

Abstract
Background and Objective:
There is compelling evidence that obstructive sleep apnoea (OSA) can affect metabolic syndrome (MetS) and cardiovascular risk, but the intermediate mechanisms through which it occurs have not been well defined. We explored the impact of OSA in morbidly obese patients with MetS on adipokines, pro-inflammatory markers, endothelial dysfunction, and atherosclerosis markers.
Methods:
We included 52 morbidly obese patients in an observational study matched for age, gender and central obesity in 3 groups (OSA-MetS, Non-OSA-MetS, and Non OSA-non-MetS). Anthropometrical, blood pressure, and fasting blood measurements were obtained the morning after an overnight polysomnography. VEGF, soluble CD40 ligand (sCD40L), TNF-α, IL-6, leptin, adiponectin, and chemerin were determined in serum by ELISA. OSA was defined as apnea/ hypopnea index ≥ 15 and MetS by NCEP-ATP III.
Results:
Cases and control subjects did not differ in age, BMI, waist circumference, and gender (43 ± 10 years, 46 ± 5 kg/m2, 128 ± 10 cm, 71% females). The cases had severe OSA with 47 (32-66) events/h, time spent < 90% SpO2 7% (5%-31%). All groups presented similar serum cytokines, adipokines, VEGF, and sCD40L levels.
Conclusions:
In a morbidly obese population with established MetS, the presence of OSA did not determine any differences in the studied mediators when matched by central obesity. Morbidly obese NonOSA-NonMetS had a similar inflammatory, adipokine VEGF, and sCD40L profile as those with established MetS, with or without OSA. Obesity itself could overwhelm the effect of sleep apnea and MetS in the studied biomarkers.
Citation:
Salord N; Gasa M; Mayos M; Fortuna-Gutierrez AM; Montserrat JM; Sánchez-de-la-Torre M; Barceló A; Barbé F; Vilarrasa N; Monasterio C. Impact of OSA on biological markers in morbid obesity and metabolic syndrome. J Clin Sleep Med 2014;10(3):263-270.
Keywords: Obstructive sleep apnoea, obesity, metabolic syndrome, adipokines, inflammatory markers, endothelial dysfunction
Metabolic syndrome (MetS) is a cluster of metabolic and cardiovascular abnormalities (central obesity, impaired glucose metabolism, hypertension, hypertriglyceridemia, and lower high-density lipoprotein cholesterol) that is associated with an increase in cardiovascular morbidity and mortality.1,2 There is increasing evidence, mainly from studies in moderately obese sleep-referred patient cohorts, that obstructive sleep apnea (OSA) may impact negatively on MetS or some of its components, independent of the body mass index (BMI).3 Furthermore, in a large cross-sectional study of non-selected morbidly obese patients with a high prevalence of MetS,4 OSA was associated with a worse metabolic profile independent of BMI, suggesting that OSA plays an important role in the pathogenesis of metabolic dysfunction, even in morbid obesity.
BRIEF SUMMARY
Current Knowledge/Study Rationale: There is evidence that obstructive sleep apnea (OSA) can affect metabolic syndrome (MetS) and cardiovascular risk even in morbid obesity, but the intermediate mechanisms through which this occurs have not been well defined.
Study Impact: In a morbidly obese population with established MetS, the presence of OSA did not determine any differences in inflammatory, adipokine, VEGF, or sCD40L profile. Obesity itself could produce a ceiling effect on sleep apnea and MetS in the studied intermediate biomarkers.
OSA stimulates, mainly through intermittent hypoxia, several intermediate mechanisms including oxidative stress, inflammation, sympathetic activation, and endothelial dysfunction that are potentially harmful to the cardiovascular system and metabolism.5 OSA could also increase visceral fat dysfunction, which could play a pivotal role in the relationship between obesity and MetS.6 Visceral fat is now considered a highly active organ that produces a variety of molecules, such as inflammatory cytokines and adipokines (e.g., leptin and adiponectin); these also modulate mechanisms related to metabolic dysfunction, such as oxidative stress, inflammation, and sympathetic activation, and they are also involved in the progression of arteriosclerosis, the key factor for cardiovascular risk.6 More specifically, one recently described adipokine, chemerin, highly expressed in adipose tissue, is thought to play an important role in metabolic lipogenesis, glucose homeostasis, and adipocyte differentiation.7
Many experimental and clinical studies have explored the possible underlying interaction between OSA, obesity and MetS, but there has been no clarification to date of the impact of each entity on the intermediate mechanisms related to pathological consequences.3 Given the concern about the confusing effect of obesity, most clinical studies match patients for BMI to assess differences between OSA patients and controls without OSA. The BMI could be insufficient, however, as abdominal visceral fat is a stronger risk factor than other fat deposits for adverse health consequences such as hypertension, insulin resistance, diabetes, and the metabolic syndrome.8,9 Waist circumference (WC) has been considered a surrogate marker of visceral adiposity.10 However, no studies matched by WC have focused on the effect of OSA on MetS and its pathogenic pathways in morbidly obese patients.
As an association between OSA and MetS has been observed even morbidly obese patients,4 in the present observational study, we investigated how the presence of OSA could be associated with alterations in three of the main known pathways involved in MetS and atherosclerosis, its underlying physiopathological process. For this purpose we compared the synthesis of several adipokines, including chemerin, pro-inflammatory markers and endothelial dysfunction markers in morbidly obese patients with MetS with and without OSA, as well as a third group of morbidly obese patients without MetS and without OSA and we matched them for waist circumference.
METHODS
Study Design, Setting, and Participants
We included patients with MetS criteria from a previous published morbid obesity cohort of 159 patients from 3 university hospitals.4 Patients with and without OSA were matched for age, gender, and waist circumference (as a measure of central obesity). The patients included were determined by the ability to match pairs from the previous cohort. To explore the effect of morbid obesity alone, we also included a third group without MetS and without OSA, matched by the same confounders. The study protocol was approved by the ethics committee of each hospital (PR052/08, 07/064/797, PI080277). All participants gave informed written consent.
