mix was added and the plates incubated at room temperature for 2 h. Fluorescence intensity (405-nm excitation, 460- and 530-nm emission) was measured using an Envision plate reader (Perkin-Elmer). Data were expressed as the ratio of 460 nm/530 nm fluorescence emissions.
MTS Viability Assay.
Each DLBCL cell line (TMD8, OCI-Ly10, OYB, HBL1, and BJAB) were cultured in triplicate in 96-well plates (∼5,000 cells per well) in the presence of vehicle or the indicated drug. On day 3, each well was replenished with freshly prepared vehicle or drug dissolved in 50 μL of culture medium. On day 5, cell viability was assayed by adding 10 μL per well of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulphophenyl)-2H tetrazolium and an electron coupling reagent (phenazine methosulfate; Promega) followed by a 2-h incubation at 37 °C and measured by the amount of absorbance (492 nm) using a 96-well plate reader (SpectraMax 250 Microplate Reader). Background was subtracted using the medium-only control wells.
Supplementary Material
Acknowledgments
We thank Drs. Jack Taunton, Shaomeng Wang, Fed Bernal, James E. Bradner, Benjamin F. Cravatt, Daniel K. Nomura, Mark Tebbe, and Kenneth Bair for kind donations to the MIPE compound library. This work was supported by the Division of Preclinical Innovation, National Center for Advancing Translational Sciences; the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research; the Intramural Research Program of the National Human Genome Research Institute; and the Intramural Research Program of the National Cancer Institute, Center for Cancer Research and the Frederick National Laboratory for Cancer Research, National Institutes of Health, including Contract HHSN261200800001E and Grant U54CA143930.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: All single-agent data reported in this paper has been deposited in the PubChem database, http://pubchem.ncbi.nlm.nih.gov (summary AID 651556).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1311846111/-/DCSupplemental.
References
- 1.Coiffier B, et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: A study by the Groupe d’Etudes des Lymphomes de l’Adulte. Blood. 2010;116(12):2040–2045. doi: 10.1182/blood-2010-03-276246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol. 2012;30(7):679–692. doi: 10.1038/nbt.2284. [DOI] [PubMed] [Google Scholar]
- 3.Kummar S, et al. Utilizing targeted cancer therapeutic agents in combination: Novel approaches and urgent requirements. Nat Rev Drug Discov. 2010;9(11):843–856. doi: 10.1038/nrd3216. [DOI] [PubMed] [Google Scholar]
- 4.Barretina J, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garnett MJ, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483(7391):570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood. 2008;112(13):4808–4817. doi: 10.1182/blood-2008-07-077958. [DOI] [PubMed] [Google Scholar]
- 7.Flaherty KT, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. US Food and Drug Administration (2010) Guidance for Industry: Codevelopment of Two or More New Investigational Drugs for Use in Combination. Available at www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM236669.pdf. Accessed June 2013.
- 9.Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat Rev Drug Discov. 2013;12(3):229–243. doi: 10.1038/nrd3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis RE, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463(7277):88–92. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lenz G, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008;319(5870):1676–1679. doi: 10.1126/science.1153629. [DOI] [PubMed] [Google Scholar]
- 12.Ngo VN, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470(7332):115–119. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Y, et al. Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell. 2012;21(6):723–737. doi: 10.1016/j.ccr.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lam LT, et al. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-κB pathways in subtypes of diffuse large B-cell lymphoma. Blood. 2008;111(7):3701–3713. doi: 10.1182/blood-2007-09-111948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borisy AA, et al. Systematic discovery of multicomponent therapeutics. Proc Natl Acad Sci USA. 2003;100(13):7977–7982. doi: 10.1073/pnas.1337088100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Small BG, et al. Efficient discovery of anti-inflammatory small-molecule combinations using evolutionary computing. Nat Chem Biol. 2011;7(12):902–908. doi: 10.1038/nchembio.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lehár J, et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol. 2009;27(7):659–666. doi: 10.1038/nbt.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lehár J, Stockwell BR, Giaever G, Nislow C. Combination chemical genetics. Nat Chem Biol. 2008;4(11):674–681. doi: 10.1038/nchembio.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellson R, et al. Transfer of low nanoliter volumes between microplates using focused acoustics—Automation considerations. J Lab Auto. 2003;8(3):29–34. [Google Scholar]
- 20.Bliss CI. The toxicity of poisons applied jointly. Ann Appl Biol. 1939;26:585–615. [Google Scholar]
- 21.Grabovsky Y, Tallarida RJ. Isobolographic analysis for combinations of a full and partial agonist: Curved isoboles. J Pharmacol Exp Ther. 2004;310(3):981–986. doi: 10.1124/jpet.104.067264. [DOI] [PubMed] [Google Scholar]
- 22.Lehár J, et al. Chemical combination effects predict connectivity in biological systems. Mol Syst Biol. 2007;3:80. doi: 10.1038/msb4100116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cokol M, et al. Systematic exploration of synergistic drug pairs. Mol Syst Biol. 2011;7:544. doi: 10.1038/msb.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. 2001;194(12):1861–1874. doi: 10.1084/jem.194.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosenwald A, Staudt LM. Gene expression profiling of diffuse large B-cell lymphoma. Leuk Lymphoma. 2003;44(Suppl 3):S41–S47. doi: 10.1080/10428190310001623775. [DOI] [PubMed] [Google Scholar]
