

Abstract
The organometallic Ni(II) chemistry of the pyrrole-based pincer ligands, (P2 RPyr)− (P2 RPyr = 2,5-(R2PCH2)2C4H2N, R = Ph or Cy) is reported. Reactions of Grignard reagents with [NiCl(P2R Pyr)] afford a variety of alkyl and aryl complexes (methyl, ethyl, benzyl, phenyl, and allyl) that all display square planar geometries about nickel. The hydride complex, [NiH(P2 CyPyr)], can also prepared either through treatment of [NiCl(P2 CyPyr)] with LiHBEt3, or by reaction of H(P2 RPyr) with [Ni(COD)2] (COD = 1,4-cyclooctadiene). Reactions of the methyl and hydride complexes with CO and CO2, respectively, evince clean migratory insertion chemistry of the Ni-C and Ni-H bonds. Both the alkyl and chloride complexes are active catalysts for the Kumada coupling of aryl chlorides and aryl or alkyl Grignard reagents at room temperature. The solid-state structures of several of the complexes are reported.
Introduction
The efficacy of pincer-type ligands in transition metal catalysis is well documented1 and continues to garner attention as new forms of reactivity are discovered for complexes containing this class of supporting ligands.2–8 The variety of pincers reported to date attests to the usefulness of these ligands and the high degree to which they can be modified9 to suit catalytic applications.10–15 In the area of nickel chemistry, complexes of pincer ligands have been demonstrated to catalyze a variety of carbon-carbon and carbon-heteroatom couplings.16 In certain instances, the stability of the pincer framework allows for detailed mechanistic information to be surmised as reactive intermediates can be observed directly.17,18
We have been interested in exploring the chemistry of the new pyrrole based pincer ligand, 2,5-bis[(diphenylphosphino)methyl]pyrrolide (abbreviated P2 PhPyr−), which was recently demonstrated to bind transition metals in a meridional fashion giving rise to square planar complexes of the type [MX(P2 PhPyr)] for Ni and Pd.19–21 These complexes were prepared in a straightforward fashion and several examples containing different X-type ligands were accessed readily through salt metatheses. We were thus intrigued to learn whether such compounds display an interesting organometallic chemistry, especially with hydrocarbyl-derived ligands. Compounds of the type [NiX(P2 PhPyr)] (X = Cl, alkyl, aryl) were envisioned to serve as active catalysts for select organic transformations based on the precedent set with other pincer systems.16 Furthermore, chelating ligands based on pyrrole motifs have been shown to produce unique catalysts when paired with certain transition metals.22 In the context of PNP ligands, the role of the pyrrolide group in modulating the catalytic activity of pincer-supported transition metal complexes remains a question.23–32
In this contribution, we extend the chemistry of the pyrrole-based PNP ligand, P2 PhPyr−, to a series of organometallic complexes of Ni(II). In addition, we report the synthesis and coordination chemistry of the new dicyclohexylphosphine-derived ligand, P2 CyPyr−. This aliphatic pyrrole-diphosphine ligand is prepared in straightforward fashion and allows access to a variety of complexes not obtainable with the diphenylphosphine analog, P2 Ph Pyr−. Nickel(II) complexes of both PNP ligands appear to be active catalysts for a variety of Kumada-type C-C cross coupling reactions.
Results and Discussion
Our initial synthesis of the pyrrole-diphosphine, H(P2 PhPyr) (1), involved reduction of the corresponding phosphine oxide to afford a BH3-protected precursor that was used directly in the synthesis of Ni(II) complexes.21 Concurrent reports by the groups of Gade19 and Mani20 demonstrated an alternative method for the preparation of 1 using the secondary phosphines (or potassium salts thereof) directly in displacement reactions with bis(aminomethyl)pyrrole precursors. We have adopted this synthetic methodology for the preparation of 1 and have extended it to include the formation of the dicyclohexylphosphine derivative (2) as shown in Scheme 1. The formation of 2 from bis(dimethylaminomethyl)pyrrole occurs in higher yield than that for 1 presumably due to the increased nucleophilicity of the secondary alkyl phosphine. 31P NMR spectra of 2 display a single peak at −7.67 consistent with trivalent phosphorus.33 Similar to compound 1, no JHP coupling is observed between the methylene protons of the pincer “arms” and the phosphorus nuclei by 1H NMR spectroscopy (see Supporting Information). Thus far compound 2 has resisted efforts at crystallization and its sensitivity to air has hampered chromatographic purification. Fortunately, however, the preparation of compound 2 directly affords material of sufficient purity for attachment to nickel(II).
scheme 1.
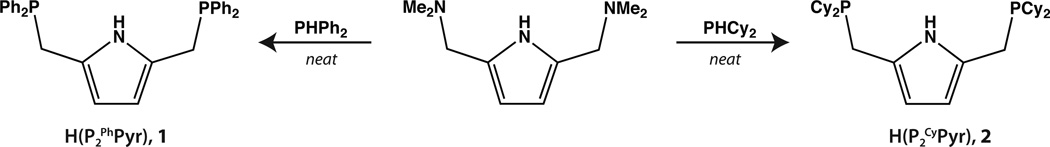
Synthesis of 1 and 2.
The reaction of 2 with anhydrous NiCl2 in the presence of Et3N affords [NiCl(P2 CyPyr)] (4) in identical fashion to the phenylphosphine analog 3. Complex 4 and all others bearing the P2 CyPyr− ligand display greater solubility in organic solvents than similar compounds containing the P2 PhPyr− ligand. Much like 3, complex 4 exists an orange-red solid (λvis = 438 nm) displaying a diamagnetic 1H NMR spectrum with the methylene protons of the ligand appearing as a pseudo-triplet, consistent with κ3-PNP coordination. The solid-state structure of 4 is shown in Figure 1. The geometry of complex 4 is square planar about nickel with a short Ni-Npyrrole distance of 1.848(2) Å. No deformation of the pyrrolic unit is apparent nor is any unexpected bond length alternation within the heterocycle. The remaining bond metrics are unremarkable, and consistent with those of other Ni(II) complexes bearing the P2 PhPyr− ligand.21
Figure 1.

Thermal ellipsoid drawing (50%) of 4. Hydrogen atoms omitted for clarity. Selected bond distances (Å) and angles (deg): Ni(1)-Cl(1) = 2.1771(8), Ni(1)-N(1) = 1.848(2), Ni(1)-Pavg = 2.200(1), P(1)-Ni(1)-P(2) = 166.01(3), N(1)-Ni(1)-Cl(1) = 177.77(7).