Each participant completed a detailed questionnaire on medical history, cardiovascular risk factors, and current medication. Anthropometric characteristics included BMI, neck circumference (at the level of the laryngeal prominence), waist circumference (WC, measured midway between the lowest rib and the iliac crest), waist/hip ratio, and percentage of body fat mass measured with electrical bioimpedance (BIA 101, Akern Bioresearch, Florence, Italy). Blood pressure was measured by a standard mercury sphygmomanometer while the subject was seated at rest; the mean value of ≥ 2 measurements was calculated.
Sleep Study
OSA was determined by a full overnight polysomnography (Siesta, Compumedics, Abbotsford, Australia). Polysomnography interpretation was assessed according to the standard criteria of Rechtschaffen and Kales. Apnea was defined as a cessation of flow ≥ 10 sec and hypopnea as 30% to 90% flow reduction for ≥ 10 sec accompanied by a fall ≥ 3% in oxyhemoglobin saturation (SpO2) and/or a microarousal. The apneahypopnea index (AHI) was the total number of such events per hour of sleep. An AHI of 15 events/h was chosen to define the presence of OSA according to the study design. The degree of nocturnal desaturation was assessed by the mean percentage of sleep time with SpO2 below 90% (Time SpO2 < 90%). Excessive daytime sleepiness was evaluated by the Epworth Sleepiness Scale (ESS).
Blood Measurements and Definition of Metabolic Syndrome
The morning after polysomnography (08:00-09:00), a venous blood sample was obtained from all patients in fasting conditions. Fasting blood glucose (FBG), percentage of glycosylated hemoglobin (HbA1c), total cholesterol, triglycerides, high-density lipoprotein (HDL) cholesterol, low-density lipoprotein (LDL) cholesterol, and very low-density lipoprotein (VLDL) cholesterol levels were determined with standard laboratory methods. Insulin was determined by the automated chemiluminescence method (Advia Centaur, Siemens, NY, USA) with a sensitivity of 0.5 mU/L and an inter-assay coefficient of variation of 7.5%. Insulin resistance was estimated using the homeostasis model assessment (HOMA) score, defined by fasting serum insulin mU/L × fasting plasma glucose mmol/L/22.5. MetS was defined according to the NCEP-ATP III modified criteria.1
Serum Special Sampling
Blood samples obtained in tubes without anticoagulant were centrifuged immediately; the resulting serum was stored in aliquots at -80° C until analysis. Measurements were always made in duplicate, and the mean values were used for analysis. Biomarker levels were determined by enzyme-linked immunosorbent assay using commercially available kits: vascular endothelial growth factor (VEGF) (Invitrogen, California, USA); soluble CD40 ligand (sCD40L) (R&D Systems, Minneapolis, USA); tumor necrosis factor α (TNF-α) (Invitrogen California, USA); interleukin 6 (IL-6) (Invitrogen California, USA); leptin (SPI-BIO, Montigny, France); adiponectin (SPI-BIO, Montigny, France) and chemerin (USCN Life Science, Wuhan, China). See online data for information about assay sensitivity and inter- and intra-assay coefficients of variation (Table S1).
Statistical Methods
Patients were matched by a weighted Manhattan distance based on the normalized variables calculated between patients. Each patient from one group was matched with another in the other group with the lowest distance from him/her. If the matching was performed in a 1:2 proportion, each patient was matched to the 2 patients in the other group with the lowest distance from him/her. Sample size was determined by the ability to match pairs from the previous cohort.
Results are shown as median and interquartile range. Due to the skewed distribution and sample size, comparisons among the 3 groups were performed with the Kruskal-Wallis test, with post hoc Bonferroni correction for multiple comparisons. Correlations between variables were explored using the Spearman test. Statistical significance was defined as p < 0.05.
RESULTS
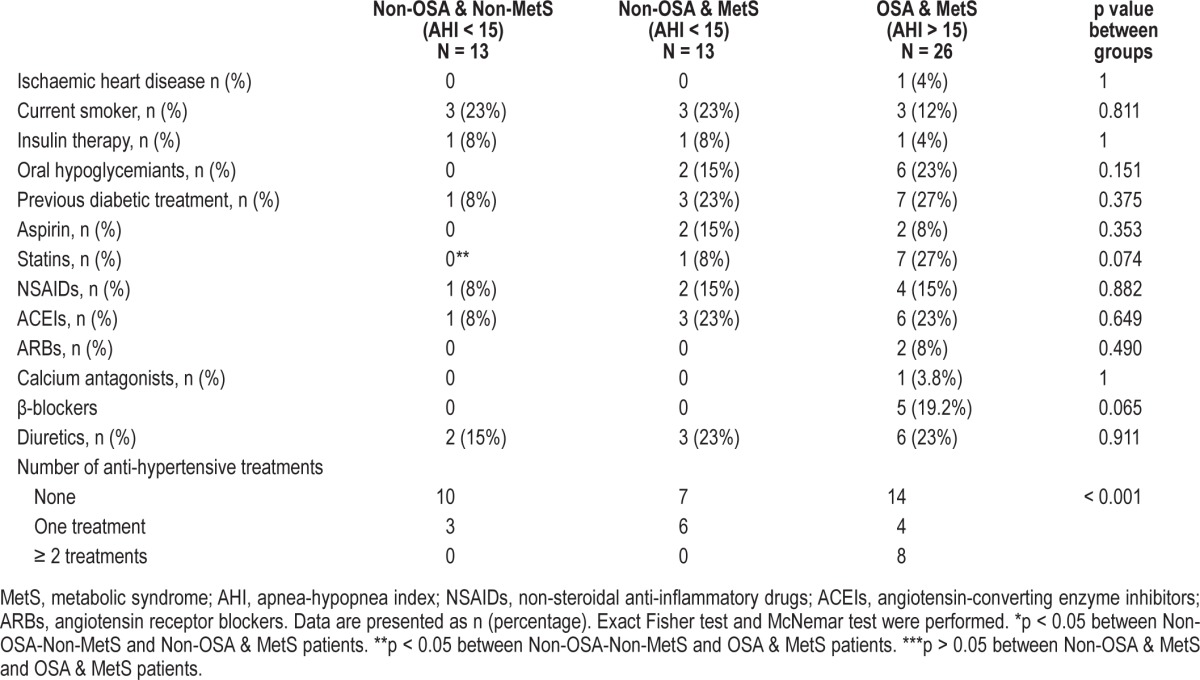
Twenty-six patients with OSA and MetS (OSA-MetS) criteria were matched with 13 non-OSA patients with MetS (non-OSA-MetS) and 13 non-OSA and non-MetS controls (non-OSA-non-MetS). Cases and control subjects were similar in age, weight, and waist circumference, as expected (Table 1). The 3 groups had similar BMI and neck circumference, and there was a nonsignificant trend toward higher fat mass in the 2 MetS groups (Table 1). As usual in morbid obesity, the cohorts were mainly composed of premenopausal women. Although the OSA-MetS patients were taking more medication and had higher previously diagnosed hypertension, there were no differences between the groups regarding drugs used or smoking status (Table 2). Post hoc analysis only showed a significant difference in the higher use of statins in OSA-MetS patients with respect to non-OSA -non-MetS patients.
