

SUMMARY
Antibodies (Abs) that preferentially target oncogenic receptors have been increasingly used for cancer therapy, but tumors often acquire intrinsic Ab-resistance after prolonged and costly treatment. Here, we arm the Ab with IFNβ and observed that it is more potent than first generation of Ab for controlling Ab-resistant tumors. This strategy controls Ab-resistance by rebridging suppressed innate and adaptive immunity in tumor microenvironment. Mechanistically, Ab-IFNβ therapy primarily and directly targets intra-tumoral dendritic cells, which re-activate CTL by increasing antigen cross presentation within the tumor microenvironment. Additionally, blocking PD-L1, which is induced by Ab-IFNβ treatment, overcomes treatment-acquired resistance and completely eradicates established tumors. Therefore, this study establishes a nextgeneration Ab-based immunotherapy that targets and eradicates established Ab-resistant tumors.
INTRODUCTION
Antibody targeting oncogenic receptors can directly inhibit tumor cell growth, providing an effective treatment option for cancer therapy(Hynes and Lane, 2005; Li et al., 2005). The major therapeutic effect of such antibody therapies is attributed to direct cytotoxicity to tumor cells by affecting oncogenic signal transduction. More recently, however, Fc receptor (FcR) signaling on immune cells is also recognized to be important for Ab mediated anti-tumor effect in vivo (Clynes et al., 2000; Musolino et al., 2008). We and others have shown that Ab-mediated tumor regression also depends on adaptive immunity in Ab-sensitive models (Abes et al., 2010; Mortenson et al., 2013; Park et al., 2010; Stagg et al., 2011; Yang et al., 2013). In Ab-sensitive tumor models, immune-activating molecules released during ADCC or by stressed tumor cells can effectively activate antigen-presenting cells (APCs), enhancing their ability to cross-prime and induce CTL responses. Recent exciting clinical trials used antibodies to block co-inhibitory signals on T cells, including CTLA-4, PD-1, and PD-L1, and demonstrated that reversing T cell suppression is another important way to improve the therapeutic effect against tumor (Brahmer et al., 2012; Sharma et al., 2011; Topalian et al., 2012; Weber, 2007). These results raise the possibility that the effect of targeted Ab cancer therapy can be further enhanced by selected immunotherapy.
Both primary and acquired resistances are major challenges for targeted therapy (Bardelli and Siena, 2010; Cobleigh et al., 1999). Most studies focus on the intrinsic resistance of oncogenic signaling, such as mutations within targeted oncogenes or in genes related to oncogenic pathways that contribute to Ab resistance (Bardelli and Siena, 2010; Misale et al., 2012; Sharma et al., 2007; Wheeler et al., 2008; Yonesaka et al., 2011). Currently, the major strategy to overcome Ab resistance in the host is to develop drugs targeting mutated oncogenes or oncogenic-pathway–related genes inside tumor cells (Bostrom et al., 2009; Fayad et al., 2013; Hurvitz et al., 2013; Krop et al., 2012; Yoon et al., 2011). Based on increasing intrinsic resistance after treatment with first generation of anti-oncogenic antibody, we propose a tumorextrinsic strategy to bypass intrinsic Ab resistance by reactivating both innate and adaptive immune cells inside the tumor. To achieve this goal, potent immune molecules that can elicit anti-tumor responses need to be identified.
Recently, an increase in type I interferons (IFNs) was found to correlate favorably with clinical immune responses against cancer (Fuertes et al., 2011). Furthermore, type I IFN signaling is essential to initiate anti-tumor T cell responses during spontaneous tumor rejection or additional various anti-tumor therapies (Burnette et al., 2011; Diamond et al., 2011; Fuertes et al., 2011; Stagg et al., 2011). These data suggest that type I IFNs are essential to initiate specific T cell responses against tumor cells. Type I IFNs have also been reported to activate memory T cells during viral infection (Kohlmeier et al., 2010). Thus far, however, systemically delivery of type I IFNs have been used cautiously in the clinic for cancer therapy due to limited potency and severe side effects (Trinchieri, 2010). Indeed, the action of this cytokine is poorly understood because it may function as either a immune activating or suppressing reagent in different disease models (Gonzalez-Navajas et al., 2012; Teijaro et al., 2013; Wilson et al., 2013). Timing, duration, and dosing of type I IFNs could be critical for determining its function as an immune activating or suppressing reagent.
Anti-CD20 coupled with IFNα showed better anti-tumor effect than anti-CD20 alone by direct and potent killing of IFNAR positive lymphoma (Xuan et al., 2010). Their data demonstrate that the IFNAR expression on tumor cell is important for the anti-tumor effect in Ab-sensitive tumor model. However, the role of IFNAR on host cells has not been well investigated. In this study, we linked IFNβ to anti-oncogenic receptor antibodies that directly target various carcinomas to test whether it can overcome Ab-resistance. We aim to investigate the detailed mechanism of how Ab-IFNβ changes the immune suppressive tumor microenvironment to induce anti-tumor immune responses and design efficient strategies to optimize targeted immune therapy.
RESULTS
Type I IFNs are required for effective tumor response to Ab therapy in vivo
Type I IFNs have emerged as potential key danger signals that initiate anti-tumor T cell responses during spontaneous tumor rejection or after various anti-tumor therapies (Burnette et al., 2011; Diamond et al., 2011; Fuertes et al., 2011; Stagg et al., 2011). We hypothesize that anti-oncogenic receptor antibodies induce type I IFN production in tumor tissues to bridge innate and adaptive immunity. To test whether the sensitivity of tumors to anti-oncogenic receptor antibodies in our models correlates with type I IFN levels after treatment, we generated two different tumor cell lines: Ab-sensitive and Ab-resistant. TUBO cells derived from Her2/neu Tg mice, in which Neu is the dominant signal for cell growth, serve as the anti-Neu Ab-sensitive tumor cell line (Rovero et al., 2000). The EGFR-transfected B16 cells, in which EGFR is unable to deliver growth signals, serve as the tumor cell line that is completely resistant to anti-EGFR treatment. We treated mice bearing these tumors with their respective antibodies and evaluated type I IFN production inside the tumor. We found that IFNα5 (Figure 1A) and IFNβ (Data not shown) production increased in the Ab-sensitive tumor model, but not in the Ab-resistant tumor models, suggesting that increased type I IFN production was caused by Ab-induced oncogenic receptor blockade and stress. Also, consistent with these results, we treated EGFR-transfected TUBO cells with anti-EGFR, in which the transfected EGFR can deliver signals and partially contribute to cell growth. The EGFR-transfected TUBO cells served as the tumor cell line that is partially resistant to anti-EGFR Ab treatment, and we observed similar reduction of type I IFN production (Figure S1). To test whether type I IFNs are required for the Ab-mediated anti-tumor effect in vivo, we treated mice with anti-IFNAR blocking Ab during anti-Neu treatment in the Ab-sensitive TUBO tumor model. We found that intratumorally blocking type I IFN signaling impaired the therapeutic effect of anti-Neu antibody (Figure 1B), suggesting that type I IFNs might be the essential cytokines for Ab-mediated tumor regression. It also raises the possibility that lower levels of type I IFNs may limit immune responses in host bearing Ab-resistant tumor. To further test whether delivering additional type I IFNs directly into tumors is sufficient to control Ab-resistant tumor growth, tumor-bearing mice were treated with Adenovirus encoding IFNβ (Adenovirus-IFNβ). As shown in Figure 1C, Adenovirus-IFNβ treatment by itself was able to sufficiently control Ab-resistant tumor growth. Taken together, these data argue that directing type I IFNs into tumors may be sufficient to overcome tumor immune evasion or antibody resistance.
Figure 1. Type I interferons are induced and necessary during antibody-mediated tumor regression.
