

Abstract
O -GlcNAc glycosylation is a dynamic protein posttranslational modification with roles in processes such as transcription, cell cycle regulation, and metabolism. Detailed mechanistic studies of O-GlcNAc have been hindered by a lack of methods for measuring O-GlcNAc stoichiometries and the interplay of glycosylation with other posttranslational modifications. We recently developed a method for labeling O-GlcNAc-modified proteins with resolvable poly(ethylene glycol) mass tags. This mass tagging approach enables the direct measurement of glycosylation stoichiometries and the visualization of distinct O-GlcNAc-modified subpopulations. Here, we describe protocols for labeling O-GlcNAc glycoproteins in cell lysates with mass tags.
Keywords: O-linked N-acetylglucosamine, glycosylation, posttranslational modifications, chemoenzymatic labeling, poly(ethylene glycol), protein subpopulations
Introduction
O-GlcNAc glycosylation (or O-GlcNAcylation) is the posttranslational modification of serine and threonine residues of proteins with β-linked N-acetyl D-glucosamine. The O-GlcNAc modification is found on over 500 intracellular proteins and affects processes such as transcription, apoptosis, glucose sensing, and metabolism (Hart et al., 2007; Love and Hanover, 2005; Rexach et al., 2008). Chemoenzymatic labeling of O-GlcNAcylated proteins with a resolvable mass tag is a method for quantitatively imaging and analyzing O-GlcNAc-modified protein subpopulations. This approach relies on attaching a poly(ethylene glycol) (PEG) polymer of defined molecular mass (e.g., 2-kDa or 5-kDa) selectively onto the O-GlcNAc sugar and resolving the labeled populations by SDS-PAGE. The method is performed in three steps: (1) enzymatic labeling of O-GlcNAc residues with a mutant galactosyltransferase and an unnatural ketogalactose sugar, (2) conjugation of an aminooxy-functionalized PEG mass tag to the ketogalactose sugar, (3) SDS-PAGE and immunoblotting for the protein(s) of interest (Scheme 1a) (Rexach et al., 2010). A variation on this approach replaces the ketogalactose sugar with an azidogalactose sugar and utilizes [3+2] azide-alkyne cycloaddition chemistry to conjugate an alkyne-functionalized PEG mass tag to the glycoproteins (Scheme 1b) (Clark et al., 2008). Notably, this mass tagging approach can be applied to endogenous proteins using standard molecular biology techniques and requires no protein purification, radiolabels, or advanced instrumentation.
Scheme 1.

Chemoenzymatic labeling of O-GlcNAc residue with mass tags. (a) O-GlcNAcylated proteins in cell lysates are labeled with an unnatural ketogalactose sugar (1) and a mutant galactosyltransferase (Y289L GalT). Labeled proteins are reacted with an aminooxy-functionalized 2-kDa or 5-kDa mass tag (2) through a bioorthogonal oxime forming reaction. Glycosylated proteins labeled with the mass tag are resolved and identified by immunoblotting. The mass tag shifts the mass of the glycosylated subpopulation by intervals of 2-kDa or 5-kDa, depending on the number of O-GlcNAc moieties attached. (b) Alternatively, O-GlcNAcylated proteins in cell lysates are labeled with an unnatural azidogalactose sugar (3) and the mutant GalT. Labeled proteins are reacted with an alkynyl-functionalized 2-kDa or 5-kDa mass tag (4) through a bioorthogonal [3+2] azide-alkyne cycloaddition chemistry reaction.
The ability to visualize distinct O-GlcNAc-modified subpopulations and quantify in vivo glycosylation stoichiometries (Fig. 1) provides information that is not readily attainable using other methods (Rexach et al., 2012; Rexach et al., 2010; Yi et al., 2012). O-GlcNAc glycosylation stoichiometries can be rapidly measured on endogenous proteins and compared across different cellular conditions (Rexach et al., 2012; Rexach et al., 2010; Yi et al., 2012). For example, the mass tagging approach was used to measure changes in the glycosylation stoichiometry of cyclic-AMP response element-binding protein (CREB) and phosphofructokinase-1 (PFK1) in response to neuronal depolarization and hypoxia, respectively (Rexach et al., 2012; Yi et al., 2012). Additionally, the glycosylation state (e.g., mono-, di-, or tri-) of proteins can be rapidly established. A protein that exists in a non-glycosylated, monoglycosylated, and diglycosylated state will resolve into three distinct bands after mass tagging and SDS-PAGE. When used in conjunction with site-directed mutagenesis, the approach can also determine the occupancy of the O-GlcNAc sugar at specific amino acid sites (Rexach et al., 2012; Rexach et al., 2010; Yi et al., 2012). Finally, the mass tagging approach can be used to monitor the interplay between O-GlcNAcylation and other post-translational modifications (Rexach et al., 2012; Rexach et al., 2010). In previous work, neurons were activated by KCl-induced depolarization and, at defined time points, the cell lysates were mass tagged and immunoblotted with both a total CREB antibody and a phospho-Ser133-specific CREB antibody. This enabled us to visualize 4 distinct posttranslationally-modified subpopulations of CREB and probe how glycosylation affected the kinetics of CREB phosphorylation and vice versa (Rexach et al., 2012).
Figure 1.

O-GlcNAc stoichiometries of Sp1, Nup62, synapsin IIa, and CREB. Cell lysates were labeled with the unnatural ketogalactose sugar (1), reacted with a 2-kDa or 5-kDa mass tag (2), resolved by SDS-PAGE, and identified by immunoblotting. A 2-kDa mass tag was used to label CREB and a 5-kDa mass tag was used to label Sp1, Nup62, and synapsin Ia (upper band) and synapsin IIa (lower band). (…) denotes the non-glycosylated protein fraction; (←) denotes shifted protein fraction.