Table 1.
Patients' general characteristics: matched 1:1 and 1:2 by waist circumference, age and sex.
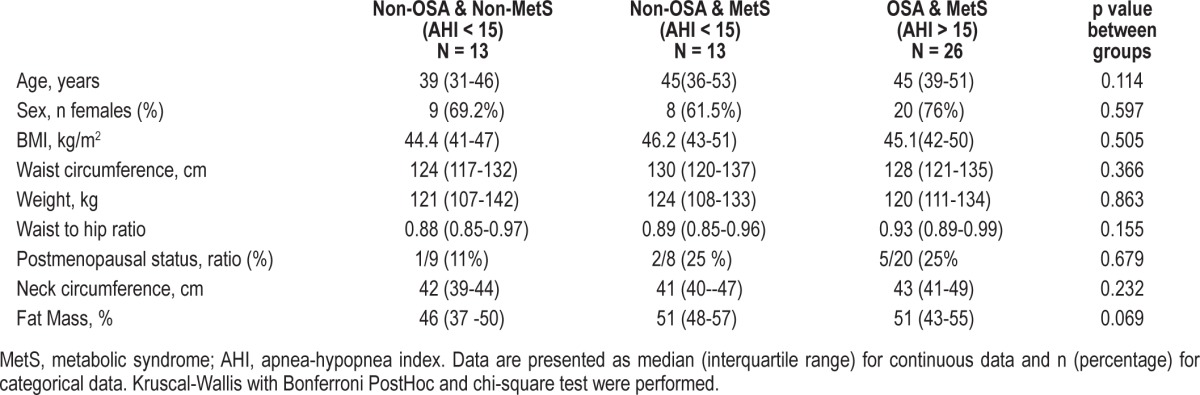
Table 2.
Previous diseases and treatments: matched 1:1 and 1:2 by waist circumference, age and sex.
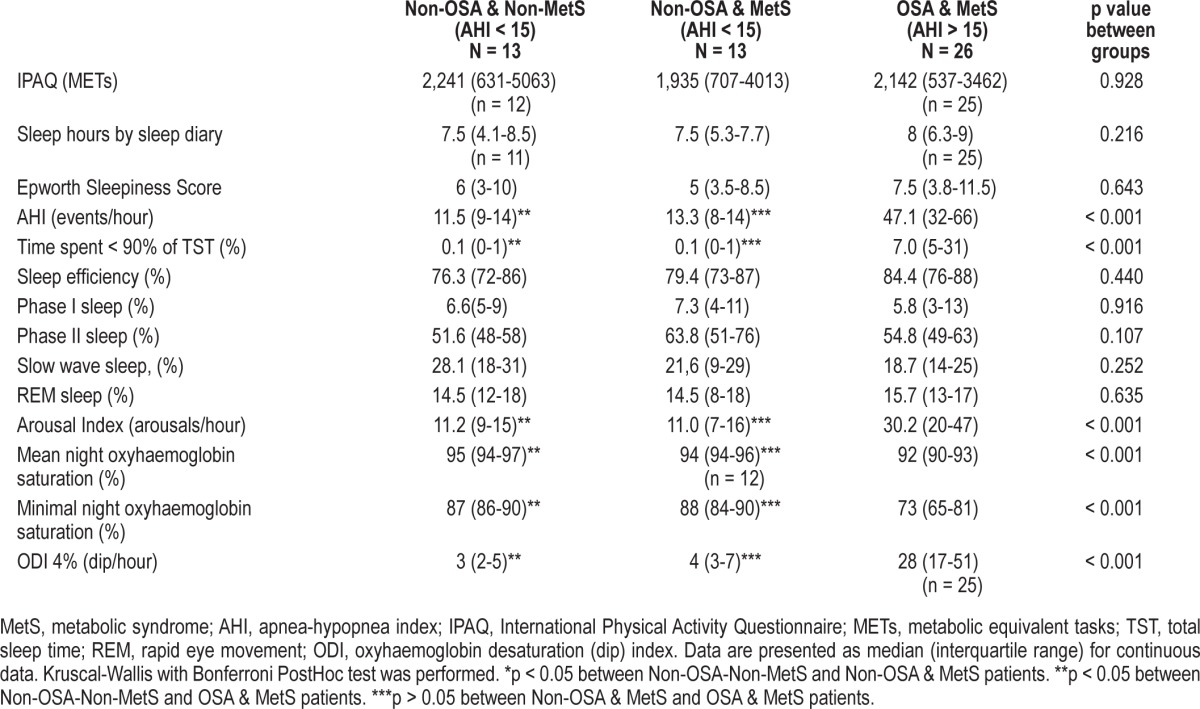
The OSA-MetS patients had a high AHI with a median and interquartile range (IQR) of 47.1 (32-66) events/h, Time SpO2 < 90% 7% (5%-31%), and a mean and minimal night oxyhemoglobin saturation of 92% (90%-93%) and 73% (65%-81%), respectively (Table 3). Although OSA patients had higher arousal indexes, all the groups had similar sleep efficiency and sleep stage percentages. No differences between the groups were encountered in physical activity measured by the IPAQ questionnaire or sleep hours measured by the sleep diary (Table 3).
