

Abstract
Staphylococcus aureus is responsible for a variety of human infections, including life-threatening, systemic conditions. Secreted proteome, including a range of proteases, constitutes the major virulence factor of the bacterium. However, the functions of individual enzymes, in particular SplA protease, remain poorly characterized. Here, we report development of specific inhibitors of SplA protease. The design, synthesis, and activity of a series of α-aminoalkylphosphonate diaryl esters and their peptidyl derivatives are described. Potent inhibitors of SplA are reported, which may facilitate future investigation of physiological function of the protease. The binding modes of the high-affinity compounds Cbz-PheP-(OC6H4−4-SO2CH3)2 and Suc-Val-Pro-PheP-(OC6H5)2 are revealed by high-resolution crystal structures of complexes with the protease. Surprisingly, the binding mode of both compounds deviates from previously characterized canonical interaction of α-aminoalkylphosphonate peptidyl derivatives and family S1 serine proteases.
Keywords: Staphylococcus aureus, SplA protease, protease inhibitor, α-aminoalkylphosphonate
Introduction
Staphylococcus aureus is a versatile human pathogen responsible for a variety of infections, ranging from minor superficial skin lesions to life-threatening systemic conditions.1 The virulence of S. aureus relies on a number of extracellular factors, including secreted proteases. Evidence collected since the early 1970s supports various roles of aureolysin, staphopains, V8 protease, and epidermolytic toxins (ETs) in infection by S. aureus.2 Much less is known about six family S13 serine proteases encoded in an spl operon, yet the location of the spl operon on a staphylococcal pathogenicity island vSaβ among well-established virulence factors4 suggests possible role in pathogenesis.
Specific inhibitors are valuable tools in assessing physiological functions of target enzymes. Selective protease inhibitors are often obtained by grafting an inhibitory function on a peptide scaffold of known substrates. The substrate preference of SplA was previously thoroughly characterized. Screening of libraries of tetrapeptide fluorescent substrates demonstrated exclusive preference for tyrosine and phenylalanine at P1 (notation according to Ref.5). In turn, evaluation of a large combinatorial protein library demonstrated extended preference for substrates with a consensus sequence of (W/Y)-L-Y*(T/S) (asterisks indicates the site of hydrolysis).6 Limited substrate specificity of SplA suggests that it should be possible to develop specific peptidyl inhibitors by using α-aminoalkylphosphonate diaryl ester inhibitory function.
Peptidyl derivatives of α-aminoalkylphosphonate diaryl esters are irreversible serine protease inhibitors. Nucleophilic attack by the active-site serine hydroxyl oxygen on a moderately electrophilic phosphorus atom leads to the formation of a phosphonate ester. The initial enzyme–inhibitor complex is unstable. Thereafter, slow hydrolysis of the aryl ester (with a half-life ranging from few hours to few days) leads to the formation of a so-called aged complex (Supporting Information Fig. S1). Known crystal structures of aged complexes of family S1 serine proteases with phosphonate inhibitors demonstrate a tetrahedral geometry of the phosphorus atom, which resembles the transition state observed during peptide-bond hydrolysis, while interaction of the peptidyl part is reminiscent of a binding mode of a substrate.7–8
In this work, we set out to develop specific inhibitors of SplA protease to provide efficient tools for future characterization of its physiological role. We synthesized multiple peptidyl phosphonate derivatives designed, among others, based on the sequences of efficiently recognized substrates and determined their effects on SplA activity. Crystal structures of two of the most efficient inhibitors were solved in complex with SplA, which demonstrate unusual mode of binding, unlike that observed for canonical inhibitors complexed with family S1 proteases.
Results
Initial assessment of phosphonate scaffold for SplA inhibitor development
Simple Cbz-protected phosphonic analogs of amino acids often show considerable selectivity for particular serine proteases according to their P1 preference.9 Our preliminary evaluation of this assumption and investigation of the influence of different substituents at the phenyl ester rings by evaluating the activity of multiple available inhibitors against SplA protease indicated that none of the alanine, valine, valine-related, methionine, lysine, arginine, or arginine-related phosphonate derivatives inhibited the enzyme when used at a concentration of 200 μM (1:100 enzyme:inhibitor molar ratio). At the same time, two (30 and 31) of the six phenylalanine derivatives tested inhibited the activity of the enzyme (Table1 and Fig. 1). This is in agreement with the previously determined substrate specificity of SplA, which will accept Phe at the P1 subsite despite a strong preference for Tyr. What is more, one of the six leucine derivatives tested (19) also inhibited the enzyme. This is surprising, given that such specificity was not observed in earlier substrate preference studies.6 Nevertheless, the activity of 19 may be explained by the hydrophobic nature of the leucine side chain, which has a hydrophobicity roughly comparable to that of the benzyl group of phenylalanine. In addition, 19 is a strongly activated compound owing to the electron-withdrawing substituents at para position of the ester rings. The same is true for the active derivatives of phenylalanine (30 and 31), where potency is increased by the electron-withdrawing properties of ester ring substituents. Overall, this confirms that the phosphonate scaffold is adequate for the development of selective SplA inhibitors and that inhibitor potency can be adjusted not only at the peptidyl side but also by varying the ester substituents on phosphorus atom.
Table 1.