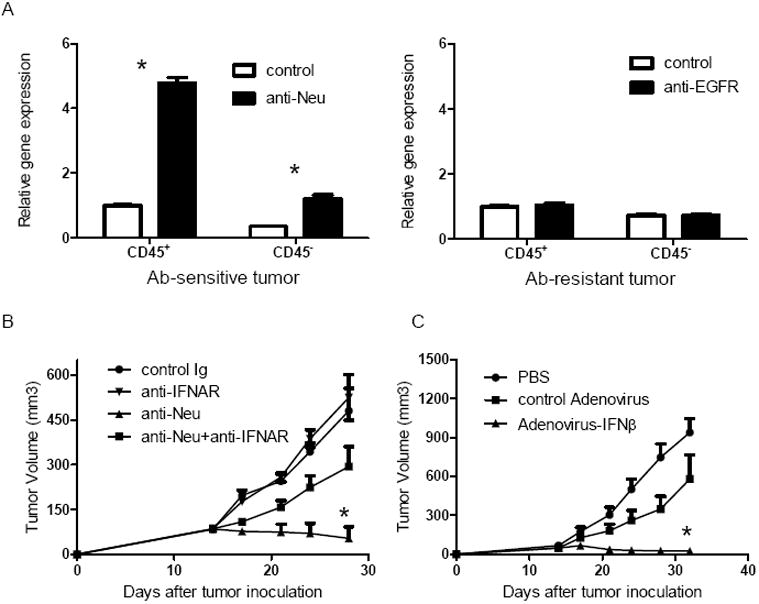
A) WT BALB/c mice (n=4/group) were injected subcutaneously with 5×105 TUBO cells, then 100 μg of anti-Neu (left panel) or control IgG was administered on day 14. WT B6 mice (n=4/group) were injected subcutaneously with 7×105 B16-EGFR cells, then 100 μg of anti-EGFR (right panel) or control IgG was administered on day 14. Four days after the treatment, tumor was digested and sorted to CD45+ and CD45- population. Quantitive real-time PCR was performed to detect the expression of mRNA levels of mIFNα5. Mean + SD are shown. B) WT BALB/c mice (n=5/group) were injected subcutaneously with 5×105 TUBO-EGFR cells, then 100 μg of anti-Neu was administered on days 14 and 21. 200ug of anti-IFNAR or control Ig was intratumorally administrated on the same days. The growth of tumor was measured and compared twice a week. Mean + SEM are shown. C) WT B6 mice (n=5/group) were injected subcutaneously with 5×105 B16-EGFR cells, then 2×109 Adenovirus-IFNβ or control virus was intratumorally administrated on days 14 and 21. The growth of tumor was measured and compared twice a week. *, Mean + SEM are shown. p < 0.05 compared with control group. One representative experiment of three is depicted. See also Figure S1.
Targeted delivery of IFNβ enhances the Ab-mediated therapeutic effect
Our data suggest that targeting tumor with type I IFNs has a potentially therapeutic effect, even for Ab-resistant tumors. There are two issues about using type I IFNs directly for cancer therapy: 1) Local delivery of type I IFNs might not be feasible for many patients. 2) In the clinic, systemic type I IFN administration has dose-dependent side effects, and its limited ability to shrink tumors may be associated with a failure to achieve a high enough concentration within tumor tissues. To better increase type I IFN concentration selectively inside tumors, we generated an anti-EGFR-IFNβ (Ab-IFNβ) fusion protein to deliver IFNβ directly into EGFR-expressing tumor tissues (Figure S2A). We first checked that the functions of both anti-EGFR and IFNβ remained intact in this fusion protein. Both functions were well maintained in this fusion protein, since it was able to bind to EGFR+ cells (Figure S2B) and activate the IFNAR signal pathway (Data not shown). We then examined whether anti-EGFR-IFNβ could specifically deliver IFNβ to the EGFR+ tumor site in vivo. Indeed, the concentration of anti-EGFR-IFNβ in tumor tissues remained high for at least 7 days (Figure S2C), while it dramatically decreased in other tissues less than one week after initial injection. Furthermore, we investigated the side effect of the anti-EGFR-IFNβ by measuring serum cytokines, ALT and AST levels (biomarkers for tissue injury). Among the cytokines we have checked, including TNF, IL-12, IFNγ, MCP-1, IL-6 and IL-10, there are slightly increased levels of IFNγ and MCP-1 expression at 6 hours and one day after injection (Figure S2D). We observed no increase of ALT and AST levels above base line after the treatment (Data not shown).
We treated mice bearing established EGFR+ tumors with our next generation anti EGFR-IFNβ fusion protein or the first generation anti-EGFR Ab, Cetuximab, in order to compare the potency of the fusion protein against its predecessor. Impressively, we observed that the therapeutic effect of anti-EGFR-IFNβ was much more effective at lower doses and shorter duration than anti-EGFR Ab alone in the partially resistant TUBO-EGFR tumor model (Figure 2A). Similarly elevated anti-tumor effects were observed from intra-mammary fat pat tumor injection model (Data not shown). To further test whether Ab-IFNβ could control tumor growth in a completely resistant tumor model, we tested the efficacy of anti-EGFR-IFNβ in B16-EGFR, an Ab-resistant model. As predicted, while anti-EGFR treatment alone could not inhibit B16-EGFR tumor growth in vivo, anti-EGFR-IFNβ treatment was again able to greatly control tumor growth (Figure 2B). KRAS mutations have been reported to be key factors contributing to anti-EGFR resistance in many patients bearing EGFR+ tumors (Misale et al., 2012). To test whether anti-EGFR-IFNβ is effective in a KRAS-mutation-induced Ab-resistant tumor model, we treated a KRAS-mutated H460 human tumor with anti-EGFR-IFNβ in our previously established adaptive immune-reconstituted Rag1-/- mice (Lee et al., 2009; Yang et al., 2013). We adoptively transferred 2 million of unpurified LN cells from OT-I Tg mice, which consist about 3~5% non OT-I CD8 T cells of total CD8 T cells, into Rag1-/- mice bearing established human H460 tumor. Therefore, only a few hundred T cells will react to human antigens, which is similar to the number of tumor-reactive T cells observed in human patients. In this model, the presence of 95% of OT-I T cells was used to prevent rapid homeostasis of extremely low number of CD8+ T cells in Rag1-/- mice. Anti-tumor T cell responses could be initiated for tumor regression if proper activation is triggered by targeted treatment in this tumor model (Lee et al., 2009; Yang et al., 2013). Although murine IFNβ cannot suppress human tumor cells in vitro, anti-EGFR-murine IFNβ showed superior therapeutic effect compared to anti-EGFR Ab alone in vivo (Figure 2C). To test whether anti-EGFR-IFNβ could control tumor metastasis, we injected B16-EGFR intravenously to WT mice in order to mimic tumor metastasis. We found that anti-EGFR-IFNβ could control metastatic tumor better than anti-EGFR alone and also prolong the survival of mice (Data not shown).
Figure 2. Ab-IFNβ dramatically enhances the anti-tumor effect of Ab.
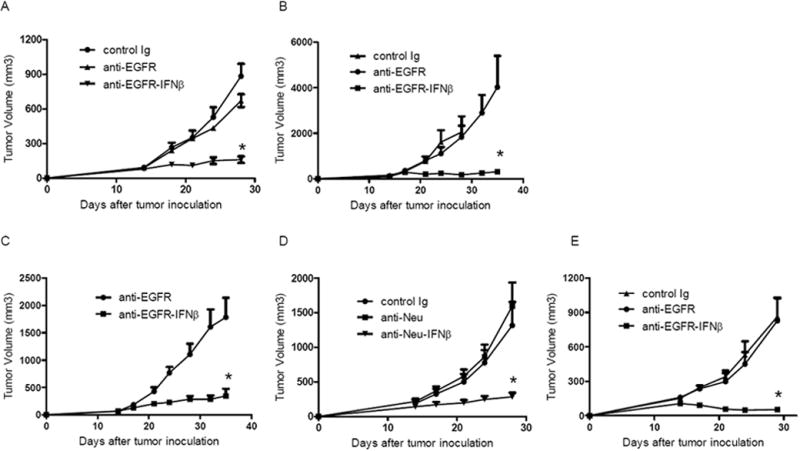
A) WT BALB/c mice (n=5/group) were injected subcutaneously with 5×105 TUBO-EGFR and treated with 25 μg of anti-EGFR, anti-EGFR-IFNβ or control Ab on days 14, 18, and 22. The tumor growth was measured and compared twice a week. B) WT B6 mice (n=5/group) were injected subcutaneously with 7×105 B16-EGFR and treated with 25 μg of anti-EGFR, anti-EGFR-IFNβ or control Ab on days 14, 18, and 22. The tumor growth was measured and compared twice a week. C) Rag1-/- mice (n=5/group) were injected subcutaneously with 3×106 H460 cells, and 2×106 OTI LN cells were adoptively transferred on day 13. Twenty five μg of anti-EGFR-IFNβ or control Ab was administered on days 14, 18, and 22. The growth of tumor was measured and compared twice a week. D) NeuOTI/OTII-Tg female mice (n=5/group) were injected subcutaneously with 1×106 NOP23 and treated with 25 μg of anti-Neu, anti-Neu-IFNβ or control Ab on days 14, 18, and 22. The tumor growth was measured and compared twice a week. E) EGFR-Tg mice (n=5/group) were injected subcutaneously with 5×105 B16-EGFR and treated with 25 μg of anti-EGFR, anti-EGFR-IFNβ or control Ab on days 14, 18, and 22. The tumor growth was measured and compared twice a week. *, p < 0.05 compared with control group. Mean + SEM are shown. One representative experiment of three is depicted. See also Figure S2 and Table S1.