Previous electrochemical experiments with complex 3 demonstrated that an electrocatalytic process occurs in methylene chloride at low potentials.21 This process most likely corresponds to the abstraction of a chlorine atom by the dimeric Ni(I) species, [NiI(μ2-P2 PhPyr)]2 (13),19 which is formed by reduction of 3 around −2.1 V versus the ferrocene/ferrocenium couple (Scheme 2). To examine whether a similar process occurs for 4, its cyclic voltammogram was recorded in CH2Cl2. In contrast to 3, the CV of 4 in CH2Cl2 displays no cathodic events within the solvent window. Such a result is consistent with the more reduced nature of 4 with respect to 3 due to the presence of the more electron-donating dicyclohexylphosphine groups of the ligand. In THF, the CV of 4 does display an irreversible cathodic event at low potentials (see SI). Although not as well resolved as that observed for complex 3, the cathodic events are somewhat comparable (see SI). Whether this process corresponds to formation of an analogous Ni(I) dimeric species is not known at this time. However, treatment of 4 with hydride sources does not result in formation of an analogous NiI dimer as observed with 3 (vida infra).19
Scheme 2.

Electrocatalytic behavior of complex 3 in dichloromethane.
Much like 3, complex 4 also displays an irreversible anode process corresponding to oxidation of the Ni(II) complex. For complex 4 this event occurs at +0.23 V (vs Fc/Fc+), lower than that observed for 3 (+0.45 V) as expected. A quasi-reversible event is also observed for 4 at potentials higher than +0.23 V (see SI). The current response for this process is less than that expected for a one-electron process and therefore may represent a subsequent redox event of the species generated from the irreversible oxidation at +0.23 V, although further experiments are required to test this possibility. Although uncommon, Ni(III) species have been invoked as intermediates in C-C coupling reactions.11,17,34–38
Both compounds 3 and 4 react cleanly with several Grignard reagents to afford the corresponding hydrocarbyl complexes (Scheme 3). As with 3 and 4, each of the alkyl complexes is diamagnetic displaying a virtual triplet resonance for the methylene protons of the PNP ligand. The solid-state structures of compounds 7 and 11 and compounds 10 and 14 are shown in Figures 2 and 3, respectively. Additionally, the solid-state structure of 6 can be found in the Supporting Information. Relevant metric parameters for each complex appear in Table 1. Each of the structures is similar displaying square-planar planar geometries about nickel. The only structure that appears slightly distorted is that of the benzyl complex, 10. This species displays an increased contraction of the P(1)-Ni(1)-P(2) bond angle (158.8°) relative to the other structurally characterized compounds. The allyl complex, 14, displays an η1 coordination mode as expected for a 16-electron square-planar compound, displaying a C(31)–C(32) bond distance of 1.501(5) Å and a C(32)–C(33) bond distance of 1.373(5) Å consistent with single and double bonds, respectively. The 1H NMR spectrum for 14 is also indicative of η1 coordination in solution at room temperature (see SI).
Scheme 3.

Synthesis of Ni(II) alkyl complexes.
Figure 2.

Thermal ellipsoid drawings (50%) of compounds 7 (left) and 11 (right). Hydrogen atoms omitted for clarity. For compound 7, one of the two crystallographically independent molecules of the asymmetric unit is displayed. Selected bond distances and angles can be found in Table 1.
Figure 3.

Thermal ellipsoid drawings of compounds 6 and 14. Hydrogen atoms and minor components of the disorder (for 14) omitted for clarity. Selected bond distances and angles can be found in Table 1.
Table 1.
Selected bond distances (Å) and angles (deg) for nickel(II) hydrocarbyl complexes.
| Compound | Ni(1)-C(31) | Ni(1)-N(1) | Ni(1)-Pavg | P(1)-Ni(1)-P(2) | Ni(1)-C(31)-C(32) |
|---|---|---|---|---|---|
| 6 | 1.967(2) | 1.886(2) | 2.178(2) | 165.11(2) | N/A |
| 7† | 1.997(2) | 1.891(2) | 2.182(1) | 166.60(3) | 111.4(1) |
| 10 | 1.980(1) | 1.890(1) | 2.198(1) | 158.83(2) | 110.9(1) |
| 11 | 1.903(1) | 1.884(1) | 2.179(1) | 167.57(1) | 122.3(1) |
| 14 | 1.993(2) | 1.893(1) | 2.199(1) | 166.04(2) | 114.1(2) |
Data shown for one of two chemically identical but crystallographically independent molecules of 7 in the asymmetric unit. See SI for more information.
Unexpectedly, complex 14 could only be prepared with the P2 CyPyr− ligand. Attempted reactions of 3 with allyl Grignard afforded a mixture of starting material (3) and the NiI dimer, 13 (Scheme 2). The nature of the reductant in this reaction is not known at present. β-Hydride elimination of [Ni(η1-C3H5)(P2 PhPyr)] to afford allene and a putative nickel-hydride is an attractive proposal given the reported instability of “[NiH(P2 PhPyr)]” towards formation of 13.19 The stability of complex 7 towards elimination, however, argues against such a possibility (vida infra). Attempted observation of the alkylation reaction between 3 and allylmagnesium chloride by 1H NMR spectroscopy in benzene-d6 provided no further insight into the reaction, indicating peaks solely due to the starting materials. In addition to allyl Grignard, divergent reactivity between compounds 3 and 4 was also observed with hydride sources (vida infra).
All alkyl complexes described in Scheme 3 are stable indefinitely under an inert atmosphere at room temperature. Furthermore, the ethyl species, 7, proved resistant to β-H elimination even upon heating to 80 °C overnight.39 Despite their thermal stability, select alkyl complexes did react cleanly with carbon monoxide. Reaction of complex 5 with 1 atm of CO at 50 °C in benzene-d6 afforded the corresponding acyl insertion product after 24 hrs (Equation 1) as judged by 1H NMR and IR spectroscopy (see SI). Reaction of the phenyl complex (11) with CO (g), however, did not afford the benzoyl species under similar conditions.