Phosphonate Inhibitors Tested in This Study and Their Affinity for SplA Protease
| # | Compound structure | Ki (µM) | k2/Ki (M−1 s−1) | Reference (synthesis) |
|---|---|---|---|---|
| 1 | Cbz-(4-GuPhg)P-(OC6H5)2*TFA | >200 | <3 | 10 |
| 2 | Cbz-(4-GuPhg)P-(OC6H4−4-S-CH3)2*TFA | >200 | <3 | 11 |
| 3 | Cbz-(4-GuPhg)P-(OC6H4−4-SO2-CH3)2*TFA | >200 | <3 | 11 |
| 4 | Cbz-(4-GuPhg)P-(OC6H4−4-Cl)2*TFA | >200 | <3 | 11 |
| 5 | Cbz-(HomoArg)P-(OC6H5)2*HCl | >200 | <3 | 8 |
| 6 | Cbz-ArgP-(OC6H5)2*HCl | >200 | <3 | 8 |
| 7 | Cbz-LysP-(OC6H5)2*HCl | >200 | <3 | 12 |
| 8 | Cbz-MetP-(OC6H5)2 | >200 | <3 | 13 |
| 9 | Cbz-MetP-(OC6H4−4-S-CH3)2 | >200 | <3 | 13 |
| 10 | Cbz-AlaP-(OC6H5)2 | >200 | <3 | 14 |
| 11 | Cbz-AlaP-(OC6H4−4-S-CH3)2 | >200 | <3 | 14 |
| 12 | Cbz-ValP-(OC6H5)2 | >200 | <3 | 14 |
| 13 | Cbz-ValP-(OC6H4−4-S-CH3)2 | >200 | <3 | 14 |
| 14 | Cbz-ValP-(OC6H4−4-SO2-CH3)2 | >200 | <3 | 14 |
| 15 | Cbz-nValP-(OC6H5)2 | >200 | <3 | 14 |
| 16 | Cbz-nValP-(OC6H4−4-S-CH3)2 | >200 | <3 | 14 |
| 17 | Cbz-LeuP-(OC6H5)2 | >200 | <3 | 15 |
| 18 | Cbz-LeuP-(OC6H4−4-S-CH3)2 | >200 | <3 | 15 |
| 19 | Cbz-LeuP-(OC6H4−4-SO2-CH3)2 | 3.8 | 4*103 | This study |
| 20 | Cbz-LeuP-(OC6H4−4-O-CH3)2 | >200 | <3 | 15 |
| 21 | Cbz-LeuP-(OC6H4−4-Cl)2 | >200 | <3 | 15 |
| 22 | Cbz-LeuP-(OC6H4−4-CH(CH3))2 | >200 | <3 | 15 |
| 23 | Boc-Val-Pro-LeuP-(OC6H5)2 | 14 | 21 | This study |
| 24 | Boc-Val-Pro-LeuP-(OC6H4−4-S-CH3)2 | 9.9 | 222 | This study |
| 25 | Boc-Val-Pro-LeuP-(OC6H4−4-SO2-CH3)2 | <0.2 | >8*103 | This study |
| 26 | Boc-Val-Pro-LeuP-(OC6H4−4-O-CH3)2 | 17 | 60 | This study |
| 27 | Boc-Val-Pro-LeuP-(OC6H4−4-Cl)2 | 6.1 | 871 | This study |
| 28 | Boc-Val-Pro-LeuP-(OC6H4−4-CH(CH3)2)2 | >200 | <3 | This study |
| 29 | Cbz-PheP-(OC6H5)2 | >200 | <3 | 15 |
| 30 | Cbz-PheP-(OC6H4−4-S-CH3)2 | 83 | 25 | 15 |
| 31 | Cbz-PheP-(OC6H4−4-SO2-CH3)2 | 3.5 | 2.3*103 | 15 |
| 32 | Cbz-PheP-(OC6H4−4-O-CH3)2 | >200 | <3 | 15 |
| 33 | Cbz-PheP-(OC6H4−4-Cl)2 | >200 | <3 | 15 |
| 34 | Cbz-PheP-(OC6H4−4-CH(CH3)2)2 | >200 | <3 | 15 |
| 35 | Boc-Val-Pro-PheP-(OC6H5)2 | 2.3 | 2.5*103 | This study |
| 36 | Boc-Val-Pro-PheP-(OC6H4−4-S-CH3)2 | 2.5 | 7*103 | This study |
| 37 | Boc-Val-Pro-PheP-(OC6H4−4-SO2-CH3)2 | <0.2 | >8*103 | This study |
| 38 | Boc-Val-Pro-PheP-(OC6H4−4-O-CH3)2 | 34 | 563 | This study |
| 39 | Boc-Val-Pro-PheP-(OC6H4−4-Cl)2 | 5.1 | 718 | This study |
| 40 | Boc-Val-Pro-PheP-(OC6H4−4-CH(CH3)2)2 | 10 | 371 | This study |
| 41 | Suc-Val-Pro-PheP-(OC6H5)2 | 10 | 1*103 | 13 |
| 42 | Boc-Phe-Leu-PheP-(OC6H5)2 | 31 | 88 | This study |
| 43 | Boc-Phe-Leu-PheP-(OC6H4−4-S-CH3)2 | 45 | 311 | This study |
| 44 | Boc-Phe-Leu-PheP-(OC6H4−4-SO2-CH3)2 | 5.1 | 830 | This study |
| 45 | Boc-Phe-Leu-PheP-(OC6H4−4-O-CH3)2 | >200 | <3 | This study |
| 46 | Boc-Phe-Leu-PheP-(OC6H4−4-CH(CH3)2)2 | >200 | <3 | This study |
Figure 1.
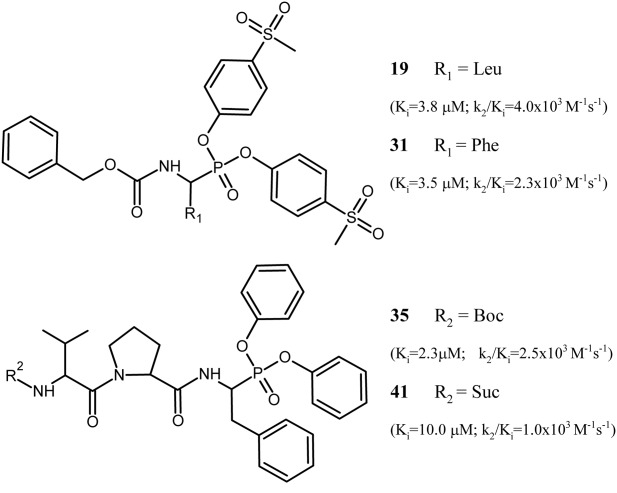
Structures and binding characteristics of selected SplA inhibitors used in the study. A full list of evaluated inhibitors and their affinities toward SplA is provided in Table1.
Optimization of phosphonate inhibitors of SplA protease
In general, extension of the peptidyl fragment of a phosphonate derivative improves the selectivity and potency of the inhibitor, by providing a larger surface of interaction with the enzyme.16 This was first tested using tripeptide derivatives of phosphonic leucine (compounds 23–28). A significant improvement over 17–22 was observed even though the peptidyl moiety selected was poorly compatible with the previously identified preferred substrates of SplA. Again, inhibitor potency depended on the electron-withdrawing properties of the ester ring substituents. Unfortunately, owing to this effect, the high reactivity and susceptibility to hydrolysis of the most potent derivatives of the series (25), as well as 31 of the previous series, render them of no practical value.
The modification of phosphonic phenylalanine analogs with peptide chains was especially interesting in light of the previously determined substrate preference of SplA.6 Although the presence of tyrosine at the P1 and P3 positions was strongly favored among substrates, Phe was also accepted at these positions and was used for construction of the inhibitors owing to the choice of synthetic reaction used. We evaluated the activity of five phosphonate derivatives of the FLF tripeptide with different substituents at the para positions of the ester rings (42–46). Surprisingly, the most potent derivative of this series (44) was less potent than the most potent derivative of the VPLP series (25), although the former was apparently a much better mimic of a preferred substrate. Therefore, we also examined known phosphonate inhibitors of chymotrypsin (35–41). Despite the failure of the peptidyl moiety to fulfill the substrate preference of SplA at the P2 position, the inhibitors of this series were among the most potent compounds tested in the study. The potency of the best VPFP inhibitor (37) was comparable to that of the best inhibitor of VPLP series (25); however, both were at least 10 times more potent than the best inhibitor of the FLFP series (44). To further address this issue, we evaluated the kinetics of hydrolysis of substrates based on the above-mentioned inhibitor sequences, namely Boc-Phe-Leu-Phe-AFC and Boc-Val-Pro-Phe-AFC. The respective KM values of 60 and 22 μM are roughly consistent with the different affinities of SplA for the two inhibitors.