Anti-tumor immunity mediated by anti-Neu Ab treatment is reduced in neu Tg mice, as high Neu expression (as self and tumor-associated antigens) in all mammary glands tolerizes host immune cells during early life due to the nature of transgene expression. The NEUOT-I/OT-II transgenic mice model, in which oncogenic rat neu is highly expressed in both mammary tumor and normal mammary gland, is an important model for monitoring the T cell response during spontaneous tumor development and adoptive transfer cancer therapy (Wall et al., 2007; Yang et al., 2009). NOP23 is one of the Neu+ mammary tumor cell lines generated from NEUOT-I/OT-II transgenic mice (Yang et al., 2009). Here we use NOP23 tumor bearing NEUOT-I/OT-II transgenic mice as a Neu-tolerized host model. Impressively, anti-Neu-IFNβ more profoundly inhibited tumor growth than anti-Neu Ab alone even in a tolerized model (Figure 2D). To test whether anti-EGFR-IFNβ can induce a similar tumor regression in EGFR tolerized host, we crossed EGFR Tg mice in mixed background (Politi et al., 2006) to B6 background for 10 generations to allow the growth of syngeneic B16-EGFR tumor. We treated B16-EGFR bearing EGFR Tg mice with anti-EGFR-IFNβ. Consistent with anti-Neu-IFNβ, anti-EGFR-IFNβ shows a better anti-tumor effect than anti-EGFR alone (Figure 2E). We also observed similar increased anti-tumor effect by arming another anti-EGFR Ab clone (Cetuximab, C225) with IFNβ (Data not shown) Collectively, these data suggest that Ab-IFNβ fusion protein therapy used at low doses and for short duration is superior compared to first generation Ab therapy for controlling tumors, even in Ab-resistant tumor models and in tolerized hosts (Table S1).
The therapeutic effect of anti-EGFR-IFNβ fusion protein depends on adaptive immunity
Since a previous study demonstrated that type I IFNs can directly and potently induce apoptosis of tumor cell for tumor regression (Xuan et al., 2010), we speculated that the anti-EGFR-IFNβ should show similar anti-tumor effect in Rag1-/- mice compared with WT mice if the direct killing of tumor cells is the major mechanism. However, anti-EGFR-IFNβ was surprisingly unable to inhibit tumor growth in these immune-compromised Rag1-/- mice at all (Figure 3A). Thus, these data support that the anti-EGFR-IFNβ-mediated therapeutic effect requires adaptive immunity, and not direct killing of tumor cells.
Figure 3. Anti-EGFR-IFNβ can induce anti-tumor specific CTL responses, which are responsible for the tumor regression.
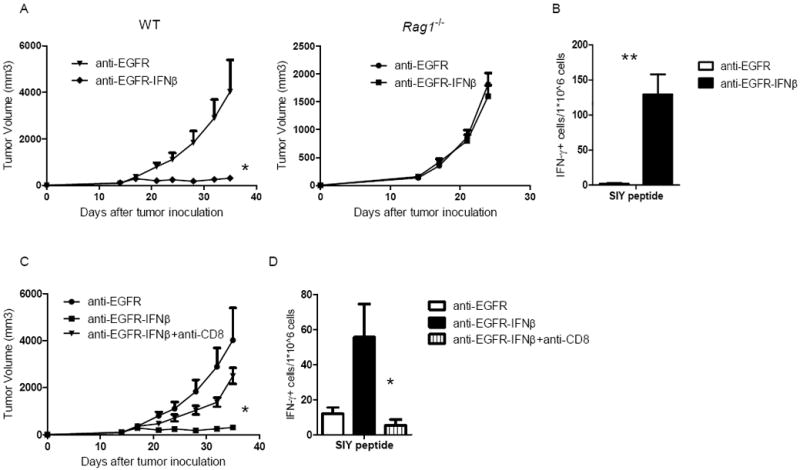
A) WT and Rag1-/- B6 mice (n=5/group) were injected subcutaneously with 5×105 B16-EGFR-SIY cells and treated with 25 μg of anti-EGFR-IFNβ or control Ab on days 14, 18, and 22. The tumor growth was monitored twice a week. B) Seven days after last treatment, dLN cells were collected and IFNγ ELISPOT assay was performed with SIY peptide restimulation. C) WT B6 mice (n=5/group) were injected subcutaneously with 5×105 B16-EGFR-SIY and treated with 25 μg of anti-EGFR-IFNβ or control Ab on days 14, 18, and 22. A CD8-depleting antibody (200μg/mouse) was administered on the same day as anti-EGFR-IFNβ. The tumor growth was measured and compared twice a week. D) Seven days after last treatment, dLN cells were collected and IFNγ ELISPOT assay was performed with SIY peptide restimulation. *, p < 0.05 compared with control group. Mean + SEM are shown. One representative experiment of three is depicted. See also Figure S3.
CD8+ T lymphocytes are the major cell population involved in controlling the growth of many tumors. To determine whether they are also involved in the anti-EGFR-IFNβ-mediated anti-tumor effect, we tracked the anti-tumor T cell response during the priming phase. We established a B16-EGFR-SIY tumor cell line in which the SIY peptide (SIYRYYGL) was linked to EGFR molecule; this SIY peptide serves as a surrogate marker that can be specifically recognized by endogenous or 2C Tg CD8+ T cells. After anti-EGFR-IFNβ treatment, drain lymph node (dLN) lymphocytes were isolated from tumor-bearing mice and stimulated with SIY peptide, and IFNγ production was measured as an effector-function readout of activated T cells. As shown in Figure 3B, anti-EGFR-IFNβ treatment increased IFNγ production from tumor-antigen specific T cells compared with anti-EGFR treatment alone. To address whether CD8+ cells are essential for the therapeutic effect of anti-EGFR-IFNβ, we administered a CD8-depleting antibody during anti-EGFR-IFNβ treatment in B16-EGFR-bearing WT B6 mice and measured tumor growth. CD8+ cell depletion eliminated the therapeutic effect of anti-EGFR-IFNβ (Figure 3C), suggesting that adaptive immune responses are required for controlling tumor growth. Consistent with this finding, we found that the T cell response ex vivo dramatically decreased after anti-CD8 depletion (Figure 3D). The depletion of other cells, including NK and B cells, did not affect the anti-tumor effect of anti-EGFR-IFNβ (Figure S3). Taken together, these data suggest that anti-EGFR-IFNβ treatment can increase T cell priming for tumor regression.
The therapeutic effect of anti-EGFR-IFNβ requires IFNAR expression on host bone marrow-derived cells
IFNAR is widely expressed on almost all cell types, thus both normal and tumor cells are potential targets of anti-EGFR-IFNβ. The efficacy of anti-CD20-IFNα fusion protein has been reported to require IFNAR expression on tumor cells (Xuan et al., 2010). We therefore hypothesized that host cell expressed IFNAR may not be required for the therapeutic effect of anti-EGFR-IFNβ. To this end, we compared the anti-tumor effect in tumor-bearing Ifnar1-/- mice, which lack IFNAR expression on host cells, but not on tumor cells. To our surprise, the therapeutic effect of anti-EGFR-IFNβ was abolished in Ifnar1-/- mice (Figure 4A). To study whether the IFNAR signaling on host cells is associated with adaptive immune responses, we checked the T cell responses after treatment and found that there was no increased CTL response in Ifnar1-/- mice (Figure 4B). These results suggest that the therapeutic effect and related CTL responses by anti-EGFR-IFNβ requires IFNAR-mediated activation in host cells, but not tumor cells. Since all host tissues express IFNAR, we constructed IFNAR bone marrow chimeric (BMC) mice to further dissect whether IFNAR-expressing bone marrow-derived cells or stromal cells of host are required for the anti-tumor effect. We found that IFNAR expression on bone marrow-derived cells is required, since the anti-tumor effect of anti-EGFR-IFNβ is dramatically impaired in Ifnar1-/- BM-reconstituted mice (Figure 4C). These data suggest that the anti-EGFR-IFNβ mediated its anti-tumor effect not by directly inhibiting tumor cell growth, but by activating host bone marrow-derived cells to change the tumor microenvironment.
Figure 4. The anti-tumor effect of anti-EGFR-IFNβ requires IFNAR expression on host bone marrow-derived cells.