Basic Protocol I: Labeling O-GlcNAc residues in cell lysates with a resolvable mass tag using oxime chemistry
Materials
Sodium dodecyl sulfate (SDS)
Complete Protease Inhibitor Tablet (Roche); dissolve to 50X concentration in 1 mL of H2O
Protein concentration assay, such as the bicinchoninic acid (BCA) protein detection assay
Methanol
Chloroform
H2O (Ultrapure or Milli-Q grade)
Triton X-100 (20% w/v in H2O)
4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) (100 mM, pH 7.9)
100 mM MnCl2
5 M NaCl
UDP-ketogalactose (Uridine 5′-diphospho-2-acetonyl-2-deoxy-α-D-galactopyranose diammonium salt; synthesis described in (Khidekel et al., 2003))
Mutant Y289L galactosyltransferase (Y289L GalT; 2 mg/mL; (Khidekel et al., 2003); see Support Protocol I)
Urea (ultra pure)
3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS)
Dithiothreitol (DTT)
Aminooxy-functionalized poly(ethylene glycol) (aminooxy-functionalized PEG; 60 mM in H2O; synthesis described in (Rexach et al., 2010))
Sodium acetate buffer (1.8 M, pH 3.89)
pH indicator paper (pH 1–6)
Protocol
Lysing the cells or tissue pellet and measuring protein concentration
-
1
Prepare a cell or tissue pellet using standard techniques. Lyse it by adding an equal volume of boiling Lysis Buffer (2% SDS (w/v) in H2O, 1X Roche Complete Protease Inhibitor Cocktail; see Reagents and Solutions) to resuspend the sample.
Lysing the cell or tissue sample quickly under denaturing conditions is critical to maintain the protein glycosylation states. In those cases where non-denaturing buffers will be used, prepare the sample on ice using ice-cold buffers and supplement each buffer with a β-N-acetylglucosaminidase inhibitor such PUGNAc (O-(2-acetamindo-2-deoxy-D-glucopyranosylidene)amino N-phenylcarbamate, 10 μM) and the phosphatase inhibitors NaVO3 (1 mM), NaF (20 mM), and Na2MoO4 (0.5 mM). Occasionally it may be desirable or advantageous to perform a subcellular fractionation (see Troubleshooting). -
2
Sonicate the cell lysate until the solution is no longer viscous.
-
3
Boil the sample for 5 minutes.
-
4
Centrifuge the sample for 10 minutes at 21,885 x g, 4 °C. Promptly remove the sample from the centrifuge and place at room temperature to avoid SDS precipitation. Transfer the solubilized lysate in the supernatant into a fresh 1.5-mL microcentrifuge tube and discard any remaining pellet.
-
5
Measure the protein concentration of your sample.
We generally prefer measuring protein concentrations using the BCA protein detection assay due to its compatibility with high concentrations of SDS. However, other protein detection assays that are compatible with the chosen lysis buffer may also be acceptable.
Precipitating the protein
-
6
Aliquot 100 μg of protein into a 1.5-mL microcentrifuge tube. If the volume of the 100 μg protein solution is less than 100 μL, add Lysis Buffer to 100 μL. If the volume of the 100 μg protein solution is greater than 100 μL, then scale the volume of the following solutions accordingly.
For each set of experimental conditions, it is important to also prepare a separate control sample that will be incubated with everything except for the Y289L GalT enzyme. -
7
To the 100 μL of protein lysate, add 300 μL of methanol, and vortex the solution. Next, add 100 μL of chloroform, and vortex the solution again. Finally, add 225 μL of water and vortex. Each vortex step should be carried out for approximately 10 seconds.
The solution should quickly separate into two phases, with a clear bottom phase and a cloudy top phase. -
8
Centrifuge the samples for 10 minutes at 21,885 x g, 4 °C.
Two separate phases should appear after centrifuging the samples and a white pellet (the precipitated protein) should reside at the interface of those phases. -
9
Immediately remove and discard most of the top phase without disturbing the pellet.
The top layer can usually be removed down to within a millimeter or two of the pellet without disturbing it. These steps should be done at room temperature. -
10
Add 225 μL of methanol and gently disrupt the protein pellet by briefly vortexing or inverting the tube several times until the top and bottom phases are fully mixed.
Be careful not to lose your pellet in the tube cap at this step. -
11
Centrifuge the sample for 10 minutes at 21,885 x g, 4 °C.
After centrifuging the sample, the solution should be in only one phase and the white pellet should now reside at the bottom of the tube. -
12
Remove the liquid phase and allow the pellet to air dry.
It is important that the solution phase fully evaporates without over-drying the pellet. Over or under-dried pellets will be difficult to resuspend. An ideal point is typically when the pellet is still somewhat translucent and no liquid puddle remains. If all of the supernatant has been carefully removed from the pellet, the pellet will usually dry within 30 minutes.
Labeling the samples with ketogalactose and mutant Y289L galactosyltransferase
-
13
Resuspend the pellet in 20 μL of Ketone Labeling Resuspension Buffer (1% SDS (w/v) in H2O, 1X Roche Complete Protease Inhibitor Cocktail; see Reagents and Solutions).
Resuspending the protein sample after precipitation is often difficult, particularly if it is over or under dry. Vortexing the sample (being careful not to cause foam), sonicating the sample, boiling the sample, and agitating the sample by pipetting may all improve the speed and efficiency of protein resuspension. -
14
To each sample, place the 20 μL of resuspended protein (5 mg/mL) on ice and add in the following order: 9 μL of 20% Triton X-100, 38 μL of H2O, 10 μL of 100 mM HEPES pH 7.9, 10 μL of 100 mM MnCl2, 2 μL of 5 M NaCl, 2 μL of 50X Roche Complete Protease Inhibitor Cocktail. Mix by pipetting up and down. Then add 5 μL of 10 mM UDP-ketogalactose substrate and mix. To all samples except the control reaction, add 4 μL of 2 mg/mL Y289L GalT. To the control reaction, add 4 μL of H2O.
In those situations where a significant number of samples will be labeled, a master mix containing H2O, HEPES, MnCl2, NaCl, and protease inhibitor cocktail can be prepared. In that case, add the Triton X-100 to the sample and pipette to fully mix, followed by the master mix, UDP-ketogalactose, and Y289L GalT (in that order).The addition of 4 μL of 2 mg/mL Y289L assumes that the Y289L GalT is sufficiently active to completely label a model glycosylated peptide at a concentration of 50 ng/μL (see Support Protocol I). If the Y289L GalT is less active, then adjust the enzyme concentration to ensure complete reaction. -
15
Incubate the reaction overnight (12 – 16 hours) at 4 °C while inverting the sample end-over-end.
-
16
Precipitate the protein as described above (‘Precipitating the protein’).