Table 3.
Patients' sleep characteristics: matched 1:1and 1:2 by waist circumference, age and sex
As regards the MetS components, there were significant differences in systolic and diastolic blood pressure, triglycerides, HDL cholesterol, and fasting glucose. These differences were, as expected, mainly the result of the more favorable metabolic profile of the non-OSA-non-MetS obese group (Table 4). Although there was a significant trend of OSA-Mets patients with higher triglycerides and lower HDL cholesterol levels, no differences were found in the post hoc analysis between MetS patients with and without OSA (Table 4). When glucose metabolism was further analyzed, no differences were found in fasting insulin, the HOMA index, or glycosylated hemoglobin (Table 4). OSA-MetS patients tended to present a pattern of more impaired liver enzymes, but this was only significant for alanine aminotransferase. Figure 1 shows the distribution of MetS components in the 3 groups.
Table 4.
Patients' metabolic characteristics: matched 1:1and 1:2 by waist circumference, age and sex.
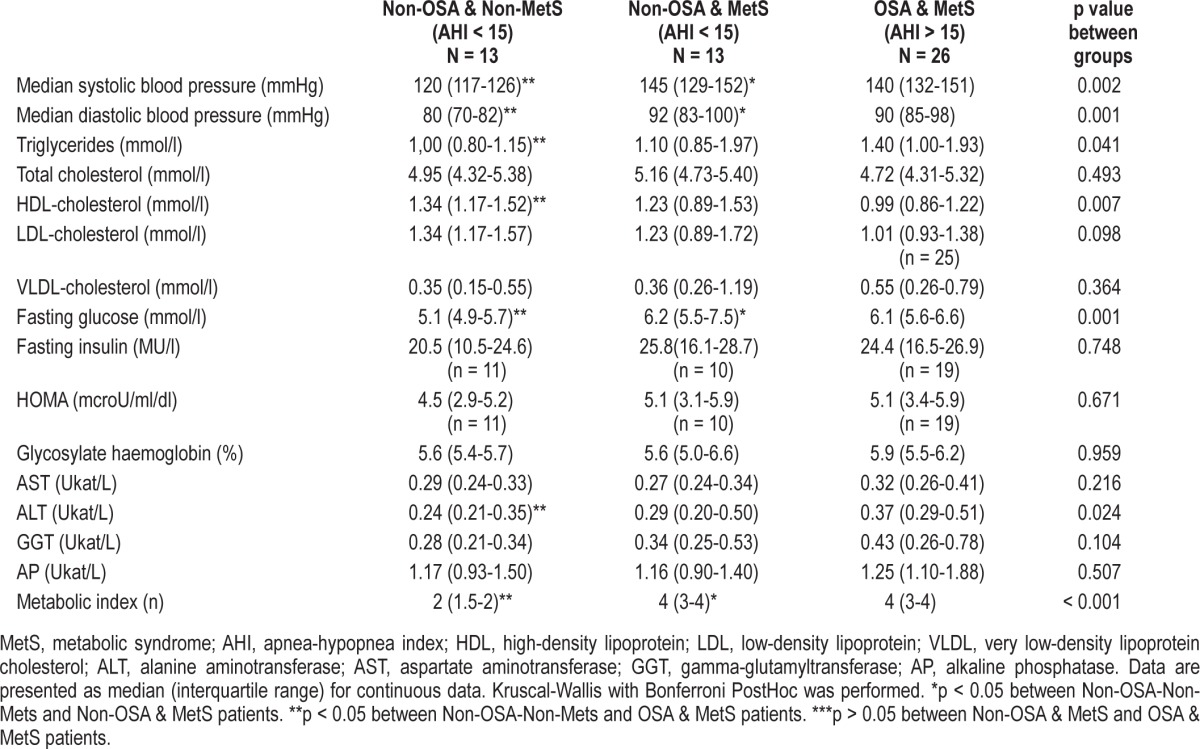
Figure 1. Distribution of metabolic syndrome components between the three groups.
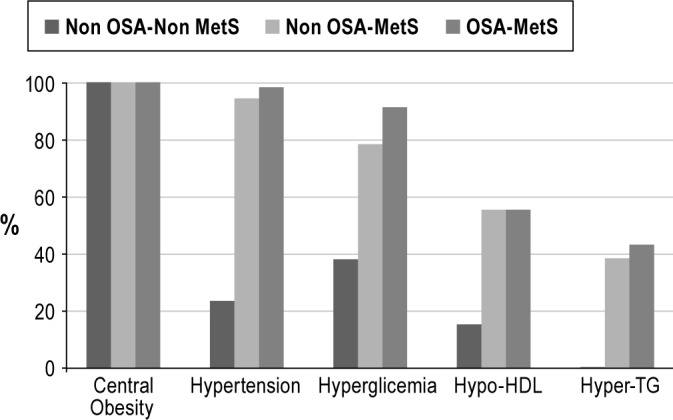
All the groups presented similar serum levels of cytokines (IL-6, TNF-α), adipokines (leptin, adiponectin, and chemerin), VEGF, and sCD40L (Table 5). No correlation was found between the biomarkers and any OSA variable (data not shown). Leptin was closely correlated with the anthropo-metric variables (BMI [r = 0.479, p < 0.001], waist-to-hip ratio [r = -0.598, p < 0.001], and fat mass [r = 0.401, p = 0.004]), while adiponectin was correlated negatively to waist-to-hip ratio (r = -0.598, p < 0.001), waist (r = -0.407, p = -0.003), and neck circumference (r = -0.406, p < 0.003) and VEGF with fat mass (r = 0.3, p = 0.021).