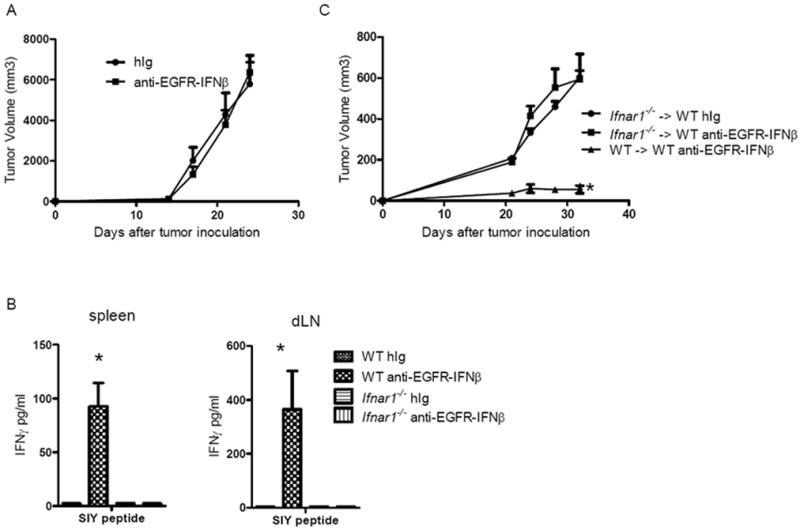
A) WT and Ifnar1-/- B6 mice (n=5/group) were injected subcutaneously with 5×105 B16-EGFR-SIY cells and treated with 25 μg of anti-EGFR-IFNβ or control Ab on days 14, 18, and 22. The tumor growth was monitored twice a week. B) Seven days after the last treatment, dLN and spleen cells were collected and IFNγ producing in supernatant was detected by CBA assay. C) Thirty days after indicated bone marrow chimera reconstitution, mice were injected subcutaneously with 7×105 B16-EGFR-SIY and treated with 25μg of anti-EGFR-IFNβ or control Ig on days 14, 18, and 22. Tumor growth was measured and compared twice a week. *, p < 0.05, compared to control group. Mean + SEM are shown. One of two representative experiments is shown.
Elevated DC cross-presentation contributes to the anti-tumor effect of anti-EGFR-IFNβ
Given that CD8+ cells are essential for the anti-tumor effect of anti-EGFR-IFNβ treatment, we further explored the mechanism underlying how anti-EGFR-IFNβ enhances CTL responses. We observed a dramatic increase of IFNγ producing CD8+ cells after anti-EGFR-IFNβ treatment even without exogenous SIY peptide stimulation, suggesting that cross-presentation of APCs is increased (Figure 5A). Since cross-presentation was proposed to be the dominant priming mechanism to activate CTLs for anti-tumor immunity (Huang et al., 1994; Kurts et al., 2010), we speculated that increase of cross-priming by fusion protein might be essential to restore DC function to re-activate CTL inside tumor and draining LN. To address the possibility that anti-EGFR-IFNβ could increase the cross-presentation of APCs, we used an antigen-specific system to track the priming and activation of tumor-antigen-specific T cells. DCs from the dLNs of anti-EGFR-IFNβ-treated B16-EGFR-SIY-bearing mice were assessed for their ability to enhance the specific anti-tumor CTL response by incubating them with SIY-reactive 2C T cells in an ex vivo assay. Indeed, DCs from anti-EGFR-IFNβ-treated mice induced more IFNγ production from 2C T cells (~33 times) compared to anti-EGFR treatment, even without re-stimulation by exogenous SIY peptide (Figure 5B). Cumulatively, the data suggest that anti-EGFR-IFNβ-activated DCs enhanced CD8+ T cell activation through increased cross-priming function. Furthermore, these results indicate that the IFNβ component of the fusion protein is responsible for activating the DC cross-presentation pathway, as anti-EGFR alone did not induce strong DC activation.
Figure 5. Anti-EGFR-IFNβ restores the cross-presentation ability of DCs.
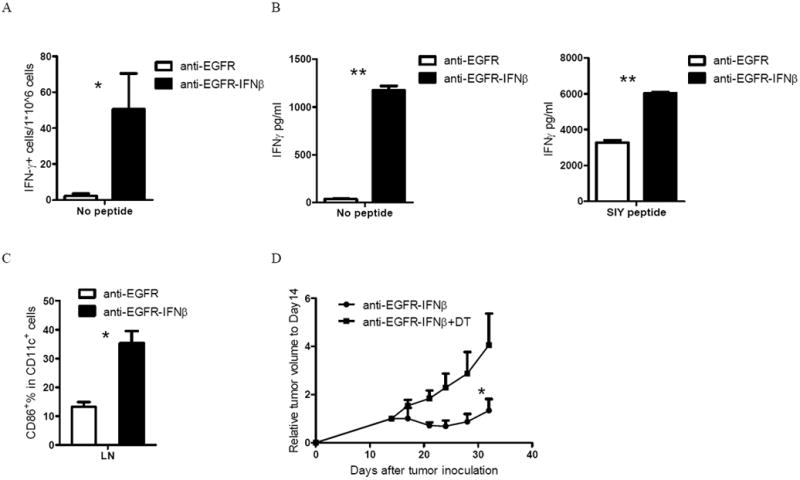
A) WT B6 mice (n=5/group) were injected subcutaneously with 5×105 B16-EGFR-SIY and treated with 25 μg of anti-EGFR-IFNβ or control Ab on days 14 and 18. Seven days after the last treatment, dLN cells were collected and IFNγ ELISPOT assay was performed without SIY peptide restimulation. B) WT B6 mice (n=5/group) were injected subcutaneously with 5×105 B16-EGFR-SIY and treated with 25 μg of anti-EGFR-IFNβ or control Ab on days 14 and 18. Four days after the last treatment, DCs from dLNs were purified by CD11c+ positive selection and incubated with purified naïve 2C T cells with or without SIY peptide restimulation. IFNγ production was measured 2 days later. C) DCs activation markers were also measured by flow cytometry. D) About 40 days after indicated bone marrow chimera reconstitution, mice were injected subcutaneously with 5×105 B16-EGFR-SIY and treated with 25μg of anti-EGFR-IFNβ or control Ig on days 14, 18, and 22. DT or PBS was administrated on the same days as treatment. Tumor growth was measured and compared twice a week. *, p < 0.05 compared with control group. Mean + SEM are shown. One representative experiment of two or three is depicted. See also Figure S4.
Additionally, exogenous antigenic peptide (SIY) was added into the culture to illicit an increased SIY specific 2C T cell response for better analysis of the overall direct priming function of DCs. DCs from the anti-EGFR-IFNβ-treated mice induced ~2 times more IFNγ production from the 2C T cells compared to anti-EGFR treatment upon exogenous SIY-peptide re-stimulation (Figure 5B). These results suggest that DCs from the dLN of anti-EGFR-IFNβ-treated hosts are likely to be more activated than DCs from mice treated with anti-EGFR alone. Indeed, these DCs have high expression of activation markers, including CD86, as assessed by flow cytometry (Figure 5C). To further dissect whether anti-EGFR-IFNβ-induced DC activation is responsible for the enhanced T cell activation, tumor-bearing CD11c-DTR BMC mice were treated with DT to deplete DCs during anti-EGFR-IFNβ treatment. We found that the anti-EGFR-IFNβ-mediated therapeutic effect is impaired when DCs are not present (Figure 5D). Taken together, these data suggest that improved CD8+ CTL priming and function by increased DC cross-presentation might be the major mechanism underlying the therapeutic effect of anti-EGFR-IFNβ.
Anti-EGFR-IFNβ directly targets DCs to reverse the tolerized tumor microenvironment
Our data revealed that increased cross-priming is important for the improved anti-tumor effect by anti-EGFR-IFNβ treatment. However, the IFNAR-expressing cells directly responsible for the therapeutic effect of anti-EGFR-IFNβ need to be identified. To address this issue, Ifnar1flox/flox mice were bred to various Cre-Tg mice. When IFNAR was selectively absent in CD11c+ cells in CD11c-Cre+Ifnar1flox/flox mice, the anti-tumor effect of anti-EGFR-IFNβ disappeared (Figure 6A), suggesting that direct DC activation by anti-EGFR-IFNβ may be the major contributor to its therapeutic effect. When IFNAR was selectively absent in T cells in CD4-Cre+Ifnar1flox/flox mice, the anti-tumor effect of anti-EGFR-IFNβ was slightly impaired (Figure 6B), suggesting that direct targeting of IFNAR on T cells may further activate T cells for improved anti-tumor effect. To further test this idea, we evaluated the effect of anti-EGFR-IFNβ stimulation in both DC and T cell activation in an in vitro assay; indeed, anti-EGFR-IFNβ increased the activation of both DCs and T cells (Figure S5). These data collectively suggest that direct activation of IFNAR-expressing DCs plays a major role in the anti-EGFR-IFNβ-mediated therapeutic effect, which can be further enhanced by engaging IFNAR expressed on T cells.
Figure 6. DCs are the major cell type directly responding to the anti-EGFR-IFNβ treatment.