Labeling the samples with the aminooxy-functionalized PEG mass tag
-
17
Resuspend the precipitated protein in 43.6 μL of Mass Tag Resuspension Buffer (7 M urea, 10 mM HEPES, pH 7.9, 2% (w/v) CHAPS, 1 mM DTT, 1X Roche Complete Protease Inhibitor Cocktail; see Reagents and Solutions).
As before, this step is often difficult if the pellet is not ideally dried. It may take up to an hour for the precipitated protein to redissolve. -
18
Add 6 μL of 60 mM aminooxy-functionalized PEG, 1.422 μL of 1.8 M sodium acetate buffer, pH 3.89, and mix.
The aminooxy reaction requires a pH of 4.5. Check the final pH of your solution on pH paper to ensure that it is 4.5. -
19
Incubate at room temperature for 20–24 hours. Gently rock the samples by placing them in a tube rack on a table-top rocker.
-
20
After the 20–24 hour incubation, add 50 μL of Neutralization Buffer (1% (w/v) SDS, 100 mM HEPES, pH 7.9; see Reagents and Solutions), mix, and precipitate the reaction as described above (‘Precipitating the protein’).
-
21
Resuspend the pellet in 30 μL of 1% (w/v) SDS to a final concentration of 3.3 mg/mL. Perform gel electrophoresis and immunoblotting using standard techniques. During initial experiments, we typically run the entire sample in a single gel lane.
Best results are generally achieved by resolving 50–75% of the labeled protein dissolved in SDS-PAGE loading dye on a 4–12% Bis-Tris NuPAGE gradient gel (Invitrogen). To adequately resolve the shifted from the non-shifted protein subpopulations, the gel is typically run until the protein of interest has migrated through at least 50% of the gel.As Nup62 and Sp1 are heavily O-GlcNAc glycosylated, they serve as excellent positive controls for the labeling experiments (Fig. 1).See Data Analysis for how to calculate protein glycosylation stoichiometries.
Basic Protocol II: Labeling O-GlcNAc residues in cell lysates with a resolvable mass tag using [3+2] azide-alkyne cycloaddition chemistry
Basic Protocol I describes labeling O-GlcNAc residues in cell lysates with a resolvable mass tag by reacting a ketone moiety with an aminooxy PEG reagent to form an oxime linkage. This is the best-characterized system but requires chemical synthesis of the UDP-ketogalactose and aminooxy-functionalized PEG reagents. An alternative to this approach relies on [3+2] azide-alkyne cycloaddition (“Click”) chemistry, which can be performed with commercially available materials (Scheme 1b). The “Click” protocol utilizes a Click-iT O-GlcNAc Enzymatic Labeling System (Invitrogen) for the first set of reactions, and a Click-iT Protein Reaction Buffer Kit (Invitrogen) and alkynyl-functionalized PEG (Creative PEGWorks) for the second set of reactions. While both methods apply the same principles, it is important to note that the “Click” protocol may require some validation. For example, it is essential to confirm that the cycloaddition reaction proceeds to completion under your conditions.
Materials
Sodium dodecyl sulfate (SDS)
Complete Protease Inhibitor Tablet (Roche); dissolve to 50X concentration in 1 mL of H2O
Complete Protease Inhibitor Tablet EDTA-free (Roche); dissolve to 50X concentration in 1 mL of H2O
Protein concentration assay, such as the bicinchoninic acid (BCA) protein detection assay
Methanol
Chloroform
H2O (Ultrapure or Milli-Q grade)
Click-iT O-GlcNAc Enzymatic Labeling System (Invitrogen)
Dimethylsulfoxide (DMSO)
Click-iT Protein Reaction Buffer Kit (Invitrogen)
-
Alkynyl-functionalized poly(ethylene glycol) (alkynyl-functionalized PEG; mPEG-Alkyne, 2-kDa or 5-kDa, Creative PEGWorks; 10 mM in DMSO)
Before using any new batch of PEG, it is important to check the quality by mass spectrometry. The spectrum of high quality PEG has one peak centered on the correct mass (2-kDa or 5-kDa). The spectrum of low quality PEG has multiple peaks, usually centered at intervals of the correct mass. 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) (100 mM, pH 7.9)
Protocol
Follow Steps 1 – 12 of Basic Protocol I.
Labeling the samples with azidogalactose and mutant Y289L galactosyltransferase (modified from the ‘Click-iT O-GlcNAc Enzymatic Labeling System’ manual)
-
13
Resuspend the pellet in 20 μL of Azide Labeling Resuspension Buffer (1% SDS (w/v) in 20 mM HEPES pH 7.9, 1X Roche Complete Protease Inhibitor Cocktail; see Reagents and Solutions).
Resuspending the protein sample after precipitation is often difficult, particularly if it is over or under dry. Vortexing the sample (being careful not to cause foam), sonicating the sample, boiling the sample, and agitating the sample by pipeting may all improve the speed and efficiency of protein resuspension. -
14
Add 144 μL of 10 mM HEPES buffer, pH 7.9 to the UDP-GalNAz (Component A of the ‘Click-iT O-GlcNAc Enzymatic Labeling System’) and mix well.
Keep on ice. Aliquot any remaining UDP-GalNAz and store at −80 °C until further use. -
15
For each resuspended protein sample (Step 13), place the 20 μL of resuspended protein on ice and add, in the following order: 22.5 μL of H2O, 40 μL of Labeling buffer (Component C of the ‘Click-iT O-GlcNAc Enzymatic Labeling System’), 5.5 μL of 100 mM MnCl2, and 2 μL of 50X Roche Complete Protease Inhibitor Cocktail. Mix by pipetting up and down. Then add 5 μL of 0.5 mM UDP-GalNAz and mix. Finally, to each sample except the control reaction, add 5 μL of Y289L GalT (labeled as Gal-T1 (Y289L), Component B of the ‘Click-iT O-GlcNAc Enzymatic Labeling System’). To the control reaction, add 5 μL of H2O.
-
16
Incubate the reaction overnight (12 – 16 hours) at 4 °C while inverting the sample end-over-end.
-
17
Precipitate the protein as described above (‘Precipitating the protein’).