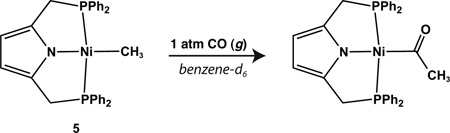 |
(1) |
In addition to hydrocarbyl species, we also examined the synthesis of a Ni(II) hydride complex. Previous attempts to prepare a nickel hydride complex by treatment of 1 with [Ni(COD)2] were reported by Gade to yield the dimeric NiI species, 13.19 We too confirmed this result with 3. Upon switching to the P2 CyPyr− ligand, however, the hydride species 15 was obtained in good yield either through reaction of 2 with [Ni(COD)2] or by treatment of 4 with LiHBEt (Equations 2 and 3, respectively). The former procedure was found to be preferable as a means of preparing 15. The dichotomy in reactivity between 1 and 2 with hydride sources is consistent with PCP systems,40 but notable given the recent finding that a related carbazole-based pincer ligand containing diphenylphosphine groups is capable of stabilizing a nickel(II) hydride.41 Thus, the observed propensity for formation of a Ni(I) dimer with compounds containing the P2 PhPyr− ligand in the presence of certain nucleophiles (hydride and allyl sources) may be both a function of the more electron deficient nature of the ligand and its smaller steric profile with respect to P2 CyPyr− (cone angles of diphenylphosphinoethane and dicyclohexylphosphinoethane = 125° and 142°, respectively).42
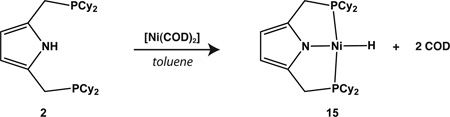 |
(2) |
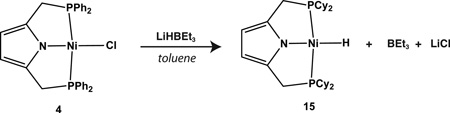 |
(3) |
We have not yet been able to grow crystals of 15 suitable for X-ray diffraction, but NMR data are consistent with a monomeric, square planar Ni(II) species. Compound 15 displays a triplet resonance for the hydride ligand at −17.61 ppm in benzene-d6 with a coupling constant of 59.3 Hz to 31P (see SI). Unlike the alkyl complexes described above, 15 does not react cleanly with CO (g) as judged by NMR spectroscopy. The compound does insert CO2 readily, however, to give the putative metal formate species, [Ni(O2CH)(P2 CyPyr)] (Equation 4; see also SI).43,44
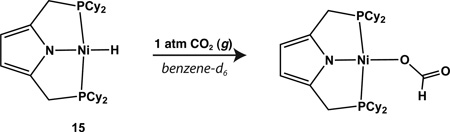 |
(4) |
Catalytic Reactions
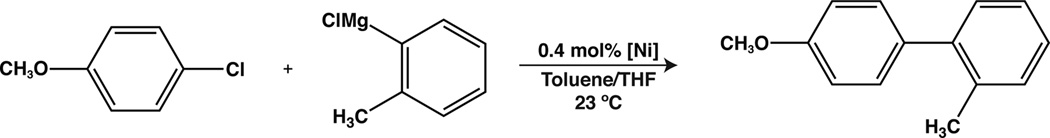
The nickel(II) complexes described above were examined as catalysts for the Kumada coupling of aryl chlorides with a variety of Grignard reagents. Each complex examined gave comparable results for the representative coupling of 4-chloroanisole with o-tolylmagnesium chloride (Table 2). The couplings were found to proceed in high yield at room temperature with low catalyst loadings (< 1 mol%). Even the dimeric Ni(I) complex, 13, was found to give productive coupling (Table 2, entry 7). No significant differences were found for the P2 PhPyr− ligand versus P2 CyPyr−, and therefore compound 3 was employed in a screen of different coupling partners. In addition, complex 3 is stable under ambient conditions in the solid state facilitating its use as a precatalyst.
Table 2.
Kumada coupling of 4-chloroanisole and o-tolylmagnesium chloride employing various nickel(II) precatalysts.‡
 | ||
|---|---|---|
| Entry | Catalyst | Yield |
| 1 | 3 | 92 |
| 2 | 4 | 89 |
| 3 | 5 | 90 |
| 4 | 6 | 87 |
| 5 | 11 | 82 |
| 6 | 12 | 82 |
| 7 | 13 | 84 |
| 8 | 14 | 91 |
Conditions: 1.0 mmol 4-chloroanisole; 1.3 mmol o-tolyl Grignard; 3 mL toluene; 12 h. Yields reported for isolated product.
Table 3 displays the results of several Kumada couplings catalyzed by complex 3. As is evident from the table, the catalyst system exhibited good substrate scope for aryl chlorides containing both electron-releasing and electron-withdrawing substituents. Added steric bulk to the electrophilic coupling partner resulted in slightly lower yields as demonstrated by couplings using 2-chlorotoluene (Entry 3). In contrast, added bulk to the nucleophilic coupling partner actually lead to a slight increase in reaction yield (Entries 7 – 12). This result is most likely due to suppression of the small amount of homocoupling product observed with phenylmagnesium chloride (< 5%). Couplings employing alkyl Grignard reagents were also successful with catalyst 3 (Entries 14 and 15). In sum, complex 3 appears to demonstrate good substrate scope and catalytic efficacy at room temperature for a variety of Kumada couplings involving aryl chlorides.
Table 3.
Kumada coupling of various aryl chlorides using 3 as precatalyst.
| Entry | Electrophile | Nucleophile | Product | Time | Yield |
|---|---|---|---|---|---|
| 1 |  |
 |
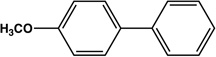 |
12 | 89 |
| 2 | 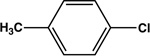 |
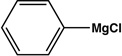 |
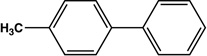 |
12 | 87 |
| 3 | 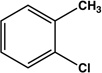 |
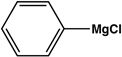 |
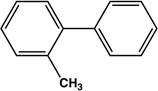 |
12 | 83 |
| 4 | 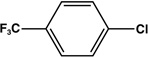 |
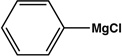
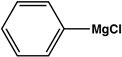 |
 |
6 | 85 |
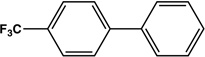
| 5 | 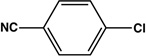 |
 |
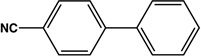 |
6 | 93 |
| 6 | 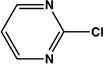 |
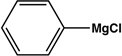 |
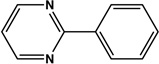 |
6 | 90 |
| 7 | 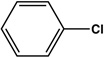 |
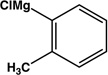 |
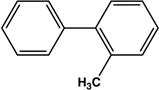 |
12 | 90 |
| 8 | 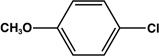 |
 |
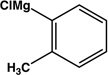
 |
12 | 92 |
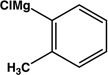
| 9 | 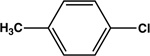 |
 |
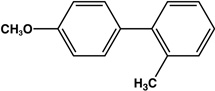
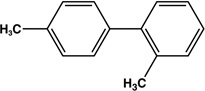 |
12 | 87 |
| 10 | 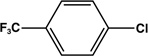 |
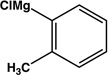 |
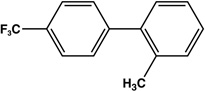 |
8 | 86 |
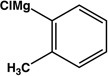
| 11 | 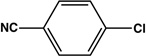 |
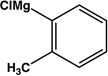 |
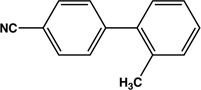 |
8 | 90 |
| 12 | 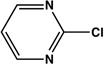 |
 |
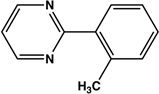 |
8 | 94 |
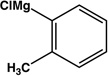
| 13 | 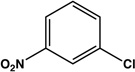 |
 |
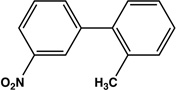 |
12 | 76 |
| 14 | 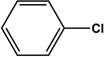 |
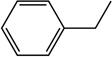 |
8 | 78 | |
| 15 | 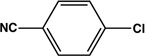 |
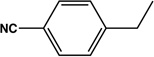 |
8 | 84 |
Conditions: 1.0 mmol aryl chloride; 1.3 mmol Grignard; 0.4 mol% 3; 3 mL toluene; 23 °C. Yields reported for isolated product.