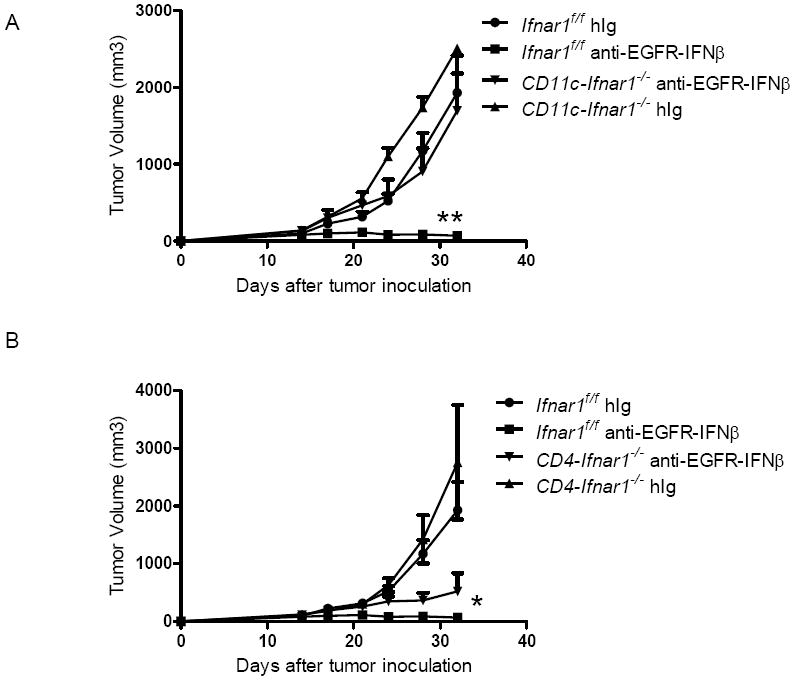
A) Ifnar1flox/flox and CD11c-Cre Ifnar1flox/flox mice (n=5/group) were injected subcutaneously with 5×105 B16-EGFR-SIY and treated with 25 μg of anti-EGFR-IFNβ or control Ab on days 14, 18 and 22. Tumor growth was measured and compared twice a week. B) Ifnar1flox/flox and CD4-Cre Ifnar1flox/flox mice (n=5/group) were injected subcutaneously with 5×105 B16-EGFR-SIY and treated with 25 μg of anti-EGFR-IFNβ or control Ab on days 14, 18 and 22. Tumor growth was measured and compared twice a week. *, p < 0.05 compared with control group. Mean + SEM are shown. One representative experiment of two is depicted. See also Figure S5.
Antagonizing anti-EGFR-IFNβ-induced PD-L1 expression achieves tumor-free outcome
Although anti-EGFR-IFNβ fusion achieved a more effective anti-tumor effect than anti-EGFR Ab alone, residual tumor eventually relapsed. We wondered whether anti-EGFR-IFNβ treatment might induce inhibitory molecule expression on tumor cells to prevent tissue damaging immune responses, ultimately weakening the anti-tumor effect over time. Although IFNγ is known to be the major inducer of PD-L1 on tumor cells (Blank et al., 2004), it raises possibility that type I IFNs have a similar effect. We accordingly evaluated PD-L1 expression after anti-EGFR-IFNβ treatment and clearly observed increased PD-L1 expression on tumor cells both in vivo and in vitro (Figure 7A and 7B). The data confirmed that type I IFNs could induce the expression of this inhibitory molecule directly. To test whether blocking PD-L1 expression can potentiate IFN-mediated tumor rejection, we then combined anti-PD-L1 with anti-EGFR-IFNβ treatment and indeed observed that anti-PD-L1 blockade could further enhance the long-term efficacy of anti-EGFR-IFNβ; mice remained tumor-free for at least 60 days after treatment (Figure 7C). To test whether such treatment can have a prolonged protective effect in preventing the growth of dormant residual cancer, mice without detectable tumor were rechallenged with lethal tumor dose. Impressively, all of them were completely resistant to tumor rechallenge. Moreover, blocking the PD-L1 pathway by anti-PD-L1 treatment further enhanced the specific anti-tumor T cell response (Figure 7D). To test whether IFN-induced PD-L1 is a dominant mechanism for immune evasion, we tested whether blockade of two major inhibitory pathways on T cells, CTLA-4 and BTLA, could synergic with anti-EGFR-IFNβ treatment. In contrast to anti-PD-L1, we did not observe similar synergistic effect when combining anti-EGFR-IFNβ with anti-CTLA-4 or anti-BTLA (Figure S6). Collectively, our data indicate that antagonizing anti-EGFR-IFNβ-induced PD-L1 expression can maximize the anti-tumor effect of anti-EGFR-IFNβ and achieve an impressive tumor-free outcome, even for Ab-resistant tumors. This combination-based strategy will likely increase the overall response and cure rates of Ab-resistant hosts, even in hosts that initially fail to respond to anti-PD-1 or anti-PD-L1 antibodies.
Figure 7. Antagonizing PD-L1 expression induced by anti-EGFR-IFNβ achieves tumor-free outcome.
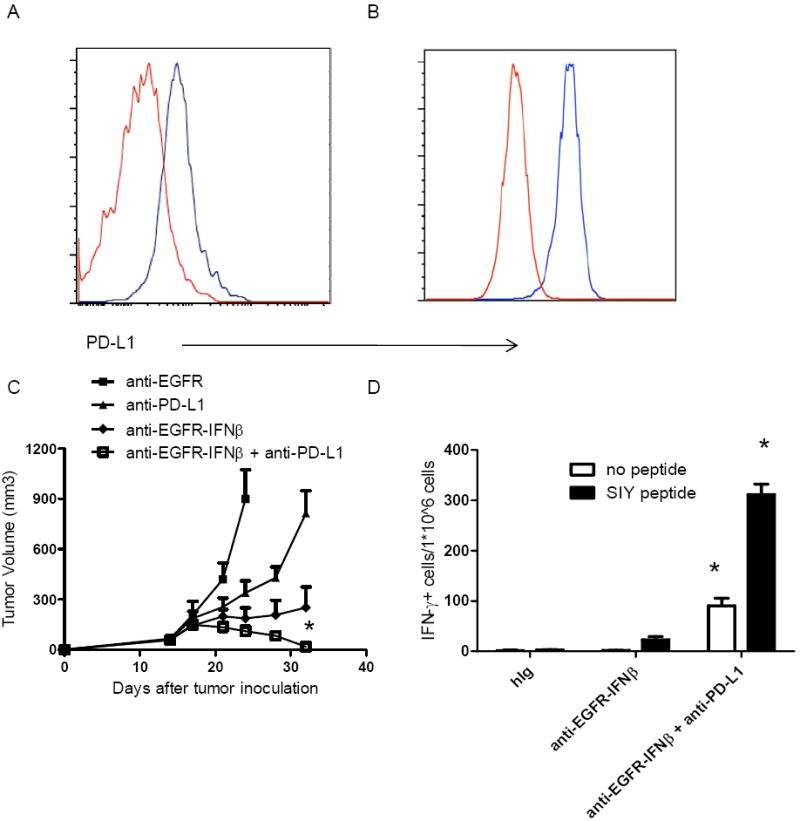
A) WT B6 mice were injected subcutaneously with 5×105 B16-EGFR-SIY cells and treated with 25 μg of anti-EGFR-IFNβ or control Ab on days 14. Two days later, tumor cells were collected and PD-L1 expression was analyzed by flow cytometry. Red line indicates control Ig treated and blue line indicates anti-EGFR-IFNβ treated group. B) B16-EGFR-SIY cells and treated with 0.02μg/ml of anti-EGFR-IFNβ during in vitro cell culture. One day later, tumor cells were collected and PD-L1 expression was analyzed by flow cytometry. Red line indicates control Ig stimulated and blue line indicates anti-EGFR-IFNβ stimulated cells. C) WT B6 mice (n=5/group) were injected subcutaneously with 5×105 B16-EGFR-SIY cells and treated with 25 μg of anti-EGFR-IFNβ or control Ab on days 14, 18, and 22. A PD-L1-blocking antibody (400μg/mouse) was administered on the same day as anti-EGFR-IFNβ. The tumor growth was measured and compared twice a week. D) Fourteen days after last treatment, splenocytes were collected and IFNγ ELISPOT assay was performed. *, p < 0.05 compared with control group. Mean + SEM are shown. One representative experiment of three is depicted. See also Figure S6.
DISCUSSION
First generation of antibodies to oncogenic receptors has been increasingly used; but intrinsic and extrinsic resistance has become major clinical challenges. We now reveal that type I IFNs play an essential and sufficient role to bridge innate and adaptive anti-tumor immune responses during antibody-based anti-tumor therapy. Clinically relevant, we have developed Ab-IFNβ as a representative next generation of Ab-based therapy, which is superior to first generation Ab therapy, even in Ab-resistant tumor cells. Our study demonstrated the essential and sufficient role of type I IFNs for Ab mediated tumor control: 1) Type I IFN production is elevated after antibody-based anti-tumor treatment. Blocking type I IFN signaling impairs Ab-mediated tumor regression. 2) Targeted delivery of IFNβ inside the tumor site by Ab-IFNβ fusion protein greatly amplifies the therapeutic effect of Ab. 3) Such anti-tumor effect depends on DC and CTL. 4) Ab- IFNβ increases DC cross-presentation and anti-tumor CTLs by directly targeting IFNAR on DCs, but not on tumor cells or T cells.