Labeling the samples with the alkynyl-functionalized PEG mass tag (modified from the ‘Click-iT Protein Reaction Buffer Kit’ manual)
-
18
Resuspend the precipitated protein in 25 μL of Alkynyl Labeling Resuspension Buffer (1% SDS (w/v) in 50 mM Tris-HCl, pH 7.5, 1X Roche Complete Protease Inhibitor Cocktail EDTA free; see Reagents and Solutions).
As before, this step is often difficult. It may take up to an hour for the precipitated protein to redissolve. -
19
Add the alkynyl-functionalized PEG (10 mM) to the Click-iT reaction buffer (Component A of the ‘Click-iT Protein Reaction Buffer Kit’) to a final concentration of 1 mM.
Store any unused reaction buffer at −20 °C for up to a year. -
20
Add 500 μL of H2O to Click-iT reaction buffer additive 1 (Component C of the ‘Click-iT Protein Reaction Buffer Kit’) and mix thoroughly.
Store any unused reagent at −20 °C for up to a year. Discard if the reagent turns brown. -
21
Add 540 μL of H2O to Click-iT reaction buffer additive 2 (Component D of the ‘Click-iT Protein Reaction Buffer Kit’) and mix thoroughly.
Store any unused reagent at 4 °C for up to a year. -
22
To the resuspended protein (Step 18), add 50 μL of the alkynyl-poly(ethylene glycol) in Click-iT reaction buffer (Step 19), 2 μL of 50X Roche Complete Protease Inhibitor Cocktail EDTA free, and 3 μL of H2O.
-
23
Vortex for 5 seconds.
-
24
Add 5 μL of CuSO4. Vortex for 5 seconds.
-
25
Add 5 μL of Click-iT reaction buffer additive 1 solution (Step 20). Vortex for 5 seconds and wait 3 minutes (but not longer than 5 minutes).
-
26
Add 10 μL of Click-iT reaction buffer additive 2 (Step 21). Vortex for 5 seconds.
The solution should turn orange at this point. -
27
Incubate at room temperature overnight (12 – 16 hours). Gently rock the samples by placing them in a tube rack on a table-top rocker.
-
28
After the 20–24 hour incubation, add 50 μL of Neutralization Buffer and precipitate the reaction as described above (‘Precipitating the protein’).
-
29
Resuspend the pellet in 30 μL of 1% (w/v) SDS. Perform gel electrophoresis and immunoblotting using standard techniques. During initial experiments, we typically run the entire sample in a single gel lane.
Best results are generally achieved by resolving 50–75% of the labeled protein on a 4–12% Bis-Tris NuPAGE gradient gel (Invitrogen). To adequately resolve the shifted from the non-shifted protein subpopulations, the gel is typically run until the protein of interest has migrated through at least 50% of the gel.As Nup62 and Sp1 are heavily O-GlcNAc glycosylated, they serve as excellent positive controls for the labeling experiments.See Data Analysis for how to calculate protein glycosylation stoichiometries.
Support Protocol I: Expression and isolation of Y289L GalT
Introduction
Active Y289L GalT can be expressed and isolated from an E. coli expression system (Ramakrishnan and Qasba, 2002). Upon overexpressing Y289L GalT, E. coli sequester the protein in inclusion bodies. This protocol describes the isolation and lysis of these inclusion bodies, followed by refolding of the Y289L GalT protein to its active conformation. Y289L GalT is also available commercially as part of the Click-iT O-GlcNAc Enzymatic Labeling System.
Materials
Y289L GalT cDNA in a pET23a plasmid backbone (Ramakrishnan and Qasba, 2002)
2-Nitro-5-(sulfothio)-benzoate (NSTB; must be made fresh as described in (Thannhauser et al., 1984)).
Ampicillin
Agar bacteria plate containing 100 μg/mL of ampicillin
Luria-Bentani (LB) broth
Isopropylthio-β-galactoside (IPTG)
Phosphate buffered saline (PBS)
25% (w/v) sucrose in PBS
Guanidine hydrochloride
Sodium sulfite
L-Arginine
Tris-HCl
Ethylenediaminetetraacetic acid (EDTA)
Cysteamine
Cystamine
Dialysis tubing, 10-kDa Nominal Molecular Weight Limit (NMWL)
Centricon centrifugal filter unit, 10-kDa NMWL
Coomassie brilliant blue R-250
Methanol
Acetic acid
SDS-PAGE reagents
MnCl2
4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES)
UDP-ketogalactose (Uridine 5′-diphospho-2-acetonyl-2-deoxy-α-D-galactopyranose diammonium salt; synthesis described in (Khidekel et al., 2003))
Click-iT O-GlcNAc Peptide LC/MS standard (Invitrogen)
2,5-dihydroxybenzoic acid
2-hydroxy-5-methoxybenzoic acid
Acetonitrile
Trifluoroacetic acid
Protocol
Inducing protein expression
-
1
Electroporate BL21(DE3) cells with 0.25 μg of Y289L GalT DNA in a pET23a plasmid, and plate the bacteria on an agar plate containing 100 μg/mL of ampicillin. Incubate the plate overnight at 37 °C.
-
2
Pick three colonies and place each separately into 100 mL of LB broth containing 100 μg/mL of ampicillin. Incubate the LB broth overnight at 37 °C with shaking at 235 RPM.
-
3
Choose the densest colony and dilute it to 1 liter with LB broth containing 100 μg/mL of ampicillin. Incubate the bacteria culture at 37 °C with shaking at 235 RPM. Periodically measure the absorbance of the culture at 600 nm.
We generally measure the absorbance of the culture every 45 minutes after diluting with LB. -
4
When the absorbance of the culture at 600 nm has reached 0.7 compared to an LB control, add IPTG to a final concentration of 1 mM. Return the bacteria culture to 37 °C with shaking at 235 RPM for an additional 4 hours.
-
5
After the 4 hours, centrifuge the bacteria for 10 minutes at 600 x g, 4 °C. Discard the supernatant and save the pellet.
After the supernatant has been removed, the bacteria can be frozen at −20 °C for up to six months.
Isolating the Y289L GalT enzyme
Unless otherwise noted, throughout the GalT isolation and refolding procedure, the GalT protein is kept on ice.
-
6
Resuspend the bacteria in 10 mL of ice-cold PBS.
It is important to ensure that all the bacteria have been resuspended before moving on to the sonication step. -
7
Sonicate thoroughly on ice to lyse the bacteria.