In summary, we developed phosphonate peptide analogs that inhibit SplA with high potency. To pinpoint the structural determinants of SplA inhibitor specificity and explain the increased potency of inhibitors with extended peptidyl moieties, we next determined high-resolution crystal structures of SplA complexed with 31 and 41 (Fig. 1).
Overall crystal structure of SplA in complex with 31 and 41
The structures of SplA complexed with 31 and 41 were solved at 1.7 and 1.8 Å resolution, respectively (Supporting Information Table SI). The overall topology of all molecules contained in the asymmetric unit of each crystal is essentially identical (Supporting Information Table SIII). The average RMSD over all Cα atoms equals 0.90 ± 0.21 Å for the 31 complex (four molecules) and 1.04 Å for the 41 complex (two molecules). Furthermore, the overall structure of the protease after it is complexed with an inhibitor is essentially identical to that of the previously reported free SplA (PDB ID: 2W7S), with average RMSD calculated for all atoms and all molecules contained in the asymmetric unit of 0.63 ± 0.30 Å for 31 complex and 0.63 ± 0.27 Å for 41 complex. The arrangements of molecules in the crystal lattice of the 31 complex and the 2W7S structure are superimposable. This enables structural comparisons with minimal bias associated with crystal packing.
Electron density of the inhibitor is well defined in both reported structures. Given that no electron density accounts for the aryl ester moiety, the structures represent “aged” complexes. Both inhibitors bind to the active site of SplA and assume an overall orientation similar to that observed in the structure of cathepsin G complexed with Suc-Val-Pro-PheP-(OC6H5)2 (PDB ID: 1CGH17) and previously predicted for SplA by molecular modeling.6 A covalent bond between a phosphorus atom of the phosphonate group of the inhibitor and Oγ of catalytic triad Ser154, together with interactions in S1 pocket, anchor the inhibitor in the active site. However, a detailed analysis immediately demonstrates marked differences between the mode of binding involved in these interactions compared to the interactions by which canonical inhibitors and other known phosphonates bind at the active site.17–18
Despite multiple attempts, we were unable to obtain crystals of SplA in complex with 42, the closest mimetic of the consensus sequence substrate evaluated in this study. The premade complex crystallized readily after several days under many different conditions, although none of many solved structures had an electron density that accounted for the inhibitor, most probably owing to hydrolysis of the enzyme–inhibitor complex. We also attempted to soak premade SplA crystals with 42, but were not successful. Soaking of premade SplA crystals with 31 and 41 was also unsuccessful, most probably owing to a relatively large molecular weight of the inhibitors.
Binding of inhibitor 31
The entire molecule of 31 is clearly defined by electron density (Supporting Information Fig. S3). The distance between Oγ of the catalytic triad Ser154 and the phosphorus atom of the phosphonate group of the inhibitor (1.69 ± 0.03 Å) as well as the continuous electron density clearly indicate covalent binding. The phosphorus atom adopts a tetrahedral conformation, with both of the phosphonate oxygen atoms pointing away from the protease surface (Fig. 2, right panel). This is unexpected for a transition-state analog and not observed in the structures of complexes of phosphonate inhibitors with serine proteases of family S1, where one of the phosphonate oxygen atoms occupies the oxyanion hole. Instead, the oxyanion hole in the structure of 31 is occupied by a water molecule that is tightly stabilized by a nearly tetrahedral network of hydrogen bonds. Two hydrogen bonds are contributed by Gly152 and Ser154 backbone amides, and are characteristic for oxygen bound at an oxyanion hole. The water molecule, in turn, donates two hydrogen bonds to the carbonyl oxygen of Gly23 and the phosphonate oxygen of the inhibitor. Tight spatial constraint of the water molecule is further supported by its low B-factor, which roughly corresponds with the B-factors of surrounding residues. In all molecules contained in the asymmetric unit, the catalytic triad histidine side chain is rotated out of its canonical orientation. The second phosphonate oxygen of the inhibitor forms a hydrogen bond with Nε2 of the catalytic triad histidine (except molecule D), hydrogen bonds with adjacent water molecules, and a weak hydrogen bond with the backbone carbonyl oxygen of Tyr170. The side chain of the phenylalanine residue at the P1 position occupies a deep, relatively hydrophobic S1 pocket of the protease in an orientation roughly comparable to that predicted by previous modeling studies and found in the crystal structures of comparable inhibitors. However, unlike the situation for canonical inhibitors or substrates, only the (S)-diastereoisomer (corresponding to the d-amino acids in stereochemical terms) is observed in the complex despite the use of racemic inhibitor preparations. The conformation of an amide bond proceeding P1 residue and its interaction with the enzyme are also unlike any of those found in canonical inhibitors. The amide hydrogen, which forms a part of an antiparallel sheet in other known structures, points away from the protease molecule and is not involved in hydrogen bonding. In turn, the carbonyl oxygen of the inhibitor forms a hydrogen bond with the main-chain amide of Gly172. The aromatic ring of the Cbz-protective group is located on the surface of the protease. It associates with loop II via van der Waals interactions. The entire region in the vicinity of the Cbz group is dynamic, as exemplified by high B-factors and undefined electron density for parts of loop II. The conformation of this region is similar in molecules B, C, and D, but a different conformation is found in molecule A (Supporting Information Fig. S3). This demonstrates that besides intrinsic flexibility, crystal packing further affects the orientations of the benzyl moiety and loop II. Apart from the Cbz moiety, the conformation of the remaining part of the inhibitor and its interaction with the protease remain unaffected by crystal packaging, and is identical in all molecules contained in the asymmetric unit.
Figure 2.
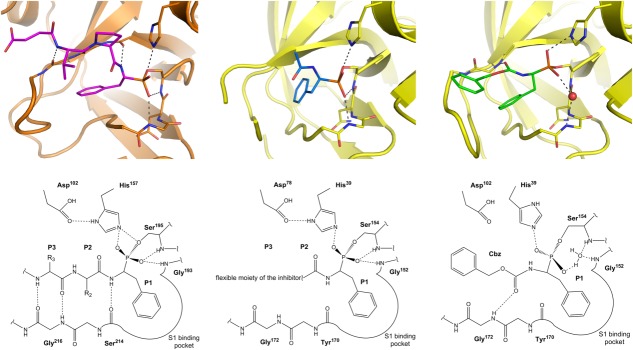
Comparison of phosphonate inhibitor binding mode to cathepsin G and SplA. In all cases the side chain of P1 residue resides in the pronounced S1 specificity pocket. (Left panel) Canonical binding mode of α-aminoalkylphosphonate inhibitors exemplified by the structure of Suc-Val-Pro-PheP-(OC6H5)2-bound cathepsin G (PDB ID: 1CGH). The peptidyl part of the inhibitor forms three canonical hydrogen bonds in the nonprimed sites of the protease active site. One of phosphonate oxygens is coordinated in the oxyanion hole. (Middle panel) In case of 41, one of phosphonate oxygens is canonically coordinated in the oxyanion hole but, the peptidyl part does not form any hydrogen bonds with the enzyme being flexible and undefined by electron density beyond P1 subsite. (Right panel) In case of 31 the oxyanion hole is occupied by a water molecule (red sphere) instead of a phosphonate oxygen. Furthermore, the peptidyl part forms a noncanonical hydrogen bond within the enzyme nonprimed side. Unlike in the cathepsin complex, both in case of 31 and 41 SplA selects (S)-diastereoisomer (corresponding to the d-amino acids) out of a racemic mixture. An interactive view is available in the electronic version of the article.