Table 5.
Biological markers: matched 1:1 and 1:2 by waist circumference, age and sex.

DISCUSSION
The present study is the first to explore the effect of OSA on the many known mechanistic pathways that may contribute to MetS in a non-selected morbidly obese population. In this extremely obese population with established MetS, the presence of OSA did not produce any differences in serum cytokines (IL-6, TNF-α), adipokines (leptin, adiponectin and chemerin), or VEGF and sCD40L levels, as no difference was found between OSA and non-OSA patients with MetS matched by central obesity. Morbidly obese patients without both established MetS and OSA already have a similar biomarker-altered profile.
The relationship between OSA and MetS or some of its components has been established, mainly through cohorts referred by sleep units with concomitant mild-to-moderate obesity.3 The major limitation of these studies is the control of obesity itself, as this is the main confounding factor and is often adjusted by multivariate analysis. To address this issue, Barceló et al. excluded patients with obesity and matched a sleep unit-referred cohort by BMI; OSA patients showed higher levels of free fatty acids, CRP, and oxidative stress markers.11 Trombetta et al. matched MetS patients by BMI with and without OSA; they demonstrated that in this high-risk cardiovascular population with moderate obesity, OSA was associated with greater sympathetic drive.12 Moreover, there have been few case-control studies that matched patients by central adiposity to study the effect of OSA on the different components of MetS13 and the different biomarkers14 related to both conditions. It is therefore important to establish whether OSA and its associated metabolic abnormalities are independent of central obesity.
Our study focused on an extremely obese population, composed mainly of premenopausal women, in which we previously showed an association of OSA with a worse metabolic profile, mainly through systolic blood pressure, triglycerides, and Hb1Ac, after adjusting by BMI and waist circumference.4 In order to explore the possible mechanistic pathways explaining this association, we matched MetS patients according to their OSA status by waist circumference as a surrogate marker of central obesity. Patients in the OSA-MetS group had greater metabolic impairment than non-OSA-Non-MetS patients. Moreover, when we compared the two MetS groups, as well as fulfilling MetS criteria, the MetS-OSA group had a tendency toward higher hypertriglyceridemia, lower HDL-cholesterol levels, higher levels of liver transaminases, and greater requirements of antihypertensive treatment. This fact did not determine any differences in the studied biomarkers between OSA and non-OSA in MetS patients. In the study by Barceló et al., in the non-obese population only OSA patients without MetS had higher levels of free fatty acids, as no significant differences were found in biomarkers between OSA and non-OSA in patients with established MetS.11 Surprisingly, when we reviewed the non-OSA-non-MetS patients, there were no differences in the biomarkers explored. Those patients who supposedly constituted the metabolically healthier group in fact presented lower blood pressure, serum triglyceridemia, and fasting glucose, while their HDL cholesterol was higher, although they already had the same biomarker profile. Metabolically healthy obesity is defined by a normal sensitivity to insulin and a normal lipid profile, and it is related to lower levels of inflammation and higher adiponectin concentrations.15,16 While American studies have shown that 30% of the obese population can be considered metabolically healthy,17 only 5% of morbidly obese patients were found to be metabolically healthy.18 In our study only three non-OSA-non-MetS patients had obesity as the sole criterion for MetS, and they also presented high HOMA-R indices. We could thus speculate that the lack of differences in the studied biomarkers could be evidence of an already adverse pre-pathological status, even in patients without established MetS in morbid obesity. Therefore, in this extremely obese population, patients without OSA or MetS already have an altered mediator pattern; if we assume the previously demonstrated effect of OSA on metabolic abnormalities, then OSA must stimulate another specific mechanism to trigger MetS, independent of those activated by obesity.
Inflammation has been recognized as an intermediary mechanism associated with insulin impairment, MetS, and cardiovascular disease.19,20 Many cross-sectional, case-control, and non-randomized interventional studies have suggested that the levels of the pro-inflammatory cytokines and C-reactive protein are elevated in OSA patients.21,22 These results have not been consistent, however, as contradictory findings have been reported in studies by both our group and others, in which this association was attributed mainly to obesity.23,24 Furthermore, obese patients are resistant to leptin, which mainly serves to regulate appetite and energy expenditure. In clinical studies, leptin has been correlated with insulin levels and BMI, and it predicts the development of MetS.25,26 Leptin was the first adipokine associated with OSA and its levels have proved to be higher in OSA patients. Vogontzas et al. found, however, that elevated leptin levels were more closely related to visceral fat and other pro-inflammatory markers than to AHI.27 This could explain the ceiling effect in the morbidly obese population in our work. Our study does not reveal any new information about adiponectin. Hypoadiponectinemia has been postulated as playing a role in the development of MetS,28 as adiponectin regulates insulin sensitivity and has an anti-inflammatory and protective vascular effect. The studies of OSA to date are not conclusive, as some have found lower levels in OSA patients, but others such as ours have failed to show this down-regulation.29,30 As an original contribution, we explored chemerin as a novel adipokine. Clinical studies have shown that patients with MetS have higher levels of chemerin than those without MetS and present an association with BMI, serum triglycerides, and blood pressure.31 A recent case-control study with a moderately obese Asian population showed that OSA patients had higher chemerin levels and found a weak but positive correlation with sleep apnea severity and WC and BMI32; in this study chemerin was also correlated with inflammation and hypothesized as a possible a link between adipose tissue, OSA, and inflammation. These results could not be confirmed in our morbidly obese population with high indices of insulin resistance, where we found that central obesity had a stronger effect than OSA.