The time and intensity of sonication will depend on the type of sonicator used. We generally sonicate 10 times for 30 seconds at 40% amplitude with 30 second pauses in between to keep the sample from heating. -
8
Add 70 mL of ice-cold PBS and centrifuge for 30 minutes at 14,000 x g, 4 °C. Discard the supernatant.
-
9
Resuspend the pellet in 50 mL of ice-cold 25% (w/v) sucrose in PBS. Centrifuge for 30 minutes at 14,000 x g, 4 °C.
Resuspending the pellet in this and subsequent steps is often difficult and laborious. However, it is critical to resuspend the pellet fully at each step to ensure adequate purity of the Y289L GalT. -
10
Repeat Step 9 three more times.
With each subsequent wash, the pellet color should change from the straw color of LB to a more ivory color.You can store the pellet after the final wash in 5 mL of 25% (w/v) sucrose in PBS overnight at 4 °C. Be certain to remove the 25% (w/v) sucrose in PBS before the next step.
Refolding the Y289L GalT
-
11
Resuspend the pellet in 20 mL of ice-cold Y289L Resuspension Buffer (5 M guanidine hydrochloride, 0.3 M sodium sulfite; see Reagents and Solutions).
-
12
Add 2 mL of NTSB and shake vigorously at room temperature for approximately 45 minutes.
The NTSB should initially be a pale yellow color. When added to the solution, the NTSB should turn the solution red. The reaction is complete when the solution turns from red back to pale yellow. This usually takes approximately 45 minutes. -
13
Add 180 mL of ice-cold H2O to precipitate the protein and centrifuge the sample for 10 minutes at 10,000 x g, 4 °C.
-
14
Discard the supernatant and resuspend the pellet in 10 mL of ice-cold H2O. Centrifuge the sample for 10 minutes at 10,000 x g, 4 °C.
-
15
Repeat Step 14 two more times.
-
16
Resuspend the pellet in 5 M guanidine hydrochloride to a protein concentration of 1 mg/mL.
1 mg/mL of Y289L GalT has an absorbance of 1.9 to 2.0 at 275 nm. -
17
Dilute the protein ten fold in ice-cold Refolding Solution (0.5 M L-arginine, 50 mM Tris-HCl, pH 8.0, 5 mM EDTA, 4 mM cysteamine, 2 mM cystamine; see Reagents and Solutions) at 4 °C over the course of 15 minutes with slow mixing.
It is important to add the Refolding Solution slowly with shaking. We usually place the protein solution on an orbital shaker and add 1/15 of the Refolding Solution each minute over the course of 15 minutes.Some protein typically precipitates as a white precipitate during the refolding. The precipitate can be removed by centrifugation (10 min, 10,000 x g, 4 °C) and discarded. Alternatively, the precipitate can be carried on to Step 18 and removed at Step 19. -
18
Dialyze the refolded protein three times for 4 hours with 4 liters of Dialysis Solution (50 mM Tris-HCl, pH 8.0, 5 mM EDTA, 4 mM cysteamine, and 2 mM cystamine; see Reagents and Solutions) at 4 °C using 10-kDa NMWL dialysis tubing.
Here again protein will precipitate. Precipitation should not be interpreted as a failed isolation. -
19
Collect the dialyzed solution and centrifuge for 10 minutes at 10,000 x g at 4 °C.
-
20
Retain the supernatant and concentrate it using a Centricon filter unit with a 10-kDa cutoff.
We usually concentrate the GalT until it has reached a concentration of 2 mg/mL, which usually requires an approximately 100-fold concentration. -
21
Check the purity of the GalT by performing SDS-PAGE on 7.5 μg of the concentrated protein and by staining the gel with Coomassie blue stain.
The approximate molecular weight of Y289L GalT is 32-kDa. We regularly obtain Y289L GalT in high purity (Fig. 2). -
22
Check the activity of the Y289L GalT in an O-GlcNAc peptide labeling reaction. Make up separate 30 μL reactions containing 0, 10, 25, 50, or 100 ng/μL of Y289L GalT and 5 mM MnCl2, 10 mM HEPES pH 7.9, 1.5 μL of 10 mM UDP-ketogalactose, and 4 μL of 100 pmole/μL Click-iT O-GlcNAc Peptide LC/MS Standard. Incubate the reactions overnight at 4 °C.
-
23
Monitor the addition of the ketogalactose to the O-GlcNAc residue on the peptide by mass spectrometry analysis (Fig. 3).
We usually analyze the reactions with matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry using a dihydroxybenzoic acid (DHB) matrix (see Reagents and Solutions), although other mass spectrometry methods also work. The unmodified peptide has a molecular weight of 1118.5 daltons. The ketogalactose adds 202 daltons to the molecular weight of the peptide. Typically complete labeling of the peptide occurs using 50 ng/μL of GalT.Once isolated, the Y289L GalT is stable for approximately one year at 4 °C.
Figure 2.
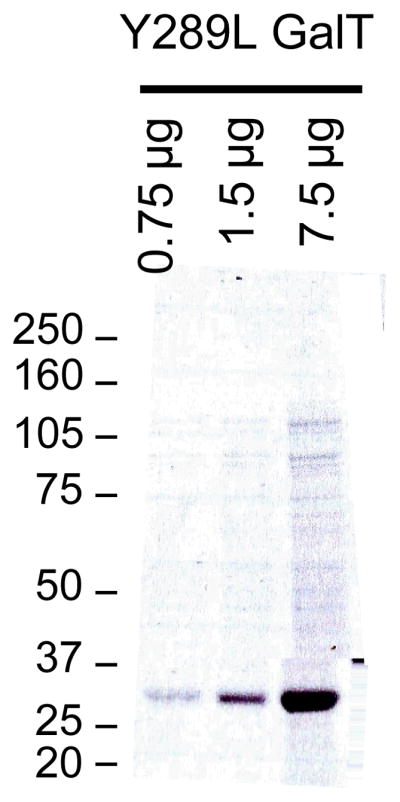
Y289L GalT. 0.75, 1.5, and 7.5 μg of expressed and refolded Y289L GalT was resolved on a 4–12% Bis-Tris NuPAGE gel and stained with Coomassie brilliant blue.
Figure 3.