Conclusions
In conclusion, we have prepared and characterized a variety of alkyl species containing the new PNP ligands, P2 PhPyr− and P2 CyPyr−. Each of the complexes demonstrates good thermal stability, including those containing alkyl ligands with β-hydrogen atoms. In addition to alkyls, a nickel hydride complex was isolated with the P2 CyPyr− ligand. This species is inaccessible with the P2 PhPyr− ligand, demonstrating the significant differences between pyrrole-based pincer ligands containing alkyl and aryl phosphine groups. Both alkyl and hydride complexes demonstrate insertion chemistry of unsaturated C–O bonds. The methyl complex, [Ni(CH3)(P2 PhPyr)], was found to insert CO cleanly at elevated temperatures to generate an acyl complex. In contrast, the hydride complex did not react cleanly with CO, but was found to insert CO2 readily at ambient temperature affording a Ni(II) formate. Finally, nickel(II) complexes of both PNP ligands are efficient catalysts for the Kumada coupling of aryl chlorides and Grignard reagents at room temperature. Similar results with a variety of different precatalysts suggest that a common active species is responsible for catalytic activity. Future work will further probe the catalytic potential of these nickel(II) complexes and investigate in detail their catalytic mechanisms.
Experimental
General Comments
Manipulations of air- and moisture-sensitive materials were performed under an atmosphere of nitrogen gas using standard Schlenk techniques or in a Vacuum Atmospheres glovebox. Tetrahydrofuran, diethyl ether, pentane, and toluene were purified by sparging with argon and passage through two columns packed with 4 Å molecular sieves or activated alumina (THF). Benzene and benzene-d6 were dried over sodium ketyl and vacuum-distilled prior to use. NMR spectra were recorded in benzene-d6 on a Varian INOVA spectrometer operating at 500 MHz (1H) and referenced to the residual 1H (7.16 ppm) or 13C (128.39 ppm) resonance of the solvent. For 31P NMR spectra an external standard of neat 85% H3PO4 (aq) was used (0.00 ppm). All reported coupling constants are in units of Hz. UV-vis spectra were recorded on a Cary-60 spectrophotometer in air-tight Teflon-capped quartz cells. Infrared spectra were recorded as thin films on a KBr plate using a Nicolet iS10 FTIR spectrometer. Cyclic voltammetry was performed at 23 °C on a CH Instruments 620D electrochemical workstation. A 3-electrode set-up was employed comprising a glassy carbon working electrode, platinum wire auxiliary electrode, and a Ag/AgCl quasi-reference electrode. Triply recrystallized Bu4NPF6 was used as the supporting electrolyte. All electrochemical data were referenced internally to the ferrocene/ferrocenium couple at 0.00 V. Elemental analyses were performed by Atlantic Microlab, Inc. of Norcross, GA or Midwest Microlab, LLC of Indianapolis, IN.
Materials
H(P2 PhPyr) (1),19–21 [Ni(CH3)(P2 PhPyr)] (5),21 and bis(dimethylaminomethyl)pyrrole45 were prepared according to literature procedures. Anhydrous nickel(II) chloride, bis-cyclooctadiene nickel(0), dicyclohexylphosphine, Grignard reagents (as solutions in THF or Et2O), lithium triethylborohydride, and catalytic substrates were purchased from Strem Chemicals or Sigma-Aldrich and used as received. Carbon monoxide and carbon dioxide were purchased from Praxair and used as received.
2,5-Bis((dicyclohexylphosphino)methyl)pyrrole, H(P2 CyPyr), 2
This compound was prepared in similar fashion to the phenyl derivative.20 A flask was charged with 1.08 g (5.96 mmol) of 2,5-bis(dimethylaminomethyl)pyrrole and 2.4 mL of dicyclohexylphosphine (12 mmol). The reaction mixture was heated to 140 °C and allowed to stir for 20 h. After cooling, all volatiles were removed in vacuo to afford 2.77 g (95%) of a white residue. The material was used directly without further purification. 1H NMR: δ 8.24 (s, 1 NH), 6.05 (d, 2 pyr-CH), 2.67 (s, 4 CH2), 1.79 (m, 4 Cy-CH), 1.69 (m, 12 Cy-CH2), 1.61 (m, 4 Cy-CH), 1.51 (m, 4 Cy-CH), 1.17 (m, 20 Cy-CH2). 13C{1H} NMR: δ 128.28 (d, obscured by solvent), 107.36 (d, JCP = 4.8), 34.25 (d, JCP = 15.3), 30.64 (d, JCP = 13.3), 29.71 (d, JCP = 8.7), 28.09 (d, JCP = 14.4), 28.02 (d, JCP = 11.4), 27.27, 21.71 (d, JCP = 19.6). 31P{1H} NMR: δ −7.67. ESI-MS (positive mode): Calcd for [M + H]+: m/z 488.4; [M(O) + H]+: m/z 504.3. Found for [M + H]+: m/z 488.1; [M(O) + H]+: m/z 504.1.