Most current strategies to improve the therapeutic effect of Ab through tumor intrinsic angle focus on how to increase tumor cell cytotoxicity by Ab and cytotoxic drugs conjugates (ADC) (Fayad et al., 2013; Hurvitz et al., 2013; Krop et al., 2012). Similar to first generation of Ab, host resistance can develop after prolonged ADC treatment. Indeed, many patients still undergo relapse or develop metastasis despite initial regression by potent cytotoxic effect of ADC. We propose that this resistance develops because ADC relies on directly killing specific tumor cells and thus cannot target all tumor cells, causing some to be selected or to acquire the ability to escape direct killing. The next generation of Ab-based treatment we describe here, however, can overcome these types of resistance by revitalizing innate and adaptive immune cells inside the tumor and generating specific T cell responses that target multiple shared tumor-mutated antigens. Thus, this strategy might be able to eradicate residual tumor cells that Ab cannot directly target. Our study further argues that improving the effectiveness of anti-tumor effector and memory T cell responses will be important for preventing relapse.
Type I IFNs have multiple potential effects on tumor growth, including inhibiting proliferation, inhibiting angiogenesis, activating innate cells, bridging innate and adaptive immunity, and directly activating adaptive immune responses. The reported role of type I IFNs on tumor cells and immune cells is complex and sometimes controversial. In the pioneer study shown by Xuan et al, anti-CD20-IFNα clearly showed that type I IFNs can have direct cytotoxic effect in lymphoma. Indeed, lymphomas, including human lymphoma, are very sensitive to type I IFN-mediated direct killing not only in vitro but also in vivo. Unexpectedly, we have shown that our Ab-IFNβ can control various carcinomas by different modes of action. 1) Rag1-/- mice bearing carcinomas fail to respond to Ab-IFNβ, suggesting the major role of adaptive immune cells. 2) CD8 depleted mice showed early relapse, suggesting essential role of CTL. 3) IFNAR+carcinomas in Ifnar1-/- mice fail to respond to Ab-IFNβ, suggesting that host IFNAR signaling is essential. 4) We have further pinpointed that IFNAR on DCs is essential for tumor regression. Ab-IFNβ cannot kill carcinoma cell line effectively in vitro as they are less sensitive to type I IFNs than lymphoma cells. Therefore, the two studies are complementary and cover two different modes of action for IFN-mediated tumor regression, depending on the sensitivity of targeted tumor to type I IFNs.
Endogenous type I IFNs have been shown to be required for rejection of highly immunogenic tumor cells, which cannot grow in WT mice (Diamond et al., 2011; Fuertes et al., 2011). The studies also showed that type I IFNs are only required for the first 6 days after tumor inoculation before tumor is established. Once the tumor is established past the first 6 days, the role of endogenous type I IFNs is diminished. Complementarily, our study showed that endogenous type I IFNs can still regain their essential role after blockade of oncogenic addiction on established tumor that subsequently triggers innate and adaptive immune responses. It is worthwhile to mention that our therapeutic effect of Ab-IFNβ is dependent on DCs, but not on CD8α+ DCs as shown in Diamond’s study since our Ab-IFNβ is effective in Batf3-/- mice (Figure S4), which might imply different mechanisms for enhancing the efficacy of antibody-based therapeutics.
It is currently unclear whether the more effective strategy to reactivate the anti-tumor T cell response within the tumor is to target the activation of T cells or APCs. We recently observed that while ScFv (Neu)-IL-2 and -IL-15 could expand T cells, even inside tumor tissues, these T cells then failed to suppress tumor growth (Data not shown); thus, directly targeting T cells without increasing APC function may not initiate tumor specific immune responses and instead unselectively amplify all pre-existed T cell responses. Recent clinical trials testing antibodies that block co-inhibitory signals in T cells (CTLA-4, PD-1, and PD-L1) have demonstrated that reversing T cell suppression is important for effective cancer immune therapy, as it impressively controlled tumor growth in 15–25% of cancer patients who failed to respond to conventional treatment (Brahmer et al., 2012; Sharma et al., 2011; Topalian et al., 2012; Weber, 2007). However, since these anti-inhibitory receptor Abs target all T cells within the body rather than targeting T cells within tumor tissues directly, they may cause unwanted tissue damage or even autoimmune disease. Our current study demonstrates that selective combination of Ab-IFNβ and anti-PD-L1 can have synergistic effect on tumor regression, raising interest in developing other combinations in the future.
Cross-presentation is the dominant priming mechanism to activate CTL responses in anti-tumor immunity (Huang et al., 1994; Kurts et al., 2010). Since DCs are the most important APCs for cross-presentation (Steinman, 2012), targeting DCs with Ab- IFNβ will confer several advantages. First, Ab-IFNβ does not directly kill tumor cells, which reduces side effects from Ab-IFNβ targeting normal cells outside tumor tissues. If DCs in normal tissues do happen to get activated by this immune therapy, they cannot activate Ag-specific T cells due to the lack of mutated neo-antigens and pre-existed Ag-specific T cells. Second, Ab-IFNβ mobilizes DCs to present a variety of tumor-derived antigens to T cells, initiating T cell responses against multiple mutated neo-tumor antigens from all tumor cells, even those not originally targeted by Ab therapy; this will ultimately help prevent the appearance of resistant clones. Third, Ab-IFNβ therapy is a rather short-term and low-dose treatment, unlike the prolonged and high-dose treatment of Ab alone, and should have lower toxicity than Ab-cytotoxic drug conjugates.
Overall, this study has several important implications for the cancer immunotherapy field. First, it establishes a way to create the next generation of Ab-based treatment, such as the Ab-IFNβ fusion protein, that elicits the adaptive arm of the immune response to more effectively deal with Ab resistance and relapse. Enhancing the CTL response can then, in turn, kill more tumor cells to create a positive-feedback loop. Second, our study provides strong evidence that type I IFNs, which link innate and adaptive anti-tumor immunity, are key players for Ab-mediated tumor regression and therefore provide an important target for cancer immunotherapy. Third, this study reveals that DCs are the major tolerized cell type in tumors, implying that they play a major role in determining the immunosuppressive tumor microenvironment. Therefore, targeting DCs will be another important strategy to improve the efficacy of cancer immunotherapy. Fourth, blocking inhibitory PD-L1 up-regulated by Ab-IFNβ treatment further enhanced the anti-tumor effect, which puts forth the concept that antagonizing immunotherapy-induced adaptive resistance will maximize the therapeutic effect of immune therapy and could guide future clinical treatment. Collectively, the strategies used in this study open up several avenues for optimizing targeted immune therapy that may have great impact in anti-tumor drug discovery and clinical cancer therapy.
EXPERIMENTAL PROCEDURES
Mice
C57BL/6J and BALB/c mice were purchased from Harlan. Rag1-/-, 2C CD8+ TCR transgenic, CD11c-DTR transgenic, Batf3-/-, CD11c-Cre and CD4-Cre transgenic mice were purchased from JAX. Ifnar1-/- mice were kindly provided by Dr. Anita Chong at the University of Chicago. Ifnar1flox/flox mice were kindly provided by Dr. Ulrich Kalinke at Institute for Experimental Infection Research, Hannover, Germany (Kamphuis, Junt et al. 2006). NeuOT-I/OT-II transgenic mice in B6 background were kindly provided by Dr. Brad H. Nelson at Trev & Joyce Deeley Research Centre, British Columbia, Canada (Wall, Milne et al. 2007). Tet-on-EGFR Tg mice were kindly provided by Harold Varmus (NCI) in 2009 and crossed to B6 background up to 10 generations (Politi et al., 2006). All mice were maintained under specific pathogen-free conditions. Animal care and use were in accordance with institutional and NIH protocols and guidelines, and all studies were approved by the Animal Care and Use Committee of the University of Chicago.
Cell lines and reagents
H460 was purchased from ATCC. TUBO was cloned from a spontaneous mammary tumor in a BALB/c Neu-transgenic mouse (Rovero et al., 2000). TUBO-EGFR was selected after transfection of pSEB-EGFR (L858R) plasmid with 2 μg/mL of Blasticidin (InvivoGen). B16-EGFR and B16-EGFR-SIY were selected for single clone after transduced by lentivirus expressing human EGFR (L858R) or EGFR (L858R)-SIY. NOP23 was cloned from a spontaneous mammary tumor in a B6 NeuOT-I/OT-II-transgenic mouse and kindly provided by Dr. Brad H. Nelson at Trev & Joyce Deeley Research Centre, Canada. H460, TUBO, B16 and its derivatives were cultured in 5% CO2 and maintained in vitro in DMEM supplemented with 10% heat-inactivated fetal bovine serum (Sigma), 2 mmol/L L-glutamine, 0.1 mmol/L MEM nonessential amino acids, 100 units/mL penicillin, and 100 μg/mL streptomycin. Anti-EGFR mAb Cetuximab was purchased from Imclone. Anti-CD8 (YTS 169.4.2), anti-NK1.1 (PK136), anti-Neu (7.16.4) Abs were produced in-house. Anti-PD-L1 (10F.9G2) and anti-Ly-6G (1A8) Abs were purchased from BioXcell. Anti-CD20 (5D2) Ab was kindly provided by Ouyang Wenjun (Genentech, San Francisco). Anti-IFNAR (MAR-5A3) Ab was kindly provided by Dr. Robert Schreiber (Washington University, St. Louis). Adenovirus expressing murine IFNβ was produced as previously described (Burnette et al., 2011).