Binding of inhibitor 41
In the structure of the complex formed by Suc-Val-Pro-PheP-(OC6H5)2 (41) and SplA, only the phosphonate moiety, P1 phenylalanine residue, and Pro–Phe peptide bond are clearly defined by electron density (Supporting Information Fig. S3). Formation of a covalent bond between Oγ of the catalytic triad Ser154 and the phosphorus atom of the inhibitor is evident. Unlike 31, but exactly as expected for a transition-state analog and observed in the structures of other phosphonate inhibitors, one of the phosphonate oxygens occupies the oxyanion hole (Fig. 2, middle panel). It accepts canonical hydrogen bonds from the backbone amides of Gly152 and Ser154. The second phosphonate oxygen of the inhibitor is hydrogen bonded with Nε2 of the catalytic triad histidine (in canonical orientation contrary to 31 structure) and via a water molecule with the backbone carbonyl oxygen of Gly23. The phenylalanine side chain of 41 in the S1 enzyme pocket adopts an orientation comparable to that of the corresponding residue of 31, although a shift by roughly 1 Å and rather poor definition by electron density suggest some flexibility. Similar to 31 and unlike the known structures of canonical inhibitors, only the (S)-diastereoisomer is found in the complex structure, again despite the use of racemic mixtures for complex formation. Beyond P1 residue only the P1–P2 peptide bond is defined by electron density. Its orientation is again not reminiscent of canonical inhibitors. One of the two models contained in the asymmetric unit suggests that, unlike the situation for canonical inhibitors, the backbone carbonyl oxygen of the P2 residue of the inhibitor forms a hydrogen bond with a water molecule, which in turn accepts a hydrogen bond from the amide hydrogen of Gly172. There is no indication of such arrangement in the second molecule present in the asymmetric unit. Beyond the P1–P2 peptide bond, the remainder of the inhibitor (Suc-Val-Pro moiety) is not defined by electron density. This suggests that this part of the inhibitor is flexible and presumably does not interact with the enzyme or interacts only transiently. Such observation is again different from what one would expect based on analysis of multiple available crystal structures of serine protease inhibitor complexes. Moreover, it does not allow explaining the observed increase of affinity upon elongation of the peptidyl moiety of the inhibitor.
The S1 specificity pocket
Although the characterized structures demonstrate noncanonical binding mode of inhibitors, they allow some conclusions concerning the likely mechanism of substrate recognition by SplA at S1 subsite. The S1 specificity pocket of SplA is delineated by parts of loop I (Ala149–Ser154)19 and loop II (Ser173–Glu179), and strands Leu169–Gly172 and Lys180–Phe182. The cavity is lined with nonpolar side chains of Ala149, Pro151, and Leu169, the Cβ atoms of Asn153 and Ser178, the Cα atom of Gly172, and the main-chain atoms of the above-mentioned secondary structures, except for the main chain of Lys180–Phe182, which is buried and only stabilizes the pocket. Apart from Gly174 and Lys175, both of which are located at the outer rim of the pocket, all atoms of the main chain that are available for hydrogen bonding either participate in intramolecular hydrogen bonding or are located such that hydrogen bonds with the side chain of the P1 residue of the inhibitor (or substrate) are not feasible. Therefore, the surface of the S1 subsite is apolar, with the exception of the side chain of Asn181 located at the bottom of the cavity, where a single lone pair on Oδ1 is available for hydrogen bonding. The S1 cavity architecture is virtually identical to that previously described for apo-SplA,6 indicating that no significant enzyme rearrangement occurs upon inhibitor binding. The only small differences are seen in the arrangement of the otherwise flexible loop II.
Binding of the benzyl moieties of both of the inhibitors at the S1 subsite of SplA relies exclusively on hydrophobic interactions supported by oxygen–aromatic ring interaction. The aromatic ring fills the space of the cavity and is situated within van der Waals distance of multiple pocket atoms. In all molecules, the aromatic rings are almost coplanar because they are restrained by the pocket geometry (i.e., positioned by aromatic ring interaction with the side-chain oxygen of Asn181). Nonetheless, the orientations (rotation in the ring plane) of the phenylalanine side chain differ between the structures of 31 and 41. This is directly caused by the different arrangements of the phosphonate oxygens. Insertion of the phosphonate oxygen into the oxyanion hole in the structure of 41 pulls the phosphorus atom by more than 1 Å relative to 31. The positions of the Cα, Cβ, and Cγ atoms of the P1 residue differ by less than 1 Å, but the differences are sufficient to change the configuration of the bonds such that the shift in the position of the entire aromatic ring means that the positions of the Cζ atoms differ by ∼3 Å in the two most divergent molecules contained in the respective asymmetric units. Different orientations of the P1 residue slightly affect the conformation of Gly172 and adjacent residues. The described differences in P1 side-chain orientation observed in SplA complexes with 31 and 41 demonstrate the overall spatial preference, rather than the exact geometric selectivity, of the pocket. This conclusion is further confirmed by the ability of the 19 and the 23–28 series to inhibit SplA.
Discussion
Inhibition of SplA by α-aminoalkylphosphonate diaryl esters and their peptidyl derivatives
Phosphonate derivatives of amino acids and peptides were first developed as serine protease inhibitors in the late 1980s.16 Since then, the structural bases of the interactions between a range of potent and selective inhibitors and various proteases of the S1 family have been provided. In this study, we evaluated the feasibility of using phosphonate scaffold to develop staphylococcal SplA protease inhibitors. It was previously demonstrated that even simple phosphonate derivatives of amino acids show significant selectivity, which is determined by the P1 substrate preference of the protease.9 Given that SplA shows exclusive specificity for Tyr or Phe at the P1 position,6 we anticipated that phosphonate derivatives of single amino acids would be highly selective inhibitors of SplA activity. Accordingly, our initial experiments demonstrated that, of the Cbz-protected phosphonic analogs of amino acids that we tested, only the phenylalanine and leucine derivatives had inhibitory activity. All of the derivatives of alanine, valine, methionine, lysine, and arginine that we tested were inactive. Although leucine is not one of the preferred P1 substrate residues,6 the reactivity of LP (phosphonic leucine; 19) is explained by the hydrophobicity of its side chain, which resembles that of the side chain of phenylalanine. Hydrophobic pockets generally select moieties with a particular overall shape and a combination of physicochemical properties rather than relying on more specific interactions. For example, chymotrypsin poorly distinguishes between leucine and either Phe or Tyr at the P1 subsite (merops.sanger.ac.uk). Screening of a large library of peptide substrates indicated that SplA preferred Tyr or Phe over other residues.6 Nonetheless, as we demonstrate here, SplA may also accept leucine at P1, at least in the context of phosphonate inhibitors.