[NiCl(P2 CyPyr)], 4
A flask was charged with 1.46 g of H(P2 CyPyr) (2.99 mmol) and 20 ml of THF. To resulting solution was added 0.388 g of anhydrous NiCl2 (2.99 mmol) and 0.83 mL of Et3N (6.0 mmol). The mixture was allowed to stir for 12 h at room temperature during which time it became dark red. All volatiles were removed in vacuo leaving a dark red colored residue that was extracted into 50 mL of benzene. The benzene extract was filtered through a pad of Celite and evaporated to dryness to afford 1.48 g (85%) of a red solid. Crystals suitable for X-ray diffraction were grown by slow cooling of a concentrated diethyl ether solution at –30 °C. Mp: 210 °C (decomp). 1H NMR: δ 6.37 (s, 2 pyr-CH), 2.64 (app t, 4 CH2, JHP = 4.5), 2.45 (m, 4 Cy-CH), 1.95 (m, 4 Cy-CH), 1.79 (m, 4 Cy-CH), 1.69 (m, 8 Cy-CH2), 1.59 (m, 12 Cy-CHn), 1.15 (m, 8 Cy-CH), 1.06 (m, 4 Cy-CH). 13C{1H} NMR: δ 138.50 (t, JCP = 7.6), 106.06 (t, JCP = 5.2), 33.11 (t, JCP = 10.5), 28.84, 28.56, 27.51 (t, JCP = 6.3), 27.34 (t, JCP = 5.2), 26.82, 23.25 (t, JCP = 10.9). 31P{1H} NMR: δ 51.10. Anal. Calcd for C30H50ClNNiP2: C, 62.04; H, 8.68; N, 2.41. Found: C, 62.55; H, 8.56; N, 2.34.
General procedure for preparation of alkyl complexes
A flask was charged with 0.200 mmol of [NiCl(P2 RPyr)] (R = Ph or Cy) and 10 mL of THF. The resulting red-orange solution was chilled to −30 °C at which point one equivalent (0.20 mmol) of the desired Grignard reagent as a solution in THF or Et2O was added dropwise. The reaction mixture was allowed to warm to room temperature and stir for 3 h during which time the colored lightened to a dark yellow-brown. All volatiles were removed in vacuo and the remaining residue was extracted into 10 mL of benzene. The benzene solution was filtered through a pad of Celite and evaporated to dryness to afford the alkyl complex as a yellow or orange microcrystalline solid that was washed with pentane. In the case of complexes containing the P2 PhPyr− ligand, the products were further washed with Et2O. Characterization data and yields for each complex appear below. For compounds 9 and 11, combustion analyses with multiple samples consistently returned low results for carbon despite satisfactory H and N values. For compound 14, analyses were repeatedly low for all elements suggesting poor or incomplete combustion. The 1H NMR spectrum of 14 is provided in the SI.
[Ni(CH3)(P2 CyPyr)], 6
Yield: 96%. Crystals suitable for X-ray diffraction were grown by slow cooling of a saturated Et2O solution at −30 °C. 1H NMR: δ 6.53 (s, 2 pyr-CH), 2.91 (app t, 4 CH2, JHP = 4.3), 2.13 (m, 4 Cy-CH), 1.83 (m, 4 Cy-CH), 1.69 (m, 8 Cy-CH2), 1.58 (m, 8 Cy-CH2), 1.44 (m, 8 Cy-CH), 1.13 (m, 4 Cy-CH), 1.08 (m 8 Cy-CH), −0.36 (t, 3 CH3, JHP = 8.5). 13C{1H} NMR: δ 135.92 (t, JCP = 7.4), 104.79 (t, JCP = 4.8), 33.43 (t, JCP = 9.9), 29.33, 28.84, 27.74 (t, JCP = 6.1), 27.47 (t, JCP = 4.7), 27.01, 25.89 (t, JCP = 10.5). 31P{1H} NMR: δ 52.11. Anal. Calcd for C31H53NNiP2: C, 66.44; H, 9.53; N, 2.50. Found: C, 66.02; H, 9.16; N, 2.29.
[Ni(CH2CH3)(P2 PhPyr)], 7
Yield 90%. Crystals suitable for X-ray diffraction were grown by vapor diffusion of pentane into a concentrated benzene solution at room temperature. 1H NMR: δ 7.62 (m, 8 Ar-H), 7.03 (m, 4 Ar-H), 6.99 (m, 8 Ar-H), 6.55 (s, 2 pyr-CH), 3.67 (app t, 4 CH2, JHP = 4.5), 0.98 (m, 2 CH2CH3), 0.83 (t, 3 CH2CH3). 13C{1H} NMR: δ 134.58 (t, JCP = 7.2), 133.89 (t, JCP = 18.4), 133.55 (t, JCP = 5.9), 130.39, 129.16 (t, JCP = 4.4), 106.28 (t, JCP = 5.2), 36.23 (t, JCP = 12.9), 15.62 (t, JCP = 3.1), −6.40 (t, JCP = 18.5). 31P{1H} NMR: δ 39.39. Anal. Calcd for C32H31NNiP2·Et2O: C, 69.25; H, 6.62; N, 2.24. Found: C, 68.66; H, 6.20; N, 2.55.
[Ni(CH2CH3)(P2 CyPyr)], 8
Yield 95%. 1H NMR: δ 6.53 (s, 2 pyr-CH), 2.92 (app t, 4 CH2, JHP = 4.3), 2.15 (m, 4 Cy-CH), 1.89 (m, 4 Cy-CH), 1.70 (m, 8 Cy-CH2), 1.59 (m, 8 Cy-CH2), 1.45 (m, 8 Cy-CH2), 1.38 (t, 3 CH2CH3), 1.15 (m, 4 Cy-CH), 1.08 (m, 8 Cy-CH2), 0.77 (m - qd, 2 CH2CH3). 13C{1H} NMR: δ 135.53 (t, JCP = 7.2), 104.80 (t, JCP = 4.8), 34.19 (t, JCP = 9.6), 29.40, 28.87, 27.83 (t, JCP = 5.9), 27.56 (t, JCP = 4.6), 26.99, 26.10 (t, JCP = 10.9), 17.82 (t, JCP = 3.3), −12.73 (t, JCP = 21.3). 31P{1H} NMR: δ 47.32. Anal. Calcd for C32H55NNiP2: C, 66.91; H, 9.65; N, 2.44. Found: C, 66.59; H, 9.68; N, 2.37.
[Ni(CH2C6H5)(P2 PhPyr)], 9
Yield 87%. 1H NMR: δ 7.49 (m, 8 Ar-H), 7.04 (m, 4 Ar-H), 6.98 (m, 8 Ar-H), 6.79 (t, 1 p-CH2C6H5), 6.74 (t, 2 m-CH2C6H5), 6.54 (d, 2 o-CH2C6H5), 6.48 (s, 2 pyr- CH), 3.57 (app t, 4 CH2, JHP = 4.5), 2.27 (t, 2 CH2C6H5, JHP = 9.3). 13C{1H} NMR: δ 151.30 (t, JCP = 2.3), 134.74 (t, JCP = 6.7), 133.68 (t, JCP = 5.7), 132.96 (t, JCP = 18.5), 130.41, 129.17 (t, JCP = 4.7), 128.52, 128.33, 122.42, 106.41 (t, JCP = 4.8), 36.31 (t, JCP = 13.2), 7.22 (t, JCP = 16.2). 31P{1H} NMR: δ 37.52. Anal. Calcd for C37H33NNiP2: C, 72.58; H, 5.43; N, 2.29. Found: C, 67.44; H, 5.64; N, 2.04.