Apart from systemic inflammation and adipokine discharge, the main direct consequences of obstructive apneas—intrathoracic pressure changes, recurrent arousal, and intermittent hypoxia—also trigger an increase in sympathetic nerve activity, reduce baroreflex sensitivity, and cause endothelial dysfunction. These changes induce increased arterial stiffness and arterial hypertension, leading to the development of atherosclerosis.33 In order to explore the latter mechanisms, we analyzed VEGF and sCD40 serum levels. VEGF is thought to promote atherogenesis through endothelial cell proliferation, cell migration, and increased vascular permeability.34 Previous clinical studies have shown higher levels of VEGF in OSA patients than in controls,35,36 mainly as a result of hypoxia. Our results were limited by the great variability in VEGF in the sample, which prevented us from finding any differences between groups. At the same time, the presence of OSA did not suppose any effect on sCD40L levels. sCD40L is considered a surrogate marker of arteriosclerosis, part of a mechanism signalling platelet activation, and it is associated with increased cardiovascular risk.37 Nevertheless, we could not rule out a role for other known intermediate mechanisms, such as increased sympathetic activity (not explored in our study), in the link between MetS, cardiovascular morbidity, and OSA in morbidly obese patients. A review of the randomized controlled interventional trials shows that Dragger et al. found an improvement in early signs of the atherosclerosis markers, C-reactive protein, and urine catecholamines in severe OSA after four months of CPAP, but their patients, unlike ours were only overweight and were otherwise free of any comorbidity.38 Further studies focusing on patients with milder obesity39,40 did not observe any changes in systemic inflammation, insulin resistance, blood lipids, leptin, or adiponectin. Similarly, West et al. also failed to find any changes in C-reactive protein and adiponectin after CPAP treatment in a population of OSA patients with established diabetes and more severe obesity.41 Taken together, these data and our own reinforce the idea of the overwhelming effect of obesity and comorbidities on the studied markers.
Our study has a number of potential limitations that should to be considered. Firstly, because of the cross-sectional descriptive nature of the study, it did not provide evidence of a cause-effect relationship. Secondly, the size of the study was limited by the prior cohort study and the need to match patients from the different groups. In order to include the maximum number of patients we performed a 2:1 match, since OSA patients were much more prevalent in our population. Although the sample size limits the power of the study, we were unable to find any trend in any biomarker explored, which makes it probable that morbid obesity itself exerts a possible ceiling effect. Finally, as mentioned above, our patients, as a clinical population, were undergoing active hypolipemia, antihypertensive, and hypoglycemia treatment that could interfere with the determination of metabolic profiles and biomarkers and minimize the difference between groups.
We could not find any distinct biomarker pattern of OSA in a morbidly obese population, nor did the studied biomarkers allow us to discriminate between patients with and without MetS in this population. Severe central obesity thus seems to trigger an array of downstream effects that overwhelm those possibly caused by intermittent hypoxia and sleep fragmentation in the mediators studied in this extremely obese population. As an independent association between metabolic abnormalities and OSA has been demonstrated not only in the general clinical population but also in high-risk cardiovascular and morbidly obese populations, we cannot rule out this being produced by other unexplored pathways. The absence of differences in the studied biomarkers between the groups does not detract from the importance of detecting OSA in morbidly obese patients, since it is a treatable condition with a great impact on health. It is therefore mandatory to perform further controlled studies to explore other pathways that could explain this relationship and enhance our understanding of the link between obesity, OSA, and cardiometabolic dysfunction, and our search for future treatment targets.
DISCLOSURE STATEMENT
Fondo de Investigación Sanitaria [Grant: FIS PI080800]; Spanish Respiratory Society SEPAR [Grant: Ayudas a la investigación 249/07]; Societat Catalana de Pneumologia SOCAP [Grants: 2052/08; 2052/09]. Fundació Catalana de Pneumologia FUCAP 2009. Study Supported by Air products. The authors have indicated no financial conflicts of interest.
ACKNOWLEDGMENTS
M. Clarke assisted with the English expression in versions of the manuscript. Advice on the statistical analysis was given by Daniel Quadras (IDIBELL, Barcelona, Spain). The authors thank the sleep unit staff of the Hospital Universitari de Bellvitge, Hospitalet de Llobregat, Spain, Tomas Brinquis, Pilar Garriga, Sandra Perez, Neus Martí, Carme Rodríguez, Rosa Miralda, Ariadna Farré and Montserrat Carreras from the Hospital de la Santa Creu i Sant Pau, Barcelona, Spain for their inestimable collaboration.