Our investigation of the influence of structurally diverse substitutions on the phenyl ester rings indicated that inclusion of electron-withdrawing moieties increases the level of inhibition of SplA, as was previously reported for other proteases.15–20
Studies on phosphonate and other transition-state-analog inhibitors, such as boronic acid derivatives and aldehydes, have demonstrated that inclusion of the sequences of efficient substrates in the peptidyl moieties of inhibitors generally increases inhibitor potency and selectivity.13 Accordingly, we report here that extension of the peptide moiety increases potency against SplA compared with single amino acid derivatives. However, unexpectedly, we observed that inhibitors with a peptidyl moiety containing residues efficiently recognized at particular positions of the substrate (FLFP) are less potent than those containing a P2 residue excluded from efficient SplA substrates (i.e., VPFP). When evaluated experimentally in this study, of analogical substrates Boc-Phe-Leu-Phe-AFC has a higher Km than the Boc-Val-Pro-Phe-AFC, even though the P2 residue of the latter substrate was not even selected among the best recognized substrates in a prior study.6 This counterintuitive phenomenon may be explained by cooperation between subsites. To facilitate synthesis, the FLF peptide moiety contained P1 residue determined as efficiently recognized by screening of a synthetic substrate library, whereas the P2 and P3 residues were selected from a proteinaceous substrate library, as described previously.6 However, neither of the previously characterized substrates contained the exact FLF sequence, and we only assumed that mixed substrates would be efficiently recognized. Therefore, the assumption that an efficient substrate may be designed based exclusively on knowledge of the specificity at particular subsites is only partially relevant because such an approach does not take into account the cooperation between subsites. Even so, the higher affinity of VPF-AFC compared with FLF-AFC does not explain the higher affinity of VPFP compared with FLFP because the mode of inhibitor binding does not resemble that of a canonical substrate.
Binding of phosphonate inhibitors
The current understanding of substrate recognition by S1 family proteases is primarily based on studies of interaction of canonical inhibitors and transition-state-analog peptide inhibitors. Both groups of molecules bind at the active site of the protease, forming an antiparallel β-sheet with the enzyme. Three conserved hydrogen bonds are formed. First, the amide hydrogen of the P1 residue donates a hydrogen bond to the carbonyl oxygen of Ser214 (chymotrypsin numbering), whereas the carbonyl oxygen and amide hydrogen of residue P3 form hydrogen bonds with Gly216 (Fig. 2, left panel). Moreover, one of the most conserved features of the interaction involves insertion of the side chain of the P1 residue into a pronounced S1 specificity pocket. Secondary, less conserved interactions of the P2 side chain and sometimes further residues and loops C and III are also observed.
The described binding mode is fully consistent with multiple known crystal structures of protein or peptide inhibitors of described classes (selected examples are provided in Supporting Information Table SII) including peptidyl derivatives of α-aminoalkylphosphonate diaryl esters. It is therefore interesting that neither of the inhibitors characterized in this study fully follows the established binding mode (Fig. 2). Although 31 inserts the P1 phenylalanine side chain into the hydrophobic S1 pocket of the protease, the enzyme selects from a racemic mixture only the (S)-diastereoisomer, which in stereochemical terms corresponds to the physiologically irrelevant d-amino acid enantiomer. The (R)-diastereoisomer, which is relevant to physiological substrates, is not observed in the co-crystal structure. Stereoselectivity of SplA toward (S)-diastereoisomers is surprising with respect to prior knowledge on phosphonate inhibitors. Although Walker et al. demonstrated that chymotrypsin and elastase are inhibited by (S)-diastereoisomers of phosphonate inhibitors, the proteases consistently preferred the (R)-diasteroisomers with at least an order of magnitude preference in k2/Ki.21 Moreover, all previously reported crystal structures of phosphonate inhibitor–protease complexes contain (R)-diastereoisomers of inhibitors. Of further differences, the amide hydrogen of P1 residue points away from the enzyme instead of forming a canonical hydrogen bond with the carbonyl oxygen of Tyr170 (equivalent of Ser214 in chymotrypsin). Instead, the carbonyl oxygen of the P2 residue forms a hydrogen bond with Gly172, whereas in the canonical case, the corresponding residue (Gly216 in chymotrypsin) interacts with P3 residue of the substrate. The observed differences are not brought about by an unnatural P2 moiety (Cbz). We know this because the crystal structure of trypsin complexed with an inhibitor that differs from 31 only at the P1 side chain (owing to differences in the substrate preference of both proteases) demonstrates canonical hydrogen bonds (PDB ID: 1MAX22). Moreover, although 41 may be treated as an analog of 31 with Cbz substituted by an extended peptide moiety, it also fails to interact with the protease in a canonical fashion. Again, SplA selects the (S)-diastereoisomer of 41 from a racemic mixture. In this case, the P1 residue is not involved in any direct hydrogen bonds with the enzyme. Moreover, the fact that the inhibitor is not defined by electron density beyond P1–P2 peptide bond suggests that this part of the inhibitor does not form a stable interaction with SplA. Therefore, the structure of the complex between 41 and SplA does not account for the increased activity of some of the inhibitors with extended peptidyl moiety tested in this study, such as the higher activity of 36 compared with 30. Given that 41 was previously crystallized in complex with cathepsin G, and demonstrated canonical hydrogen bonding in this context (PDB ID: 1CGH), the binding mode of 41 is clearly dictated by the SplA protease and is not inhibitor-specific.
Neither the structure of apo-SplA nor the structures presented in this study provide a mechanistic explanation of the observed divergence in interaction behavior of phosphonate inhibitors complexed with SplA compared to phosphonate inhibitors complexed with other family S1 proteases. The only feature of SplA in the vicinity of the active site that is pronouncedly different from the active sites of most S1A proteases is the lack of a canonical P2-binding site (short loop C; Supporting Information Fig. S5). This feature is characteristic not only of SplA but also of other S1B proteases of divergent substrate specificities, which share significant sequence and structural homology to SplA. These include the staphylococcal proteases SplB, the V8 protease, and the ETs. V8 protease lacks loop III, the loop III regions of ETA and ETB, as well as loop C of SplB, the V8 protease, and the ETs are all shorter than the corresponding structures in chymotrypsin. Loops III and C are too distant from the catalytic histidine in these proteases to support formation of the classical P2-binding pocket. Shorter versions of loops III and C and resulting lack of a classical P2 pocket would elegantly explain the undefined, flexible conformation of 41 beyond the P1 subsite were it not for the example of plasmin: although loop C of plasmin is also short and a pronounced P2 pocket is not present, the peptide inhibitor binds in a canonical orientation (PDB ID: 1BUI23).