[Ni(CH2C6H5)(P2 CyPyr)], 10
Yield 95%. 1H NMR: δ 7.45 (d, 2 o-CH2C6H5), 7.18 (t, 2 m-CH2C6H5), 7.00 (t, 1 p-CH2C6H5), 6.49 (s, 2 pyr-CH), 2.86 (app t, 4 CH2, JHP = 4.3), 2.10 (m, 4 Cy-CH), 2.07 (t, 2 CH2C6H5, JHP = 8.0), 1.64 (m, 12 Cy-CH + CH2), 1.58 (m, 8 Cy-CH2), 1.39 (m, 8 Cy-CH2), 1.08 (m, 12 Cy-CH + CH2). 13C{1H} NMR: one aromatic 13C resonance obscured by C6D6 δ 155.07 (t, JCP = 4.3), 135.86 (t, JCP = 7.0), 129.21, 122.47, 105.05 (t, JCP = 4.7), 33.80 (t, JCP = 9.4), 29.61, 28.97, 27.69 (t, JCP = 6.1), 27.62 (t, JCP = 4.2), 26.96, 25.19 (t, JCP = 11.2), 2.73 (t, JCP = 19.1). 31P{1H} NMR: δ 47.19. Anal. Calcd for C37H57NNiP2: C, 69.82; H, 9.03; N, 2.20. Found: C, 69.25; H, 8.94; N, 2.14.
[Ni(C6H5)(P2 PhPyr)], 11
Yield: 84%. Crystals suitable for X-ray diffraction were grown by vapor diffusion of pentane into a saturated benzene solution of the complex. 1H NMR: δ 7.33 (d, 2 o-C6H5), 7.29 (m, 8 Ar-H), 6.96–6.89 (m, 15 m,p-C6H5 + Ar-H), 6.64 (s, 2 pyr-CH), 3.65 (app t, 4 CH2, JHP = 4.7). 13C{1H} NMR: δ 149.97 (t, JCP = 26.2), 139.52 (t, JCP = 3.6), 135.35 (t, JCP = 7.1), 133.44 (t, JCP = 5.6), 133.07 (t, JCP = 20.0), 130.41, 128.95 (t, JCP = 4.7), 126.94, 106.76 (t, JCP = 5.2), 35.27 (t, JCP = 13.1). 31P{1H} NMR: δ 34.91. Anal. Calcd for C36H31NNiP2: C, 72.27; H, 5.22; N, 2.34. Found: C, 70.04; H, 5.32; N, 2.45.
[Ni(C6H5)(P2 CyPyr)], 12
Yield: 93%. 1H NMR: δ 7.80 (d, 2 o-C6H5), 7.19 (t, 2 m-C6H5), 6.95 (t, 1 p-C6H5), 2.94 (app t, 4 CH2, JHP = 4.5), 1.99 (m, 4 Cy-CH), 1.78 (m, 4 Cy-CH), 1.56 (m, 20 Cy-CH + CH2), 1.10 (m, 8 Cy-CH), 0.98 (m, 8 Cy-CH). 13C{1H} NMR: δ 153.84, 140.38 (t, JCP = 2.9), 136.30 (t, JCP = 7.1), 126.63 (t, JCP = 1.7), 122.04, 105.34 (t, JCP = 5.1), 32.99 (t, JCP = 10.6), 28.37, 28.05, 27.66 (t, JCP = 6.1), 27.35 (t, JCP = 5.0), 26.78, 25.18 (t, JCP = 10.6). 31P{1H} NMR: δ 48.56. Anal. Calcd for C36H55NNiP2: C, 69.46; H, 8.91; N, 2.25. Found: C, 69.09; H, 8.69; N, 2.31.
[Ni(η1-C3H5)(P2 CyPyr)], 14
Yield: 91%. Crystals suitable for X-ray diffraction were grown by slow cooling of a saturated Et2O solution at −30 °C. 1H NMR: δ 6.49 (s, 2 pyr-CH), 6.41 (m, 1 allyl-CH), 5.14 (m, 1 allyl-CH), 4.88 (m, 1 allyl-CH), 2.86 (app t, 4 CH2, JHP = 4.3), 2.17 (m, 4 Cy-CH), 1.89 (m, 4 Cy-CH), 1.71 (m, 2 allyl-CH2), 1.68 (m, 8 Cy-CH2), 1.57 (m, 8 Cy-CH2), 1.17 (m, 4 Cy-CH), 1.07 (m, 8 Cy-CH2). 13C{1H} NMR: δ 148.62 (t, JCP = 3.5), 135.75 (t, JCP = 6.7), 104.99 (t, JCP = 4.7), 103.89, 34.09 (t, JCP = 9.6), 29.31, 28.88, 27.73 (t, JCP = 6.1), 27.50 (t, JCP = 4.6), 26.95, 25.75 (t, JCP = 11.3), 2.61 (t, JCP = 17.9). 31P{1H} NMR: δ 47.43. HRMS (ESI, positive mode): Calcd for [M]+: m/z 585.3163 Found for [M]+: 585.3149.
[NiH(P2 CyPyr)], 15
A flask was charged with 0.488 g of H(P2 CyPyr) (1.00 mmol), 0.275 g of [Ni(COD)2] (1.00 mmol), and 20 mL of toluene. The yellow-brown solution was allowed to stir at room temperature for 8h. All volatiles were removed in vacuo and the resulting solid was washed with pentane to afford 0.496 g (91%) of a yellow microcrystalline solid. The material was further purified by recrystallization in Et2O at −30 °C. 1H NMR: δ 6.60 (s, 2 pyr-CH), 3.01 (app t, 4 CH2, JHP = 4.3), 2.05 (m, 4 Cy-CH), 1.76 (m, 4 Cy-CH), 1.72 (m, 4 Cy-CH), 1.66 (m, 4 Cy-CH), 1.62 (m, 4 Cy-CH), 1.54 (m, 4 Cy-CH), 1.46 (m, 4 Cy-CH), 1.35 (m, 4 Cy-CH), 1.07 (m, 12 Cy-CH + CH2), −17.61 (t, NiH, JHP = 59.3). 13C{1H} NMR: δ 136.38 (t, JCP = 7.5), 104.98 (t, JCP = 4.9), 34.52 (t, JCP = 11.6), 30.11, 29.27, 27.40 (app t, JCP = 5.4, two overlapping triplets), 26.90, 26.23 (t, t, JCP = 10.1). 31P{1H} NMR: δ 67.21. Anal. Calcd for C30H51NNiP2: C, 65.95; H, 9.41; N, 2.56. Found: C, 65.60; H, 9.22; N, 2.49.
