
Fig. 6. Proposed model of CCM pathology.
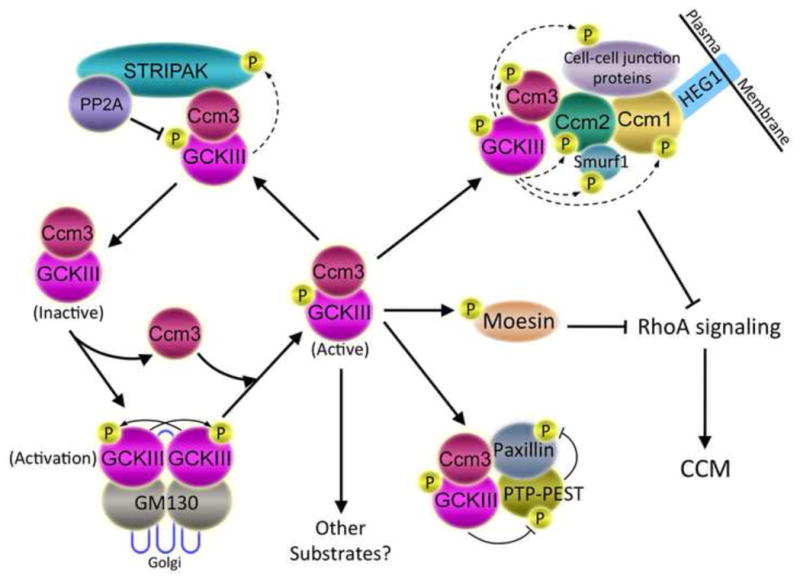
Binding of GCKIII monomers to GM130 in the Golgi induces GCKIII dimerization, autophosphorylation, and activation (lower left). Activated GCKIII kinases are bound and stabilized by Ccm3 (center) and targeted to the STRIPAK complex (upper left), the Ccm protein complex (upper right), the PTP-PEST-Paxillin complex (lower right), moesin (center right), and potentially to other unknown pathways. By binding to Ccm2 protein, Ccm3 is thought to bring GCKIII kinases to Ccm2 and Ccm1, forming a complex that regulates cell-cell junction formation, disassembly, and stability (upper right). Dashed arrows indicate potential phosphorylation of proteins by GCKIII kinases. For example, GCKIII kinases might phosphorylate STRIPAK components (upper left), Ccm proteins (upper right), Ccm protein-associated proteins (represented in the diagram by Smurf1, which is regulated by phosphorylation, but not currently known to be a substrate of GCKIII kinases), or neighboring junctional proteins. Both the Ccm1-Ccm2-Ccm3 complex and GCKIII kinase-phosphorylated moesin inhibit RhoA signaling (right), maintaining junctional homeostasis. Depletion of Ccm1, Ccm2, Ccm3, or GCKIII kinases leads to enhanced RhoA activity, junctional instability, and onset of CCM. Since STRIPAK negatively regulates Ccm3-GCKIII kinases, loss of STRIPAK function is expected to inhibit CCM-relevant pathways, and not induce CCM.