The nonpeptidyl moieties of transition-state-analog inhibitors mimic one of the reaction intermediates that arrest catalysis. In previously determined structures of complexes of phosphonate inhibitors and S1A subfamily proteases, the phosphorus atom mimics the transition tetrahedral configuration of the carbonyl carbon of the scissile peptide bond, and one of the phosphonate oxygens mimics the oxyanion and its binding in the oxyanion hole. This notion is not supported by the structure of 31, where both phosphonate oxygens point away from the oxyanion hole. The phenomenon is difficult to explain. It seems unrelated to the chemical structure of the compound, given that a comparable inhibitor binds trypsin in a canonical fashion, with one of phosphonate oxygens coordinated in the oxyanion hole (PDB ID: 1MAX). Neither is it directly brought about by the SplA protease, given that the phosphonate moiety of the second crystallized inhibitor (41) binds in a canonical fashion. Structural analysis does not resolve this issue. The inhibitor is not affected by packing interactions, and the oxyanion hole is not blocked by any means besides those involving a water molecule. The water molecule present in the oxyanion hole is, however, a conserved feature of S1 family protease crystals obtained in an inhibitor-free form, and is easily displaced by inhibitors in other known structures of protease–inhibitor complexes, including SplA structure that contains 41. It seems possible, therefore, that an elusive combination of all the factors mentioned above results in the observed orientation of phosphonate oxygens.
The S1 specificity pocket
An interesting feature of the S1 specificity pocket of SplA is the presence of an asparagine side chain at its base. This polar group violates the overall hydrophobic nature of the pocket. Given that a similarly oriented aspartic acid residue determines the S1 specificity of trypsin, we previously speculated that the carbonyl oxygen of Asn181 may interact with the hydroxyl moiety of P1 tyrosine. The issue cannot, however, be resolved by the available structures. Although the orientation of P1 phenylalanine observed in the structure of 41 supports such a predicted interaction, the orientation found in the structure of 31 does not.
The role of Asn181 is clearly defined in the case of phenylalanine binding. The asparagine side chain stabilizes the orientation of the phenyl ring through an interaction between oxygen and the aromatic ring. The interatomic distances of such interactions range from 3 to 5 Å, with an optimal distance of 3.5–3.7 Å. Importantly, the angle between the oxygen atom and the phenyl ring plane should not exceed 50°.24 In both structures provided in this study, the mutual orientation of side-chain oxygen of Asn181 and the P1 phenyl ring fulfills the above-mentioned requirements. Further supporting the proposed role of Asn181 is the fact that of all the oxygen-containing groups found in proteins, it is the amide carbonyl oxygen that forms the strongest interaction with aromatic rings.24 Moreover, in a ligand-free structure of SplA, the side chain of Asn181 is already preoriented by hydrogen bonds with the backbone oxygen atoms of Ala147 and Ser178, such that Oδ1 is exposed in S1 pocket and ready to orient the P1 residue phenyl ring, as observed in the structures of the complexes.
To conclude, the potent inhibitors of SplA protease reported here should facilitate future elucidation of its physiological function. If SplA is truly of relevance in staphylococcal pathogenesis, the noncanonical binding surface of phosphonate inhibitors at the active site of SplA opens whole new perspectives of designing inhibitors of exclusive selectivity and potential therapeutic effect.
Materials and Methods
Protein expression and purification
The SplA protease was obtained using a recombinant expression system, as described previously.6 In brief, Bacillus subtilis strain WB800 carrying the pWB980-SplA plasmid was cultured overnight at 37°C in tryptic soy broth containing kanamycin (10 µg/mL). Culture supernatant was collected by centrifugation and precipitated by the addition of ammonium sulfate (561 g/L) at 4°C. Precipitated proteins were collected by centrifugation and resuspended in 50 mM sodium acetate, pH 5.5. After overnight dialysis against the same buffer, SplA was recovered using SP Sepharose Fast Flow ion-exchange chromatography. The protein was purified to homogeneity using Source S ion-exchange chromatography in 50 mM sodium acetate, pH 5.0. The buffer was exchanged to a desired formulation using Superdex 75 pg.
Inhibitor synthesis
Compounds 23–28 and 35–46 (Table1) were synthesized as outlined in Supporting Information Figure S2. Triaryl phosphites were prepared from 1 eq of phosphorus trichloride and 3 eq of substituted phenol in refluxing dry acetonitrile for 2 h. After evaporation of the volatile component, the crude phosphite was used in next step. Racemic, Cbz-protected α-aminoalkylphosphonate diaryl esters were prepared as a starting material by α-amidoalkylation of triaryl phosphite with benzyl carbamate and an aldehyde in acetic acid. The mixture was refluxed for 2 h, and the final product was crystallized from methanol.
The peptidyl moiety of the inhibitor was extended by first removing the Cbz-protective group by exposure to 33% HBr in acetic acid. The product was crystallized from diethyl ether as a hydrobromide salt. The α-aminoalkylphosphonate diaryl ester hydrobromide salt (1.2 eq) and appropriate Boc-protected amino acid were dissolved in dry acetonitrile in the presence of triethylamine (2.5 eq). Next, 1.2 eq of the coupling reagent O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) was added for overnight reaction at room temperature. The obtained phosphonate dipeptides were purified by column chromatography. The Boc-protective group was removed using 50% trifluoroacetic acid in dichloromethane. Additional amino acids were coupled as described above. All final compounds were purified using column chromatography on silica gel. Other examined phosphonate inhibitors were synthesized as described previously: 1,10 2–4,11 5–6,8 7,12 8–9,13 10–16,20 17–18, 20–22, 29–34,15 and 41.13
The purity of intermediates and final compounds was determined using thin-layer chromatography (TLC). The identity of all compounds was confirmed using 1H and 31P NMR (Bruker AC-TM DRX 300) and MS (Waters LCT Premier XE) analyses (see Supporting Information).