[Ni(C{O}CH3)(P2 PhPyr)]
This species was observed in solution by adding ~1 atm of CO (g) to a 50 mM solution of [Ni(CH3)(P2 PhPyr)] in benzene-d6. After heating at 50 °C for 15 hrs, the acyl complex was observed to form as judged by NMR and IR spectroscopy. 1H NMR: δ 7.64 (m, 8 Ar-H), 6.97 (m, 12 Ar-H), 6.57 (s, 2 pyr-CH), 3.61 (app t, 4 CH2, JHP = 4.7), 1.91 (s, 3 C(O)CH3). 13C{1H} NMR: acyl carbon not observed δ 134.00 (t, JCP = 6.7), 133.70 (t, JCP = 19.8), 133.45 (t, JCP = 5.8), 130.65, 129.29 (t, JCP = 4.8), 106.72 (t, JCP = 4.9), 39.40 (t, JCP = 8.2, C(O)CH3), 35.20 (t, JCP = 14.4). 31P{1H} NMR: δ 32.69. IR (film, KBr, cm−1): 1614 (νCO).
[Ni(O2CH)(P2 CyPyr)]
This species was observed in solution by adding ~1 atm of CO2 (g) to a 30 mM solution of [NiH(P2 CyPyr)] in benzene-d6. After standing at room temperature for 24 hrs, the formate complex was observed to form as judged by NMR spectroscopy. 1H NMR: δ 7.91 (t, 1 CO2-H, JHP = 3.3), 6.32 (s, 2 pyr-CH), 2.61 (app t, 4 CH2, JHP = 4.5), 2.56 (m, 4 Cy-CH), 1.85 (m, 4 Cy-CH), 1.74 (m, 4 Cy-CH), 1.69 (m, 8 Cy-CH2), 1.63 (m, 4 Cy-CH), 1.56 (m, 8 Cy-CH), 1.25 (m, 4 Cy-CH), 1.16 (m, 4 Cy-CH), 1.08 (m, 4 Cy-CH). 13C{1H} NMR: δ 167.80 (O2CH), 138.39 (t, JCP = 8.5), 106.31 (t, JCP = 5.2), 33.72 (t, JCP = 9.7), 28.67, 28.52, 27.62 (t, JCP = 6.4), 27.40 (t, JCP = 4.9), 26.77, 22.17 (t, JCP = 11.6). 31P{1H} NMR: δ 47.86. IR (film, KBr, cm−1): 1631 (νCO), 1298 (νCO).
General procedure for cross-coupling reactions
A Schlenk tube was charged with 2.0 mg (4 µmol) of the desired nickel complex and 3 mL of toluene or THF. The electrophilic coupling partner (1.0 mmol) was then added followed by the Grignard reagent (1.3 mmol) as a solution in THF. The reaction mixture was allowed to stir at room temperature for 6 – 12 h before being quenched with 5 mL of deionized water. To the quenched reaction mixture was added 10 mL of diethyl ether causing separation of the organic layer. The organic layer was extracted and dried over Na2SO4. After filtration and evaporation of the solvents the crude product was purified by column chromatography on Al2O3 (3% hexanes/EtOAc).
X-ray Data Collection and Structure Solution Refinement
Crystals suitable for X-ray diffraction were mounted in Paratone oil onto a glass fiber and frozen under a nitrogen cold stream maintained by an X-Stream low-temperature apparatus. The data were collected at 98(2) K using a Rigaku AFC12/Saturn 724 CCD fitted with Mo Kα radiation (λ = 0.71073 Å). Data collection and unit cell refinement were performed using Crystal Clear software.46 The total number of data were measured in the range 2.0 < θ < 27.6° using ω scans. Data processing and absorption correction, giving minimum and maximum transmission factors, were accomplished with Crystal Clear and ABSCOR,47 respectively. All structures were solved by direct methods and refined on F2 using full-matrix, least-squares techniques with SHELXL-97.48,49 Non-hydrogen atoms were refined with anisotropic displacement parameters. All carbon bound hydrogen atom positions were determined by geometry and refined by a riding model with the exception of those attached to select carbon atoms in the structures of 6 and 8. In these cases, a positional disorder among the carbon atoms prevented satisfactory hydrogen positions to be calculated.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by a TRAC award from UTSA and by a grant from the Welch Foundation (AX-1772 to ZJT).
Footnotes
ASSOCIATED CONTENT
Supporting Information. Additional spectra, tabulated crystallographic data, and crystallographic information (cif) files. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Singleton JT. Tetrahedron. 2003;59:1837–1857. [Google Scholar]
- 2.Morales-Morales D, Jensen CGM, editors. The Chemistry of Pincer Compounds. Amsterdam: Elsevier B. V; 2007. [Google Scholar]
- 3.van der Vlugt JI, Reek JNH. Angew. Chem. Int. Ed. 2009;48:8832–8846. doi: 10.1002/anie.200903193. [DOI] [PubMed] [Google Scholar]
- 4.Selander NJ, Szabó K. Chem. Rev. 2010;111:2048–2076. doi: 10.1021/cr1002112. [DOI] [PubMed] [Google Scholar]
- 5.van Koten G, Gebbink RJMK. Dalton Trans. 2011;40:8731–8732. doi: 10.1039/c1dt90120f. [DOI] [PubMed] [Google Scholar]
- 6.Gunanathan C, Milstein D. Acc. Chem. Res. 2011;44:588–602. doi: 10.1021/ar2000265. [DOI] [PubMed] [Google Scholar]
- 7.Haibach MC, Kundu S, Brookhart M, Goldman AS. Acc. Chem. Res. 2012;45:947–958. doi: 10.1021/ar3000713. [DOI] [PubMed] [Google Scholar]
- 8.Gelman D, Musa S. ACS Catal. 2012;2:2456–2466. [Google Scholar]
- 9.Vabre B, Canac Y, Duhayon C, Chauvin R, Zargarian D. Chem. Commun. 2012;48:10446–10448. doi: 10.1039/c2cc35377f. [DOI] [PubMed] [Google Scholar]