Production of Ab-IFNβ fusion protein
The V region of anti-EGFR was cloned from anti-EGFR (LA22) hybridoma (ATCC). The V region of heavy chain and light chain were cloned into Abvec-IgG1 and Abvec-kappa, respectively (Smith et al., 2009). Then mouse IFNβ was inserted into the C terminal of heavy chain as a fusion protein with a SGGGGSGGGGSGGGGSGGGG linker. The whole heavy chain with IFNβ and light chain were cloned to pEE6.4 and pEE12.4 (Lonza), respectively. Then both vectors were digested by NotI and BamHI. The complete hCMV-MIE-heavy chain/SV40 transcription unit form digested pEE6.4 plasmid was ligated into the large NotI-BamHI fragment from the digested pEE12.4 plasmid containing the light chain expression cassette. The plasmid containing both heavy chain and light chain was transfected into CHO cell and stable clones were established according to the manual (Lonza). The fusion protein anti-EGFR-IFNβ was purified by Protein A column according to the manual (Repligen Corporation). Anti-Neu-IFNβ was produced by the same way except that the cDNA of V region was from anti-Neu (7.16.4) hybridoma.
Tumor growth and treatments
Approximately 6×105 TUBO, TUBO-EGFR, B16-EGFR, B16-EGFR-SIY or NOP23 cells were injected subcutaneously on the right flank into 6-12 week old mice. Tumor volumes were measured along 3 orthogonal axes (a, b, and c) and calculated as tumor volume = abc/2. After tumor was established (~14 days), mice were treated with 3 intratumoral injections of 25 μg of anti-EGFR-IFNβ or control antibody every four days. For CD8 depletion experiments, 200 μg of anti-CD8 antibody was injected intraperitoneally at the same time as the anti-EGFR-IFNβ treatment. Approximately 3×106 H460 cells were injected subcutaneously on the right flank into 6-12 week old Rag1-/- mice. After the tumor was established (~13 days), 2×106 LN cells from OTI TCR transgenic mice were adoptively transferred to mice by intravenous injection. One day later, mice were treated with 3 intratumoral injections of 25 μg of anti-EGFR-IFNβ or control antibody on day 14, 18 and 22 after tumor inoculation.
Type I interferons mRNA expression after antibody treatment
Approximately 6×105 TUBO, TUBO-EGFR or B16-EGFR cells were injected subcutaneously on the right flank into 6-12 week old mice. Mice were treated with 100 μg of anti-Neu, anti-EGFR or control antibody on day 14 after tumor inoculation. Four days later, tumor was digested with 1mg/ml of collagenase VIII (Sigma) and 200ug/ml DNase I (Sigma) at 37°C for 30 minutes. Live CD45+ and CD45- cell population were sorted by FACS AriaII (BD Bioscience). Total RNA was isolated by RNeasy mini kit (Qiagen) and reverse transcripted to cDNA by Sensiscript RT kit (Qiagen).The expression level of type I interferons mRNA was analyzed by quantitative real-time PCR. The primers used for the assay are the following: mIFNα5, 5’;-ATGAAGTCCATCAGCAGCTC, 5’;-AGGGGCTGTGTTTCTTCTCT; β-actin, 5’;-ACACCCGCCACCAGTTCGC, 5’;-ATGGGGTACTTCAGGGTCAGGATA.
Measurement of IFNγ-secreting T cells by ELISPOT or CBA assay
SIY peptide-reactive T cells were measured by ELISPOT assay. Spleen or lymph node cells were resuspended in RPMI 1640 supplemented with 10% FCS, 2 mmol/L L-glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin. A total of 1-4×105 spleen or lymph node cells were used for the assay. SIY peptide was added at concentration of 5 μg/mL. After 48 hours of incubation, IFNγ production was determined by IFNγ ELISPOT kit according to the manual (BD Bioscience) or CBA assay (BD Bioscience). The visualized cytokine spots were enumerated with the ImmunoSpot analyzer (CTL).
Ex vivo DC cross-presentation assay
B16-EGFR-SIY bearing mice were treated with 25 μg of anti-EGFR, or anti-EGFR-IFNβ intratumoral injection on days 14 and 17. Four days later, draining lymph node was digested with 1mg/ml of collagenase VIII (Sigma) and 200ug/ml DNase I (Sigma) at 37°C for 15 minutes. DCs were purified by CD11c positive selection Kit (Stemcell). Approximately 1×105 DCs were mixed together with purified 2×105 2C T cells with or without 5μg/mL of SIY peptide to restimulate the T cells. Two days later, the supernatants were collected, and IFNγ was measured by CBA assay (BD Bioscience).
Generation of bone marrow chimeras
WT mice were lethally irradiated with a single dose of 1000 rads. The next day irradiated mice were adoptively transferred with 2-3×106 WT, Ifnar1-/- or CD11c-DTR Tg donor bone marrow cells. Mice were maintained on sulfamethoxazole and trimethoprim (Bactrim) antibiotics diluted in drinking water for 4 weeks after reconstitution. Mice were injected with tumor cells 5-6 weeks post reconstitution.
Detection of endotoxin in mAb and fusion protein preparation
Endotoxin was measured by the limulus amebocyte lysate assay (Cambrex inc. MD). For all mAb preparations, the amount of endotoxin was determined to be < 0.2 E.U./mg mAb.
Flow cytometric analysis
Single cell suspensions of cells were incubated with anti-CD16/32 (anti-FcγIII/II receptor, clone 2.4G2) for 10 minutes and then subsequently stained with conjugated antibodies. All fluorescently labeled monoclonal antibodies were purchased from Biolegend or eBioscience. Samples were analyzed on a FACSCanto flow cytometer (BD Biosciences), and data were analyzed with FlowJo software (TreeStar, Inc.).
Statistical analysis
Mean values were compared using an unpaired Student’s two-tailed t test. Error bars represent SD or SEM. Statistically significant differences p<0.05, and p<0.01 are noted with *, and ** respectively.
Supplementary Material
Figure 8. Proposed model of how type I IFNs link the innate and adaptive anti-tumor immunity.
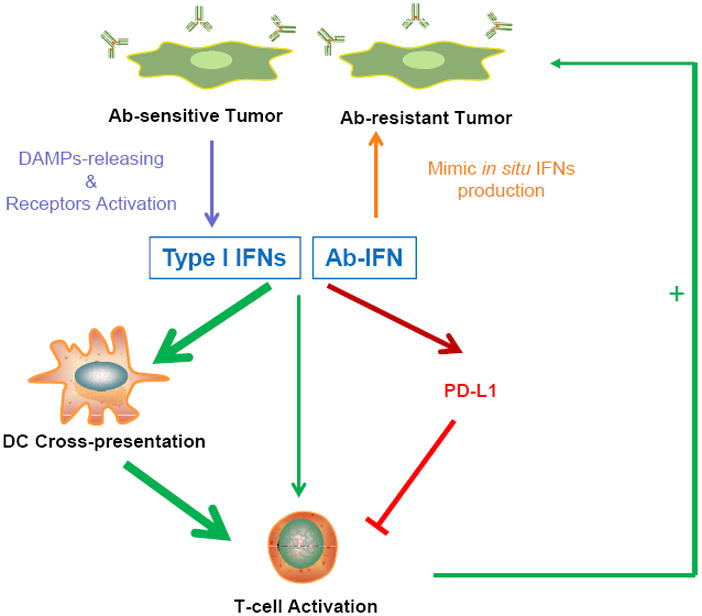
DAMPs released from Ab-sensitive tumor, but not Ab-resistant tumor after Ab therapy, will induce type I IFNs production. Ab-IFN mimics in situ type I IFN production in Ab-resistant tumor. Type I IFNs simultaneously increase DCs cross-presentation for better CTL responses, directly activate T cells and also induce PD-L1 expression to weaken the anti-tumor immunity. Ab-IFN combined with PD-L1 blockade maximizes the anti-tumor immune responses.
SIGNIFICANCE.
Ab-resistance is a major challenge for Ab-based cancer therapies. Current strategies to overcome Ab-resistance focus on targeting intrinsic resistance and improving direct killing of tumor cells. Due to the heterogeneity of tumor cells and their microenvironment, these direct killing strategies can’t target all the tumor cells and eventually Ab-resistance will develop. We now reveal that type I IFNs are essential and sufficient to bridge innate and adaptive immune responses for antibody induced tumor regression. We have designed a next generation of Ab based biologics (Ab-IFNβ fusion) to control Ab-resistance by revitalizing innate and adaptive immune cells inside the tumor. Collectively, this study opens up several avenues for optimizing targeted immune therapy that may eventually eradicate Ab-resistant tumor cells.
Highlights.