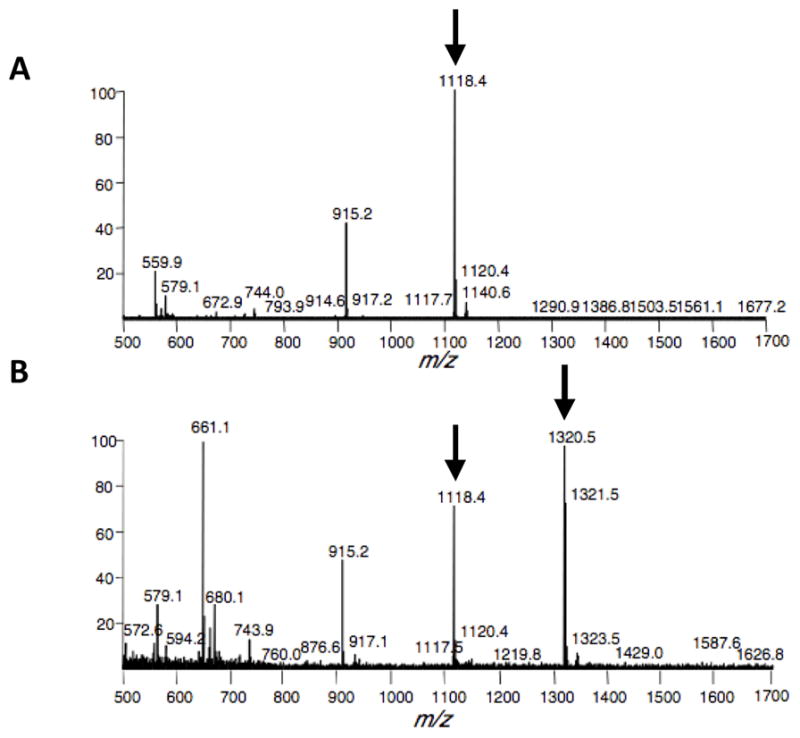
Mass spectrometry analysis of the ketogalactose labeling of an O-GlcNAc glycosylated peptide. (a) Control reaction (no GalT). The unlabeled peptide has a molecular weight of 1118.4 daltons. (b) Labeling reaction. The product of the ketogalactose labeling has a molecular weight of 1320.5 daltons. The presence of unlabeled peptide at m/z 1118.4 suggests that the reaction did not go to completion and that a higher concentration of GalT should be used for labeling reactions.
Reagents and Solutions
All reagents and solutions should be prepared with ultrapure (Milli-Q) grade H2O.
Lysis Buffer
2% SDS (w/v) in H2O
-
1X Roche Complete Protease Inhibitor Cocktail
Once the protease inhibitors have been added, the solution can be stored for up to 2 weeks at 4 °C. Storage at 4 °C may lead the SDS to precipitate. If this occurs, be certain to re-dissolve by warming the sample to room temperature before use.
Ketone Labeling Resuspension Buffer
1% SDS (w/v) in H2O
-
X Roche Complete Protease Inhibitor Cocktail
Once the protease inhibitors have been added, the solution can be stored for up to 2 weeks at 4 °C. Storage at 4 °C may lead the SDS to precipitate. If this occurs, be certain to re-dissolve by warming the sample to room temperature before use.
Mass Tag Resuspension Buffer
7 M Urea (ultra pure)
10 mM HEPES, pH 7.9
2% (w/v) CHAPS
1 mM DTT
-
1X Roche Complete Protease Inhibitor Cocktail
This solution should be prepared fresh each time.Freshly dissolved, ultrapure urea is critical in this step. A urea breakdown product is isocyanate, which may inhibit the aminooxy reaction and which is found as an impurity in urea preparations.
Neutralization Buffer
1% (w/v) SDS
100 mM HEPES, pH 7.9
Azide Labeling Resuspension Buffer
1% SDS (w/v) in 20 mM HEPES pH 7.9
-
1X Roche Complete Protease Inhibitor Cocktail
Once the protease inhibitors have been added, the solution can be stored for up to 2 weeks at 4 °C. Storage at 4 °C may lead to precipitation of the SDS. If this occurs, be certain to re-dissolve it by warming the buffer to room temperature before use.
Alkynyl Labeling Resuspension Buffer
1% SDS (w/v) in 50 mM Tris-HCl, pH 7.5
-
1X Roche Complete Protease Inhibitor Cocktail EDTA free
Once the protease inhibitors have been added, the solution can be stored for up to 2 weeks at 4 °C. Storage at 4 °C may lead to precipitation of the SDS. If this occurs, be certain to re-dissolve it by warming the buffer to room temperature before use.
Y289L Resuspension Buffer
5 M Guanidine hydrochloride
0.3 M Sodium sulfite
Refolding Solution
0.5 M L-Arginine
50 mM Tris-HCl, pH 8.0
5 mM EDTA
4 mM Cysteamine
2 mM Cystamine
Dialysis Solution
50 mM Tris-HCl, pH 8.0
5 mM EDTA
4 mM Cysteamine
2 mM Cystamine
Coomassie Brilliant Blue Stain
0.1% (w/v) Coomassie brilliant blue R-250
50% (v/v) MeOH
7.5% (v/v) Acetic acid
42.5% H2O
DHB matrix
Prepare a 20 mg/mL solution of 2,5-dihydroxybenzoic acid in an 80% acetonitrile solution containing 0.1% trifluoroacetic acid.
Prepare a 20 mg/mL solution of 2-hydroxy-5-methoxybenzoic acid in an 80% acetonitrile solution containing 0.1% trifluoroacetic acid.
Prepare a 9:1 mixture of the 2,5-dihydroxybenzoic acid solution (Step 1) and the 2-hydroxy-5-methoxybenzoic acid solution (Step 2).
