
Figure 2.
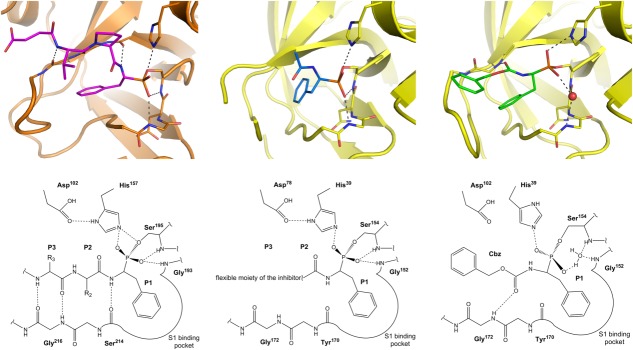
Comparison of phosphonate inhibitor binding mode to cathepsin G and SplA. In all cases the side chain of P1 residue resides in the pronounced S1 specificity pocket. (Left panel) Canonical binding mode of α-aminoalkylphosphonate inhibitors exemplified by the structure of Suc-Val-Pro-PheP-(OC6H5)2-bound cathepsin G (PDB ID: 1CGH). The peptidyl part of the inhibitor forms three canonical hydrogen bonds in the nonprimed sites of the protease active site. One of phosphonate oxygens is coordinated in the oxyanion hole. (Middle panel) In case of 41, one of phosphonate oxygens is canonically coordinated in the oxyanion hole but, the peptidyl part does not form any hydrogen bonds with the enzyme being flexible and undefined by electron density beyond P1 subsite. (Right panel) In case of 31 the oxyanion hole is occupied by a water molecule (red sphere) instead of a phosphonate oxygen. Furthermore, the peptidyl part forms a noncanonical hydrogen bond within the enzyme nonprimed side. Unlike in the cathepsin complex, both in case of 31 and 41 SplA selects (S)-diastereoisomer (corresponding to the d-amino acids) out of a racemic mixture. An interactive view is available in the electronic version of the article.