Type I IFNs are essential and sufficient for Ab-mediated tumor regression
Type I IFNs target DCs to improve cross-priming for CTLs
Type I IFNs are required for overcoming Ab resistance through extrinsic mechanism
Anti-PD-L1 Ab can synergize with IFN
Acknowledgments
We thank Dr. Harold Varmus, Dr. Anita Chong, Dr. Ulrich Kalinke, Dr. Brad H. Nelson, and Dr. Robert Schreiber for providing us with mice and reagents. This research was in part supported by U.S. National Institutes of Health grants CA141975 and CA97296, grants from Chinese Academy of Sciences and Chinese Ministry of Science and Technology grant (No.2012ZX10002006).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abes R, Gelize E, Fridman WH, Teillaud JL. Long-lasting antitumor protection by anti-CD20 antibody through cellular immune response. Blood. 2010;116:926–934. doi: 10.1182/blood-2009-10-248609. [DOI] [PubMed] [Google Scholar]
- Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol. 2010;28:1254–1261. doi: 10.1200/JCO.2009.24.6116. [DOI] [PubMed] [Google Scholar]
- Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, Gajewski TF. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004;64:1140–1145. doi: 10.1158/0008-5472.can-03-3259. [DOI] [PubMed] [Google Scholar]
- Bostrom J, Yu SF, Kan D, Appleton BA, Lee CV, Billeci K, Man W, Peale F, Ross S, Wiesmann C, Fuh G. Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen binding site. Science. 2009;323:1610–1614. doi: 10.1126/science.1165480. [DOI] [PubMed] [Google Scholar]
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, Fu YX, Auh SL. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 2011;71:2488–2496. doi: 10.1158/0008-5472.CAN-10-2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6:443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehrenbacher L, Wolter JM, Paton V, Shak S, Lieberman G, Slamon DJ. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17:2639–2648. doi: 10.1200/JCO.1999.17.9.2639. [DOI] [PubMed] [Google Scholar]
- Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208:1989–2003. doi: 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayad L, Offner F, Smith MR, Verhoef G, Johnson P, Kaufman JL, Rohatiner A, Advani A, Foran J, Hess G, et al. Safety and Clinical Activity of a Combination Therapy Comprising Two Antibody-Based Targeting Agents for the Treatment of Non-Hodgkin Lymphoma: Results of a Phase I/II Study Evaluating the Immunoconjugate Inotuzumab Ozogamicin With Rituximab. J Clin Oncol. 2013 doi: 10.1200/JCO.2012.42.7211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208:2005–2016. doi: 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012;12:125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang AY, Golumbek P, Ahmadzadeh M, Jaffee E, Pardoll D, Levitsky H. Role of bone marrow-derived cells in presenting MHC class I-restricted tumor antigens. Science. 1994;264:961–965. doi: 10.1126/science.7513904. [DOI] [PubMed] [Google Scholar]
- Hurvitz SA, Dirix L, Kocsis J, Bianchi GV, Lu J, Vinholes J, Guardino E, Song C, Tong B, Ng V, et al. Phase II Randomized Study of Trastuzumab Emtansine Versus Trastuzumab Plus Docetaxel in Patients With Human Epidermal Growth Factor Receptor 2-Postive Metastatic Breast Cancer. J Clin Oncol. 2013 doi: 10.1200/JCO.2012.44.9694. [DOI] [PubMed] [Google Scholar]
- Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- Kohlmeier JE, Cookenham T, Roberts AD, Miller SC, Woodland DL. Type I interferons regulate cytolytic activity of memory CD8(+) T cells in the lung airways during respiratory virus challenge. Immunity. 2010;33:96–105. doi: 10.1016/j.immuni.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krop IE, LoRusso P, Miller KD, Modi S, Yardley D, Rodriguez G, Guardino E, Lu M, Zheng M, Girish S, et al. A phase II study of trastuzumab emtansine in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer who were previously treated with trastuzumab, lapatinib, an anthracycline, a taxane, and capecitabine. J Clin Oncol. 2012;30:3234–3241. doi: 10.1200/JCO.2011.40.5902. [DOI] [PubMed] [Google Scholar]
- Kurts C, Robinson BW, Knolle PA. Cross-priming in health and disease. Nat Rev Immunol. 2010;10:403–414. doi: 10.1038/nri2780. [DOI] [PubMed] [Google Scholar]
- Lee Y, Auh SL, Wang Y, Burnette B, Meng Y, Beckett M, Sharma R, Chin R, Tu T, Weichselbaum RR, Fu YX. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. 2009;114:589–595. doi: 10.1182/blood-2009-02-206870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301–311. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortenson E, Park S, Zhujun J, Wang S, Fu YX. Effective anti-neu initiated anti-tumor responses require the complex role of CD4+ T cells. Clin Cancer Res. 2013 doi: 10.1158/1078-0432.CCR-12-2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, Laccabue D, Zerbini A, Camisa R, Bisagni G, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol. 2008;26:1789–1796. doi: 10.1200/JCO.2007.14.8957. [DOI] [PubMed] [Google Scholar]
- Park S, Jiang Z, Mortenson ED, Deng L, Radkevich-Brown O, Yang X, Sattar H, Wang Y, Brown NK, Greene M, et al. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell. 2010;18:160–170. doi: 10.1016/j.ccr.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006;20:1496–1510. doi: 10.1101/gad.1417406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovero S, Amici A, Di Carlo E, Bei R, Nanni P, Quaglino E, Porcedda P, Boggio K, Smorlesi A, Lollini PL, et al. DNA vaccination against rat her-2/Neu p185 more effectively inhibits carcinogenesis than transplantable carcinomas in transgenic BALB/c mice. J Immunol. 2000;165:5133–5142. doi: 10.4049/jimmunol.165.9.5133. [DOI] [PubMed] [Google Scholar]
- Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer. 2011;11:805–812. doi: 10.1038/nrc3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- Smith K, Garman L, Wrammert J, Zheng NY, Capra JD, Ahmed R, Wilson PC. Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat Protoc. 2009;4:372–384. doi: 10.1038/nprot.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagg J, Loi S, Divisekera U, Ngiow SF, Duret H, Yagita H, Teng MW, Smyth MJ. Anti-ErbB-2 mAb therapy requires type I and II interferons and synergizes with anti-PD-1 or anti-CD137 mAb therapy. Proc Natl Acad Sci U S A. 2011;108:7142–7147. doi: 10.1073/pnas.1016569108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol. 2012;30:1–22. doi: 10.1146/annurev-immunol-100311-102839. [DOI] [PubMed] [Google Scholar]
- Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, Schreiber RD, de la Torre JC, Oldstone MB. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science. 2013;340:207–211. doi: 10.1126/science.1235214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri G. Type I interferon: friend or foe? J Exp Med. 2010;207:2053–2063. doi: 10.1084/jem.20101664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall EM, Milne K, Martin ML, Watson PH, Theiss P, Nelson BH. Spontaneous mammary tumors differ widely in their inherent sensitivity to adoptively transferred T cells. Cancer Res. 2007;67:6442–6450. doi: 10.1158/0008-5472.CAN-07-0622. [DOI] [PubMed] [Google Scholar]
- Weber J. Review: anti-CTLA-4 antibody ipilimumab: case studies of clinical response and immune-related adverse events. Oncologist. 2007;12:864–872. doi: 10.1634/theoncologist.12-7-864. [DOI] [PubMed] [Google Scholar]
- Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, Gondi V, Hsu KT, Harari PM. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene. 2008;27:3944–3956. doi: 10.1038/onc.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, Aronow BJ, Karp CL, Brooks DG. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science. 2013;340:202–207. doi: 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan C, Steward KK, Timmerman JM, Morrison SL. Targeted delivery of interferon-alpha via fusion to anti-CD20 results in potent antitumor activity against B-cell lymphoma. Blood. 2010;115:2864–2871. doi: 10.1182/blood-2009-10-250555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Martin ML, Nielsen JS, Milne K, Wall EM, Lin W, Watson PH, Nelson BH. Mammary tumors with diverse immunological phenotypes show differing sensitivity to adoptively transferred CD8+ T cells lacking the Cbl-b gene. Cancer Immunol Immunother. 2009;58:1865–1875. doi: 10.1007/s00262-009-0698-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Zhang X, Mortenson ED, Radkevich-Brown O, Wang Y, Fu YX. Cetuximab-mediated tumor regression depends on innate and adaptive immune responses. Mol Ther. 2013;21:91–100. doi: 10.1038/mt.2012.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, Ercan D, Rogers A, Roncalli M, Takeda M, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med. 2011;3:99ra86. doi: 10.1126/scitranslmed.3002442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J, Koo KH, Choi KY. MEK1/2 inhibitors AS703026 and AZD6244 may be potential therapies for KRAS mutated colorectal cancer that is resistant to EGFR monoclonal antibody therapy. Cancer Res. 2011;71:445–453. doi: 10.1158/0008-5472.CAN-10-3058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.