ABBREVIATIONS
- AHI
apnea-hypopnea index
- BMI
body mass index
- ESS
Epworth Sleepiness Scale
- FBG
fasting blood glucose
- HbA1c
percentage of glycosylated hemoglobin
- HDL
high-density lipoprotein
- HOMA
homeostasis model assessment
- IL-6
interleukin 6
- LDL
low-density lipoprotein
- MetS
metabolic syndrome
- OSA
obstructive apnea-hypopnea syndrome
- sCD40L
soluble CD40 ligand
- SpO2
arterial oxygen saturation by pulse oximetry
- Time SpO2 < 90%
percentage of sleep time with SpO2 below 90%
- TNF-α
tumour necrosis factor α
- VEGF
vascular endothelial growth factor
- VLDL
very low-density lipoprotein
- WC
waist circumference
SUPPLEMENTAL MATERIAL
Assay characteristics
REFERENCES
- 1.Alberti KG, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International association for the Study of Obesity. Circulation. 2009;120:1640–5. doi: 10.1161/CIRCULATIONAHA.109.192644. [DOI] [PubMed] [Google Scholar]
- 2.Ford ES. Risks for all-cause mortality, cardiovascular disease, and diabetes associated with the metabolic syndrome: a summary of the evidence. Diabetes Care. 2005;28:1769–78. doi: 10.2337/diacare.28.7.1769. [DOI] [PubMed] [Google Scholar]
- 3.Lam JC, Mak JC, Ip MS. Obesity, obstructive sleep apnoea and metabolic syndrome. Respirology. 2012;17:223–36. doi: 10.1111/j.1440-1843.2011.02081.x. [DOI] [PubMed] [Google Scholar]
- 4.Gasa M, Salord N, Fortuna AM, et al. Obstructive sleep apnea and metabolic impairment in severe obesity. Eur Respir J. 2011;38:1089–97. doi: 10.1183/09031936.00198810. [DOI] [PubMed] [Google Scholar]
- 5.Arnardottir ES, Mackiewick M, Gislason T, Teff KL, Pack A. Molecular signatures of obstructive sleep apnea in adults: A review and perspective. Sleep. 2009;32:447–70. doi: 10.1093/sleep/32.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonsignore MR, McNicholas WT, Montserrat JM, Eckel J. Adipose tissue in obesity and obstructive sleep apnoea. Eur Respir J. 2012;39:746–67. doi: 10.1183/09031936.00047010. [DOI] [PubMed] [Google Scholar]
- 7.Ernst MC, Sinal CJ. Chemerin. at the crossroads of inflammation and obesity. Trends Endocrinol Metab. 2010;21:660–7. doi: 10.1016/j.tem.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 8.Rosito GA, Massaro JM, Hoffmann U, et al. Pericardial fat, visceral abdominal fat, cardiovascular disease risk factors, and vascular calcification in a community-based sample: the Framingham Heart Study. Circulation. 2008;117:605–13. doi: 10.1161/CIRCULATIONAHA.107.743062. [DOI] [PubMed] [Google Scholar]
- 9.Duren DL, Sherwood RJ, Czerwinski SA, et al. Body composition methods: comparisons and interpretation. J Diabetes Sci Technol. 2008;2:1139–46. doi: 10.1177/193229680800200623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pascot A, Després JP, Lemieux I, et al. Contribution of visceral obesity to the deterioration of the metabolic risk profile in men with impaired glucose tolerance. Diabetologia. 2000;43:1126–35. doi: 10.1007/s001250051503. [DOI] [PubMed] [Google Scholar]
- 11.Barceló A, Piérola J, de la Peña M, et al. Free fatty acids and the metabolic syndrome in patients with obstructive sleep apnoea. Eur Respir J. 2011;37:1418–23. doi: 10.1183/09031936.00050410. [DOI] [PubMed] [Google Scholar]
- 12.Trombetta IC, Somers VK, Maki-Nunes C, et al. Consequences of comorbid sleep apnea in the metabolic syndrome implications for cardiovascular risk. Sleep. 2010;33:1193–9. doi: 10.1093/sleep/33.9.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kono M, Tatsumi K, Saibara T, et al. Obstructive sleep apnea syndrome is associated with some components of metabolic syndrome. Chest. 2007;131:1387–92. doi: 10.1378/chest.06-1807. [DOI] [PubMed] [Google Scholar]
- 14.Lui MM, Lam JC, Mak HK, et al. C-reactive protein is associated with obstructive sleep apnea independent of visceral obesity. Chest. 2009;135:950–6. doi: 10.1378/chest.08-1798. [DOI] [PubMed] [Google Scholar]
- 15.Karelis AD, Faraj M, Bastard JP, et al. The metabolically healthy but obese individual presents a favorable inflammation profile. J Clin Endocrinol Metab. 2005;90:4145–50. doi: 10.1210/jc.2005-0482. [DOI] [PubMed] [Google Scholar]
- 16.Aguilar-Salinas CA, García EG, Robles L, et al. High adiponectin concentrations are associated with the metabolically healthy obese phenotype. Clin Endocrinol Metab. 2008;93:4075–9. doi: 10.1210/jc.2007-2724. [DOI] [PubMed] [Google Scholar]
- 17.Karelis AD, St-Pierre DH, Conus F, Rabasa-Lhoret R, Poehlman ET. Metabolic and body composition factors in subgroups of obesity: what do we know? J Clin Endocrinol Metab. 2004;89:2569–75. doi: 10.1210/jc.2004-0165. [DOI] [PubMed] [Google Scholar]
- 18.Soverini V, Moscatiello S, Villanova N, Ragni E, Di Domizio S, Marchesini G. Metabolic syndrome and insulin resistance in subjects with morbid obesity. Obes Surg. 2010;20:295–301. doi: 10.1007/s11695-009-9999-z. [DOI] [PubMed] [Google Scholar]
- 19.Frohlich M, Imhof A, Berg G, et al. Association between C-reactive protein and features of the metabolic syndrome a population-based study. Diabetes Care. 2000;23:1835–9. doi: 10.2337/diacare.23.12.1835. [DOI] [PubMed] [Google Scholar]