- 10.Benito-Garagorri D, Kirchner K. Acc. Chem. Res. 2008;41:201–213. doi: 10.1021/ar700129q. [DOI] [PubMed] [Google Scholar]
- 11.Castonguay A, Beauchamp AL, Zargarian D. Organometallics. 2008;27:5723–5732. [Google Scholar]
- 12.Leis W, Mayer HA, Kaska WC. Coord. Chem. Rev. 2008;252:1787–1797. [Google Scholar]
- 13.Castonguay A, Spasyuk DM, Madern N, Beauchamp AL, Zargarian D. Organometallics. 2009;28:2134–2141. [Google Scholar]
- 14.Schneider S, Meiners J, Askevold B. Eur. J. Inorg. Chem. 2012;2012:412–429. [Google Scholar]
- 15.He L-P, Chen T, Gong D, Lai Z, Huang K-W. Organometallics. 2012;31:5208–5211. [Google Scholar]
- 16.Wang Z-X, Liu N. Eur. J. Inorg. Chem. 2012;2012:901–911. [Google Scholar]
- 17.Hu X. Chem. Sci. 2011;2:1867–1886. [Google Scholar]
- 18.Chakraborty S, Zhang J, Patel YJ, Krause JA, Guan H. Inorg. Chem. 2012 doi: 10.1021/ic300587b. [DOI] [PubMed] [Google Scholar]
- 19.Grüger N, Wadepohl H, Gade LH. Dalton Trans. 2012;41:14028–14030. doi: 10.1039/c2dt32199h. [DOI] [PubMed] [Google Scholar]
- 20.Kumar S, Mani G, Mondal S, Chattaraj PK. Inorg. Chem. 2012;51:12527–12539. doi: 10.1021/ic301967r. [DOI] [PubMed] [Google Scholar]
- 21.Venkanna GT, Ramos TVM, Arman HD, Tonzetich ZJ. Inorg. Chem. 2012;51:12789–12795. doi: 10.1021/ic301633q. [DOI] [PubMed] [Google Scholar]
- 22.Hennessy ET, Betley TA. Science. 2013;340:591–595. doi: 10.1126/science.1233701. [DOI] [PubMed] [Google Scholar]
- 23.Mazet C, Gade LH. Organometallics. 2001;20:4144–4146. [Google Scholar]
- 24.Mazet C, Gade, Lutz H. Eur. J. Inorg. Chem. 2003;2003:1161–1168. [Google Scholar]
- 25.Li R, Larsen DS, Brooker S. New J. Chem. 2003;27:1353–1359. [Google Scholar]
- 26.Mashima K, Tsurugi H. J. Organomet. Chem. 2005;690:4414–4423. [Google Scholar]
- 27.Odom AL. Dalton Trans. 2005:225–233. doi: 10.1039/b415701j. [DOI] [PubMed] [Google Scholar]
- 28.Lin C-Y, Tsai C-F, Chen H-J, Hung C-H, Yu R-C, Kuo P-C, Lee HM, Huang J-H. Chem.-Eur. J. 2006;12:3067–3073. doi: 10.1002/chem.200500989. [DOI] [PubMed] [Google Scholar]
- 29.Tenza K, Hanton MJ, Slawin AMZ. Organometallics. 2009;28:4852–4867. [Google Scholar]
- 30.Hsu J-W, Lin Y-C, Hsiao C-S, Datta A, Lin C-H, Huang J-H, Tsai J-C, Hsu W-C. Dalton Trans. 2012 doi: 10.1039/c2dt30417a. [DOI] [PubMed] [Google Scholar]
- 31.Tian R, Ng Y, Ganguly R, Mathey F. Organometallics. 2012;31:2486–2488. [Google Scholar]
- 32.Vignesh Babu H, Muralidharan K. Dalton Trans. 2013;42:1238–1248. doi: 10.1039/c2dt31755a. [DOI] [PubMed] [Google Scholar]
- 33.Moedritzer K, Maier L, Groenweghe LCD. J. Chem. Eng. Data. 1962;7:307–310. [Google Scholar]
- 34.Jones GD, Martin JL, McFarland C, Allen OR, Hall RE, Haley AD, Brandon RJ, Konovalova T, Desrochers PJ, Pulay P, Vicic DA. J. Am. Chem. Soc. 2006;128:13175–13183. doi: 10.1021/ja063334i. [DOI] [PubMed] [Google Scholar]
- 35.Lin X, Phillips DL. The Journal of Organic Chemistry. 2008;73:3680–3688. doi: 10.1021/jo702497p. [DOI] [PubMed] [Google Scholar]
- 36.Adhikari D, Mossin S, Basuli F, Huffman JC, Szilagyi RK, Meyer K, Mindiola DJ. J. Am. Chem. Soc. 2008;130:3676–3682. doi: 10.1021/ja7108486. [DOI] [PubMed] [Google Scholar]
- 37.Kozhanov KA, Bubnov MP, Cherkasov VK, Fukin GK, Vavilina NN, Efremova LY, Abakumov GA. Journal of Magnetic Resonance. 2009;197:36–39. doi: 10.1016/j.jmr.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 38.Spasyuk DM, Gorelsky SI, van der Est A, Zargarian D. Inorg. Chem. 2011;50:2661–2674. doi: 10.1021/ic1025894. [DOI] [PubMed] [Google Scholar]
- 39.Liang L-C, Chien P-S, Lin J-M, Huang M-H, Huang Y-L, Liao J-H. Organometallics. 2006;25:1399–1411. [Google Scholar]
- 40.Boro BJ, Duesler EN, Goldberg KI, Kemp RA. Inorg. Chem. (Washington, DC, U.S.) 2009;48:5081–5087. doi: 10.1021/ic8020194. [DOI] [PubMed] [Google Scholar]
- 41.Grüger N, Rodríguez L-I, Wadepohl H, Gade LH. Inorg. Chem. 2013;52:2050–2059. doi: 10.1021/ic302454n. [DOI] [PubMed] [Google Scholar]
- 42.Tolman CA. Chem. Rev. 1977;77:313–348. [Google Scholar]
- 43.Suh H-W, Schmeier TJ, Hazari N, Kemp RA, Takase MK. Organometallics. 2012;31:8225–8236. [Google Scholar]
- 44.Schmeier TJ, Hazari N, Incarvito CD, Raskatov JA. Chem. Commun. 2011;47:1824–1826. doi: 10.1039/c0cc03898a. [DOI] [PubMed] [Google Scholar]
- 45.Kim IT, Elsenbaumer RL. Tetrahedron Lett. 1998;39:1087–1090. [Google Scholar]
- 46.Crystal Clear. The Woodlands, TX: Rigaku/MSC Inc.; Rigaku Corporation; 2005. [Google Scholar]
- 47.ABSCOR. Tokyo, Japan: Higashi; Rigaku Corporation; 1995. [Google Scholar]
- 48.SHELXTL97: Program for Refinement of Crystal Structures. Göttingen, Germany: Sheldrick, G.M., University of Göttingen; 1997. [Google Scholar]
- 49.Sheldrick GM. Acta Crystallogr., Sect. A. 2008;A64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.