Background
Selective and quantitative labeling of O-GlcNAc residues with the poly(ethylene glycol) mass tag is key to measuring O-GlcNAc stoichiometries using this chemoenzymatic approach. Previous studies have shown that GalT demonstrates greater than 11-fold selectivity for N-acetylglucosamine over glucose and glucosamine (Ramakrishnan and Qasba, 2002). The unnatural UDP-ketogalactose sugar is a selective substrate for the Y289L GalT mutant and is a poor substrate for endogenous GalT (Khidekel et al., 2003). Thus, the enzymatic labeling step allows for selective labeling of O-GlcNAc residues only in those reactions in which both the Y289L GalT and UDP-ketogalactose sugar have been included. Moreover, the oxime formation reaction between aminooxy and ketone groups is considered bioorthogonal as ketones and aldehydes are generally not found on proteins. At pH 4.5, only those proteins labeled with the unnatural ketogalactose sugar will be further elaborated with the aminooxy-functionalized PEG tag. Finally and most importantly, both the GalT reaction and subsequent oxime formation reaction proceed quantitatively. The mutant GalT quantitatively labels a model O-GlcNAcylated peptide within 6 hours at 4 °C (Khidekel et al., 2003), and the aminooxy reaction goes to completion on model peptides and proteins within 20 hours at room temperature (Khidekel et al., 2003; Rexach et al., 2010). The quantitative yield for each reaction is critical to ensure accurate calculation of glycosylation stoichiometries.
Most other methods for O-GlcNAc detection focus on relative changes in O-GlcNAc levels, rather than absolute values (Zachara, 2009). In contrast, the mass tagging approach allows for direct measurement of in vivo O-GlcNAc stoichiometries. This enables a more comprehensive analysis of the role of O-GlcNAc glycosylation in specific protein contexts. For example, we found that glucosamine stimulation of rat embryonic neurons induced a 1.4- and 3.1-fold increase in O-GlcNAcylation of CREB and Golgi reassembly-stacking protein of 55-kDa (GRASP55), respectively, suggesting that GRASP55 might be more responsive than CREB to glucosamine. However, further examination using the mass tagging approach revealed that both proteins underwent a similar change in glycosylation stoichiometry (13.1 ± 0.2% and 10.2 ± 0.5%, respectively), suggesting instead that glucosamine may uniformly induce OGT activity toward these substrates (Rexach et al., 2010). In another example, the mass tagging approach revealed that CREB is predominantly monoglycosylated in neurons despite having at least 3 distinct glycosylation sites. By combining the approach with site-directed mutagenesis, we found that CREB is glycosylated primarily at a single site, Ser40, and that glycosylation is induced by neuronal activity specifically at this site (Fig. 4) (Rexach et al., 2012). Thus, the mass-tagging approach provides a direct readout of the glycosylation state and stoichiometry, and when applied in conjunction with site-directed mutagenesis, it has the power to reveal the sugar occupancy at specific glycosylation sites. In contrast, mass spectrometry methods are valuable for identifying the O-GlcNAc sites, but they cannot readily determine their relative occupancy or interrelationship within the same molecule. This highlights the complementary information obtained using the mass tagging approach and mass spectrometric analyses.
Figure 4.
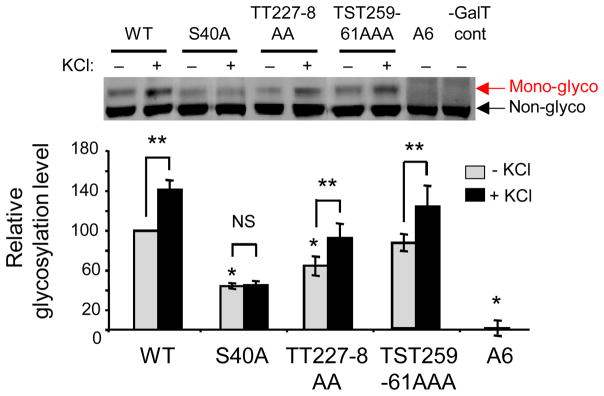
The mass tagging approach was used to determine that CREB is primarily glycosylated at Ser40 and that glycosylation is specifically induced by neuronal depolarization with KCl at Ser40. Neurons were electroporated with wild-type (WT) or mutant CREB constructs, in which the 6 potential glycosylation sites were mutated in various combinations. Neurons were then depolarized with 55 mM KCl, as indicated for 2 hours. Cell lysates were labeled with the mass tagging approach and visualized by immunoblotting for Flag-tagged CREB. Error bars, mean ± s.e.m. * P < 0.01 compared to WT, -KCl; ** P < 0.05; NS, not significant.
Knowing the glycosylation stoichiometry of a protein may provide evidence of the modification’s significance or may suggest how best to study it. For example, if the stoichiometry is low, glycosylation may be inducible, and further tests can be performed to examine the glycosylation dynamics in response to cellular stimuli. It is also worth noting that the same chemoenzymatic strategy can be used to attach other reporter groups, such as a biotin moiety. The biotin handle provides a sensitive method for the enrichment and detection of O-GlcNAcylated peptides/proteins (Tai et al., 2004), and it can facilitate the identification of O-GlcNAc modification sites by mass spectrometry (Khidekel et al., 2007; Khidekel et al., 2004).
In addition to quantifying glycosylation stoichiometries, the mass tagging approach can provide insights into the complex interplay between O-GlcNAc and other posttranslational modifications. By immunoblotting the mass-tagged lysate with both a phospho-specific antibody and an antibody against the total protein, four different subpopulations are resolved: the (1) non-glycosylated, (2) glycosylated, (3) phosphorylated, non-glycosylated, and (4) simultaneously phosphorylated and glycosylated forms (Fig. 5). This provides a direct readout of whether the two modifications are mutually exclusive on proteins of interest or whether they can coexist on the same molecule. The strategy also enables dynamic changes in glycosylation to be monitored specifically on the phosphorylated subpopulation and vice versa. For example, we determined that phosphorylation precedes glycosylation of CREB in response to KCl-induced neuronal depolarization (Rexach et al., 2012). In other studies, traditional O-GlcNAc detection methods showed that total O-GlcNAcylation levels increase on the transcriptional repressor methyl-CpG-binding protein 2 (MeCP2) in response to glucosamine (GlcN), while total Ser80 phosphorylation levels decrease, suggestive of an opposing, yin-yang relationship between glycosylation and phosphorylation. However, closer inspection using the mass tagging approach revealed that glycosylation was induced preferentially on the Ser80 phosphorylated subpopulation of MeCP2. Moreover, despite overall decreases in Ser80 phosphorylation in response to GlcN, Ser80 phosphorylation increased specifically on the glycosylated MeCP2 subpopulation. These results suggest instead a reverse yin-yang relationship and highlight the complex, potentially synergistic relationship between glycosylation and phosphorylation on MeCP2 (Rexach et al., 2010).
Figure 5.