- 20.Ridker PM, Coo N. Clinical usefulness of very high and very low levels of C-reactive protein across the full range of Framingham risk scores. Circulation. 2004;109:1955–9. doi: 10.1161/01.CIR.0000125690.80303.A8. [DOI] [PubMed] [Google Scholar]
- 21.Minoguchi K, Yokoe T, Tazaki T, et al. Increased carotid intima media thickness and serum inflammatory markers in obstructive sleep apnea. Am J Respir Crit Care Med. 2005;172:625–30. doi: 10.1164/rccm.200412-1652OC. [DOI] [PubMed] [Google Scholar]
- 22.Can M, Açikgöz S, Mungan G, et al. Serum cardiovascular risk factors in obstructive sleep apnea. Chest. 2006;129:233–7. doi: 10.1378/chest.129.2.233. [DOI] [PubMed] [Google Scholar]
- 23.Ryan S, Nolan GM, Hannigan E, Cunningham S, Taylor C. McNycolas WT. Cardiovascular risk markers in obstructive sleep apnea syndrome and correlation with obesity. Thorax. 2007;62:509–14. doi: 10.1136/thx.2006.066720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taheri S, Austin D, Lin L, Nieto F J, Young T, Mignot E. Correlates of serum C-reactive protein (CRP) - no association with sleep duration or sleep disordered breathing. Sleep. 2007;30:991–6. doi: 10.1093/sleep/30.8.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korner J, Leibel RL. To eat or not to eat - how the gut talks to the brain. N Engl J Med. 2003;349:926–8. doi: 10.1056/NEJMp038114. [DOI] [PubMed] [Google Scholar]
- 26.Galletiti F, Barbato A, Vesiero M, et al. Circulating Leptin levels predict the development of metabolic syndrome in middle-aged men: An 8-year follow-up study. J Hypertens. 2007;25:1671–7. doi: 10.1097/HJH.0b013e3281afa09e. [DOI] [PubMed] [Google Scholar]
- 27.Vogontzas AN, Papanicolaou DA, Bixler EO, et al. Sleep apnea and daytime sleepiness and fatigue: relation to visceral obesity, insulin resistance and hypercytokinemia. J Clin Endocrinol Metab. 2000;85:1151–8. doi: 10.1210/jcem.85.3.6484. [DOI] [PubMed] [Google Scholar]
- 28.Fantuzzi G. Molecular mechanisms in allergy and clinical immunology. J Allergy Clin Immunol. 2005;115:911–9. doi: 10.1016/j.jaci.2005.02.023. [DOI] [PubMed] [Google Scholar]
- 29.Wolk R, Svatikova A, Nelson CA, et al. Plasma levels of adiponectin, a novel adipocyte-derived hormone, in sleep apnea. Obesity. 2005;13:186–90. doi: 10.1038/oby.2005.24. [DOI] [PubMed] [Google Scholar]
- 30.Makino S, Handa H, Suzukawa K, et al. Obstructive sleep apnea syndrome, plasma adiponectin levels and insulin resistance. Clin Edocrinol. 2006;64:12–9. doi: 10.1111/j.1365-2265.2005.02407.x. [DOI] [PubMed] [Google Scholar]
- 31.Weigert J, Neumeier M, Wanninger J, et al. Systemic chemerin is related to inflammation rather than obesity in type 2 diabetes. Clin Endocrinol. 2010;72:342–8. doi: 10.1111/j.1365-2265.2009.03664.x. [DOI] [PubMed] [Google Scholar]
- 32.Feng X, Li P, Zhou C, Jia X, Kang J. Elevated levels of serum chemerin in patients with obstructive sleep apnea syndrome. Biomarkers. 2012;17:248–53. doi: 10.3109/1354750X.2012.658864. [DOI] [PubMed] [Google Scholar]
- 33.Kohler M, Stradling JR. Mechanisms of vascular damage in obstructive sleep apnea. Nat Rev Cardiol. 2010;7:677–85. doi: 10.1038/nrcardio.2010.145. [DOI] [PubMed] [Google Scholar]
- 34.Cherniack EP. Vascular endothelial growth factor and sleep apnea: clutching at straws in the night. Respiration. 2007;74:17–8. doi: 10.1159/000096835. [DOI] [PubMed] [Google Scholar]
- 35.De la Peña M, Barceló A, Barbe F, et al. Endothelial function and circulating endothelial progenitor cells in patients with sleep apnea syndrome. Respiration. 2008;76:28–32. doi: 10.1159/000109643. [DOI] [PubMed] [Google Scholar]
- 36.Imagawa S, Yamaguchi Y, Higuchi M, et al. Levels of vascular endothelial growth factor are elevated in patients with obstructive sleep apnea-hypopnea syndrome. Blood. 2001;98:1255–7. doi: 10.1182/blood.v98.4.1255. [DOI] [PubMed] [Google Scholar]
- 37.Szmitko PE, Wang CH, Weisel RD, Jeffries GA, Anderson TJ, Verma S. Biomarkers of vascular disease linking inflammation to endothelial activation: Part II. Circulation. 2003;108:2041–8. doi: 10.1161/01.CIR.0000089093.75585.98. [DOI] [PubMed] [Google Scholar]
- 38.Drager LF, Bortolotto LA, Figueiredo AC, Krieger EM, Lorenzi GF. Effects of continuous positive airway pressure on early signs of atherosclerosis in obstructive sleep apnea. Am J Respir Crit Care Med. 2007;176:706–12. doi: 10.1164/rccm.200703-500OC. [DOI] [PubMed] [Google Scholar]
- 39.Kohler M, Stoewhas AC, Ayers L, Senn O, et al. Effects of continuous positive airway pressure therapy withdrawal in patients with obstructive sleep apnea: a randomized controlled trial. Am J Respir Crit Care Med. 2011;184:92–9. doi: 10.1164/rccm.201106-0964OC. [DOI] [PubMed] [Google Scholar]
- 40.Hoyos CM, Killick R, Yee BJ, Phillips CL, Grunstein RR, Liu PY. Cardiometabolic changes after continuous positive airway pressure for obstructive sleep apnoea: a randomised sham-controlled study. Thorax. 2012;67:1081–9. doi: 10.1136/thoraxjnl-2011-201420. [DOI] [PubMed] [Google Scholar]
- 41.West SD, Nicoll DJ, Wallace TM, Matthews DR, Stradling JR. Effect of CPAP on insulin resistance and HbA1c in men with obstructive sleep apnoea and type 2 diabetes. Thorax. 2007;62:969–74. doi: 10.1136/thx.2006.074351. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Assay characteristics