Synthesis of SplA fluorogenic substrates (Boc-Phe-Leu-Phe-AFC and Boc-Val-Pro-Phe-AFC)
7-Amido-4-trifluoromethylcoumarin (AFC; 1 eq) and Boc-Phe-OH (1.1 eq) were dissolved in dry pyridine. The mixture was cooled to −20°C, and POCl3 (1.1 eq) was added. The reaction was allowed to warm to 0°C, and the progress of the reaction was monitored using TLC. The reaction mixture was diluted with ethyl acetate, and washed with 5% NaHCO3, 5% citric acid, and brine to afford Boc-Phe-AFC. Application of 50% trifluoroacetic acid in dichloromethane was used for Boc-deprotection. Boc-Leu/Pro-OH (1.1 eq), triethylamine (2.5 eq), and HBTU (1.2 eq) were added subsequently to the solution of the TFA salt of H-Phe-AFC (1 eq) in acetonitrile. The reaction was performed overnight at room temperature. The solvent was evaporated and the resulting oil was dissolved in ethyl acetate and washed with brine, 5% KHSO4, and 5% NaHCO3. The Boc-Leu/Pro-Phe-AFC dipeptide derivative generated was purified by column chromatography on silica gel, using CHCl3:AcOEt (4:1, v/v) as an eluent. Further, Boc-deprotection and coupling with Boc-Phe/Val-OH, which generated the final compounds, was performed as described above.
Proteolytic activity and inhibition assays
Proteolytic activity of SplA was assayed using Boc-Phe-Leu-Phe-AFC at 37°C in 0.1M Bis–Tris (pH 6.5) containing 0.01% Triton X-100. The increase in fluorescence at 505 nm was monitored following excitation at 400 nm.
The kinetics of enzyme inactivation was measured using the progress curves method.25,26 Various concentrations of inhibitor were mixed with fixed concentration of SplA (2 µM) and the substrate (Boc-Phe-Leu-Phe-AFC, 25 µM; Km = 60 µM). Care was taken to keep the concentration of dimethylsulfoxide (DMSO), which was used to prepare inhibitor and substrate stock solutions, at concentrations that do not influence the proteolytic activity of SplA. The Ki and k2/Ki values were calculated using the following equation: kobs = k2[I0]/(Ki + [I0]), where Ki is the dissociation constant of a reversible enzyme–inhibitor complex, [I0] is the initial concentration of inhibitor, and k2 is the velocity of irreversible enzyme modification.27 The pseudo-first-order rate constants (kobs) were obtained by nonlinear regression using the equation Ft = v0[1 – exp(–kobst)]/kobs + F0, where Ft is the fluorescence intensity at time t, F0 is the fluorescence intensity at time zero, and v0 is the reaction velocity at time zero. The Ki and k2/Ki values that describe SplA inactivation by the α-aminoalkylphosphonate diaryl ester derivatives are summarized in Table1. We established arbitrary activity criteria that consider inhibitors with Ki > 200 µM and k2/Ki < 3 to be inactive against SplA. All of the α-aminoalkylphosphonates used for inhibition studies were racemic mixtures, where only one of the diastereoisomers appears to be effective. Therefore, the true potency of assayed compounds could be as much as twice that reported here.16
Co-crystallization
Purified SplA (∼1 mg/mL; ∼50 μM) in 100 mM Bis–Tris (pH 6.5) containing 50 mM NaCl was titrated with aliquots containing 50 nmol of inhibitor (50 mM in DMSO) with vigorous stirring at room temperature. Proteolytic activity was monitored after addition of each aliquot until no residual activity was detected. Buffer was exchanged, and excess inhibitor was removed by gel filtration using Superdex 75 pg pre-equilibrated with crystallization buffer [5 mM Tris–HCl (pH 8.0) and 50 mM NaCl]. The complex was concentrated to 60 mg/mL and screening was performed using the sitting drop vapor-diffusion method. Crystals appeared after several weeks at room temperature in Factorial 13 (compound 31; 0.1M HEPES, pH 7.0, 0.2M calcium chloride, and 25% PEG 400028) and Factorial 41 (compound 41; 0.1M HEPES, pH 7.4, 20% PEG 4000, and 10% isopropanol). The crystals were used directly for measurements without further optimization of conditions or cryoprotection.
Data collection and structure solution
Crystals were cryo-cooled in liquid nitrogen. The high-resolution diffraction data were collected at SLS beamline X10SA. Data were indexed and integrated with MOSFLM.29 The following steps were performed using software collected in the CCP4 package.30 Data were scaled using Scala.31–32 The Matthews coefficient was analyzed to estimate the number of molecules in the asymmetric unit. Molecular replacement was performed using Phaser,33 with the alanine search model based on the structure of SplA (PDB ID: 2W7S). Initial automated model building was performed using ARP/wARP.34 The model was inspected and completed manually using Coot.35 The electron density accounting for parts of the inhibitor was clearly visible in all structures before including the inhibitor in phasing. At the final stage of the apoprotein model refinement, the inhibitors were drawn in Sketcher30 and then incorporated into the model. Appropriate library descriptions were created using the same program. Water molecules were added using ARP/wARP36 and were inspected manually. Restrained refinement was performed with Refmac5.37 Throughout the refinement, 5% of reflections were used for cross-validation analysis38 and the behavior of Rfree was used to monitor the refinement strategy. The data collection and refinement statistics are presented in Supporting Information Table SI.
Accession numbers
Coordinates and structure factors have been deposited in the Protein Data Bank with the following accession numbers: 4MVN (compound 31 complex) and 3UFA (compound 41 complex).
Additional Supporting Information may be found in the online version of this article.