- 26.Compagno M, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459(7247):717–721. doi: 10.1038/nature07968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol. 2010;2(6):a000109. doi: 10.1101/cshperspect.a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller SC, et al. Identification of known drugs that act as inhibitors of NF-kappaB signaling and their mechanism of action. Biochem Pharmacol. 2010;79(9):1272–1280. doi: 10.1016/j.bcp.2009.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alzani R, et al. Therapeutic efficacy of the pan-cdk inhibitor PHA-793887 in vitro and in vivo in engraftment and high-burden leukemia models. Exp Hematol. 2010;38(4):259–269, e2. doi: 10.1016/j.exphem.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 30.Qing G, Yan P, Xiao G. Hsp90 inhibition results in autophagy-mediated proteasome-independent degradation of IkappaB kinase (IKK) Cell Res. 2006;16(11):895–901. doi: 10.1038/sj.cr.7310109. [DOI] [PubMed] [Google Scholar]
- 31.Scheinman RI, Cogswell PC, Lofquist AK, Baldwin AS., Jr Role of transcriptional activation of I kappa B alpha in mediation of immunosuppression by glucocorticoids. Science. 1995;270(5234):283–286. doi: 10.1126/science.270.5234.283. [DOI] [PubMed] [Google Scholar]
- 32.Jane EP, Premkumar DR, Pollack IF. Bortezomib sensitizes malignant human glioma cells to TRAIL, mediated by inhibition of the NF-kappaB signaling pathway. Mol Cancer Ther. 2011;10(1):198–208. doi: 10.1158/1535-7163.MCT-10-0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahmad R, Raina D, Meyer C, Kharbanda S, Kufe D. Triterpenoid CDDO-Me blocks the NF-kappaB pathway by direct inhibition of IKKbeta on Cys-179. J Biol Chem. 2006;281(47):35764–35769. doi: 10.1074/jbc.M607160200. [DOI] [PubMed] [Google Scholar]
- 34.Kaileh M, et al. Withaferin a strongly elicits IkappaB kinase beta hyperphosphorylation concomitant with potent inhibition of its kinase activity. J Biol Chem. 2007;282(7):4253–4264. doi: 10.1074/jbc.M606728200. [DOI] [PubMed] [Google Scholar]
- 35.Alizadeh AA, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 36.Lenz G, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci USA. 2008;105(36):13520–13525. doi: 10.1073/pnas.0804295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kloo B, et al. Critical role of PI3K signaling for NF-kappaB-dependent survival in a subset of activated B-cell-like diffuse large B-cell lymphoma cells. Proc Natl Acad Sci USA. 2011;108(1):272–277. doi: 10.1073/pnas.1008969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lannutti BJ, et al. CAL-101, a p110δ selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591–594. doi: 10.1182/blood-2010-03-275305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maira S-M, et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther. 2012;11(2):317–328. doi: 10.1158/1535-7163.MCT-11-0474. [DOI] [PubMed] [Google Scholar]
- 40.Maira S-M, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7(7):1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 41.Wallin JJ, et al. GDC-0980 is a novel class I PI3K/mTOR kinase inhibitor with robust activity in cancer models driven by the PI3K pathway. Mol Cancer Ther. 2011;10(12):2426–2436. doi: 10.1158/1535-7163.MCT-11-0446. [DOI] [PubMed] [Google Scholar]
- 42. Hirai H, et al. (2010) MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 9(7):1956–1967. [DOI] [PubMed]
- 43.Atkins MB, Yasothan U, Kirkpatrick P. Everolimus. Nat Rev Drug Discov. 2009;8(7):535–536. doi: 10.1038/nrd2924. [DOI] [PubMed] [Google Scholar]
- 44.Hoellenriegel J, et al. Selective, novel spleen tyrosine kinase (Syk) inhibitors suppress chronic lymphocytic leukemia B-cell activation and migration. Leukemia. 2012;26(7):1576–1583. doi: 10.1038/leu.2012.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oltersdorf T, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 46.Souers AJ, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19(2):202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 47.Wilson WH, et al. Cancer Leukemia Group B A Cancer and Leukemia Group B multi-center study of DA-EPOCH-rituximab in untreated diffuse large B-cell lymphoma with analysis of outcome by molecular subtype. Haematologica. 2012;97(5):758–765. doi: 10.3324/haematol.2011.056531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Advani RH, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B cell malignancies. J Clin Oncol. 2012;42:7906–7915. doi: 10.1200/JCO.2012.42.7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilson WH, et al. The Bruton’s tyrosine kinase (BTK) inhibitor, ibrutinib (PCI-32765), has preferential activity in the ABC subtype of relapsed/refractory de novo diffuse large B cell lymphoma (DLBCL): Interim results of a multicenter, open-label, phase 2 study. 54th Annual Meeting and Exposition. December 2012 (American Society of Hematology, Atlanta, GA), abstr 686. [Google Scholar]
- 50. Byrd JC, et al. (2012) The Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib (PCI-32765) promotes high response rate, durable remissions, and is tolerable in treatment naïve (TN) and relapsed or refractory (RR) chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) patients including patients with high-risk (HR) disease: New and updated results of 116 patients in a phase Ib/II study. 54th Annual Meeting and Exposition, December 2012 (American Society of Hematology, Atlanta, GA), abstr 189.
- 51.Wilson WH, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11(12):1149–1159. doi: 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McCool KW, Miyamoto S. DNA damage-dependent NF-κB activation: NEMO turns nuclear signaling inside out. Immunol Rev. 2012;246(1):311–326. doi: 10.1111/j.1600-065X.2012.01101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.