References
- Lowy FD. Staphylococcus aureus infections. New Engl J Med. 1998;339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- Dubin G. Extracellular proteases of Staphylococcus spp. Biol Chem. 2002;383:1075–1086. doi: 10.1515/BC.2002.116. [DOI] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ, Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2012;40:D343–D350. doi: 10.1093/nar/gkr987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed SB, Wesson CA, Liou LE, Trumble WR, Schlievert PM, Bohach GA, Bayles KW. Molecular characterization of a novel Staphylococcus aureus serine protease operon. Infect Immun. 2001;69:1521–1527. doi: 10.1128/IAI.69.3.1521-1527.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- Stec-Niemczyk J, Pustelny K, Kisielewska M, Bista M, Boulware KT, Stennicke HR, Thogersen IB, Daugherty PS, Enghild JJ, Baczynski K, Popowicz GM, Dubin A, Potempa J, Dubin G. Structural and functional characterization of SplA, an exclusively specific protease of Staphylococcus aureus. Biochem J. 2009;419:555–564. doi: 10.1042/BJ20081351. [DOI] [PubMed] [Google Scholar]
- Salvesen G, Nagase H, RJ Beyonon, JS Bond. Proteolytic enzymes: a practical approach. Oxford, United Kingdom: Oxford University Press; 1989. pp. 83–104. [Google Scholar]
- Sienczyk M, Oleksyszyn J. A convenient synthesis of new alpha-aminoalkylphosphonates, aromatic analogues of arginine as inhibitors of trypsin-like enzymes. Tetrahedron Lett. 2004;45:7251–7254. [Google Scholar]
- Oleksyszyn J, Powers JC. Amino acid and peptide phosphonate derivatives as specific inhibitors of serine peptidases. Methods Enzymol. 1994;244:423–441. doi: 10.1016/0076-6879(94)44032-8. [DOI] [PubMed] [Google Scholar]
- Joossens J, Van der Veken P, Lambeir AM, Augustyns K, Haemers A. Development of irreversible diphenyl phosphonate inhibitors for urokinase plasminogen activator. J Med Chem. 2004;47:2411–2413. doi: 10.1021/jm0499209. [DOI] [PubMed] [Google Scholar]
- Sienczyk M, Oleksyszyn J. Inhibition of trypsin and urokinase by Cbz-amino(4-guanidinophenyl)methanephosphonate aromatic ester derivatives: the influence of the ester group on their biological activity. Bioorgan Med Chem Lett. 2006;16:2886–2890. doi: 10.1016/j.bmcl.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Jackson DS, Fraser SA, Ni LM, Kam CM, Winkler U, Johnson DA, Froelich CJ, Hudig D, Powers JC. Synthesis and evaluation of diphenyl phosphonate esters as inhibitors of the trypsin-like granzymes A and K and mast cell tryptase. J Med Chem. 1998;41:2289–2301. doi: 10.1021/jm970543s. [DOI] [PubMed] [Google Scholar]
- Oleksyszyn J, Powers JC. Irreversible inhibition of serine proteases by peptide derivatives of (alpha-aminoalkyl)phosphonate diphenyl esters. Biochemistry. 1991;30:485–493. doi: 10.1021/bi00216a026. [DOI] [PubMed] [Google Scholar]
- Sienczyk M, Winarski L, Kasperkiewicz P, Psurski M, Wietrzyk J, Oleksyszyn J. Simple phosphonic inhibitors of human neutrophil elastase. Bioorgan Med Chem Lett. 2011;21:1310–1314. doi: 10.1016/j.bmcl.2011.01.083. [DOI] [PubMed] [Google Scholar]
- Pietrusewicz E, Sienczyk M, Oleksyszyn J. Novel diphenyl esters of peptidyl α-aminoalkylphosphonates as inhibitors of chymotrypsin and subtilisin. J Enzyme Inhib Med Chem. 2009;24:1229–1236. doi: 10.3109/14756360902781512. [DOI] [PubMed] [Google Scholar]
- Oleksyszyn J, Powers JC. Irreversible inhibition of serine proteases by peptidyl derivatives of alpha-aminoalkylphosphonate diphenyl esters. Biochem Biophys Res Commun. 1989;161:143–149. doi: 10.1016/0006-291x(89)91572-6. [DOI] [PubMed] [Google Scholar]
- Hof P, Mayr I, Huber R, Korzus E, Potempa J, Travis J, Powers JC, Bode W. The 1.8 A crystal structure of human cathepsin G in complex with Suc-Val-Pro-PheP-(OPh)2: a Janus-faced proteinase with two opposite specificities. EMBO J. 1996;15:5481–5491. [PMC free article] [PubMed] [Google Scholar]
- Krowarsch D, Cierpicki T, Jelen F, Otlewski J. Canonical protein inhibitors of serine proteases. Cell Mol Life Sci. 2003;60:2427–2444. doi: 10.1007/s00018-003-3120-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perona JJ, Craik CS. Evolutionary divergence of substrate specificity within the chymotrypsin-like serine protease fold. J Biol Chem. 1997;272:29987–29990. doi: 10.1074/jbc.272.48.29987. [DOI] [PubMed] [Google Scholar]
- Sienczyk M, Winiarski L, Kasperkiewicz P, Psurski M, Wietrzyk J, Oleksyszyn J. Simple phosphonic inhibitors of human neutrophil elastase. Bioorgan Med Chem Lett. 2011;21:1310–1314. doi: 10.1016/j.bmcl.2011.01.083. [DOI] [PubMed] [Google Scholar]
- Walker B, Wharry S, Hamilton RJ, Martin SL, Healy A, Walker BJ. Asymmetric preference of serine proteases toward phosphonate and phosphinate esters. Biochem Biophys Res Commun. 2000;276:1235–1239. doi: 10.1006/bbrc.2000.3597. [DOI] [PubMed] [Google Scholar]
- Bertrand JA, Oleksyszyn J, Kam CM, Boduszek B, Presnell S, Plaskon RR, Suddath FL, Powers JC, Williams LD. Inhibition of trypsin and thrombin by amino(4-amidinophenyl)methanephosphonate diphenyl ester derivatives: X-ray structures and molecular models. Biochemistry. 1996;35:3147–3155. doi: 10.1021/bi9520996. [DOI] [PubMed] [Google Scholar]
- Parry MA, Fernandez-Catalan C, Bergner A, Huber R, Hopfner KP, Schlott B, Guhrs KH, Bode W. The ternary microplasmin-staphylokinase-microplasmin complex is a proteinase-cofactor-substrate complex in action. Nat Struct Biol. 1998;5:917–923. doi: 10.1038/2359. [DOI] [PubMed] [Google Scholar]
- Thomas KA, Smith GM, Thomas TB, Feldmann RJ. Electronic distributions within protein phenylalanine aromatic rings are reflected by the three-dimensional oxygen atom environments. Proc Natl Acad Sci USA. 1982;79:4843–4847. doi: 10.1073/pnas.79.16.4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joossens J, Ali OM, El-Sayed I, Surpateanu G, Van der Veken P, Lambeir AM, Setyono-Han B, Foekens JA, Schneider A, Schmalix W, Haemers A, Augustyns K. Small, potent, and selective diaryl phosphonate inhibitors for urokinase-type plasminogen activator with in vivo antimetastatic properties. J Med Chem. 2007;50:6638–6646. doi: 10.1021/jm700962j. [DOI] [PubMed] [Google Scholar]
- Knight GC. In: Protease inhibitors. Barrett AJ, Salvesen G, editors. Amsterdam: Elsevier; 1987. The characterization of enzyme inhibition 23–51. [Google Scholar]
- Lambeir AM, Borloo M, De Meester I, Belyaev A, Augustyns K, Hendriks D, Scharpe S, Haemers A. Dipeptide-derived diphenyl phosphonate esters: mechanism-based inhibitors of dipeptidyl peptidase IV. Biochim Biophys Acta. 1996;1290:76–82. doi: 10.1016/0304-4165(96)00012-8. [DOI] [PubMed] [Google Scholar]
- Jancarik J, Kim S-H. Sparse matrix sampling: a screening metod for crystallization of proteins. J Appl Cryst. 1991:409–411. [Google Scholar]
- Leslie AGW. Recent changes to MOSFLM package for processing film and image image plate data. Joint CCP4 and ESF-EAMCB Newsletter on Protein Crystallography. 1992;6 [Google Scholar]
- Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Evans PR. Scala. Joint CCP4 and ESF-EAMCB. Newslett Protein Crystallogr. 1997;33:22–24. [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Cryst D. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Lamzin VS, Wilson KS. Automated refinement of protein models. Acta Cryst D. 1993;49:129–147. doi: 10.1107/S0907444992008886. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Brunger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.