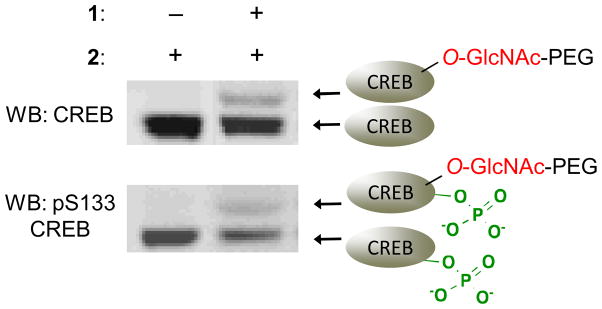
The mass tagging approach can be used to identify different posttranslationally-modified protein subpopulations. Cell lysates were labeled with the mass tagging approach, resolved by SDS-PAGE, and immunoblotted with an antibody against CREB or Ser133 phosphorylated CREB.
Critical Parameters and Considerations
The main choice to be made during this protocol is whether to use a 2-kDa or 5-kDa PEG mass tag in Step 2. The selection depends on the size of the protein of interest. In our experience, proteins smaller than 50-kDa are better visualized using a 2-kDa mass tag, whereas proteins larger than 50-kDa are better visualized using a 5-kDa mass tag. Glycosylation of the 110-kDa subunit of O-GlcNAc transferase (OGT) was readily visualized with a 5-kDa mass tag, while a 2-kDa mass tag was used for the 43-kDa protein CREB (Rexach et al., 2010). Although our studies have focused on proteins within this molecular weight range (40–110-kDa), the mass tagging approach should be amenable in principle to proteins outside of this range.
Different antibodies display different background signals. Additionally, the mass tag could limit antibody recognition of the protein, although we have never directly encountered this situation. For initial experiments, we recommend immunoblotting mass tag-labeled lysate with at least two independent antibodies for the protein of interest to ensure minimal background and adequate detection of the mass tag-labeled samples (Fig. 6).
Figure 6.

Immunoblotting with at least two independent antibodies for a specific protein helps confirm that the mass tag does not affect antibody recognition. In this example, cell lysate was labeled using the mass tagging approach, resolved by SDS-PAGE, and immunoblotted with two different antibodies against O-GlcNAc transferase (OGT).
Good positive control proteins for this reaction include specificity protein 1 (Sp1) and nucleoporin 62-kDa (Nup62). Sp1 from 293T cells and Nup62 from adult rat brain are 100% glycosylated and should resolve as a ladder of 4 and 8 shifted bands, respectively (Fig. 1). Appropriate antibodies to detect these proteins include Millipore 07-645 for Sp1 and BD Biosciences 610497 for Nup62. An incomplete shift in Sp1 and Nup62 would suggest that the labeling reaction did not go to completion. If this is the case, longer protein transfer times, longer incubation times, or more enzyme and substrate may be required (see Troubleshooting).
Chemoenzymatic labeling with mass tags is a quantitative approach for measuring the O-GlcNAcylation stoichiometry on a protein. In our experience, many proteins are O-GlcNAcylated at low stoichiometry (<10%). We have been able to detect as little as 0.8% of a glycosylated OGT standard and 2.3% of endogenous glycosylated synapsin from the adult rat brain cortex, highlighting the sensitivity of the mass tagging approach (Figs. 1 and 7). As traditional enhanced chemiluminescence (ECL)-based methods provide a limited linear detection range, we recommend analyzing the immunoblots using a more quantitative detection method, such as the Odyssey Western Blot detection system.
Figure 7.

The mass tagging approach can detect as little as 0.8% of glycosylated OGT. Labeled and unlabeled cell lysate was combined to mimic different OGT glycosylation stoichiometries. The limit of detection was determined as the lowest stoichiometry within 10% of the linear fit.
We developed the mass tagging approach to measure the stoichiometry of intracellular O-GlcNAc glycosylation. The method works by selectively labeling terminal GlcNAc residues. Terminal GlcNAc residues are characteristic of intracellular O-GlcNAc glycosylation but may also be found on complex extracellular glycans. In most cases, proteins are known to be intracellular or extracellular, making it easy to determine whether the terminal GlcNAc comprises the intracellular O-GlcNAc modification or a larger extracellular glycan. However, proteins with extracellular O-GlcNAc glycosylation have recently been identified (Matsuura et al., 2008). In those cases where the assignment is less clear, additional methods such as subcellular fractionation or mass spectrometry may be required to carefully characterize the type of glycosylation. For example, chemoenzymatic labeling of O-GlcNAc-modified proteins with a biotin tag provides a unique signature upon MS/MS analysis that allows for unambiguous assignment of the O-GlcNAc modification (Khidekel et al., 2004).
We have studied the [3+2] azide-alkyne cycloaddition labeling method in less detail than the ketone-aminoxy oxime labeling method. The Y289L GalT/UDP-GalNAz step goes to completion on a model peptide within 12 hours at 4 °C. When using this approach, it will be important to determine conditions that lead to complete reaction between the azido-galactose labeled proteins and alkynyl-functionalized PEG. O-GlcNAcylation stoichiometries should be interpreted carefully until the approach is fully validated with proteins of known stoichiometry (Rexach et al., 2010).
Anticipated Results
Chemoenzymatic labeling with mass tags can provide information on the O-GlcNAc modification state of a protein (e.g. whether the protein is predominantly mono-, di-, or triglycosylated), as well as its in vivo glycosylation stoichiometry. The expected result from a successful labeling reaction is an immunoblot containing a negative control lane (e.g., no GalT added) with a band for the protein of interest and an experimental lane containing this band shifted (either entirely or in part) by 2- or 5-kDa mass increments, depending on the mass tag used (Fig. 1).
Data Analysis
The number of extra bands in the experimental lane represents the different glycosylated subpopulations of the protein. A single band shifted higher by 2- or 5-kDa suggests that the protein is predominantly monoglycosylated. The presence of two bands, one shifted higher by 2- or 5-kDa and the other by 4- or 10-kDa, suggests that the protein is both mono- and diglycosylated. It is important to note that although a protein may be monoglycosylated, it can still have multiple sites of glycosylation. The number of mass-shifted bands only describes the distinct glycosylated subpopulations that have a total of one, two, three, etc. O-GlcNAc sugars per protein. As such, the monoglycosylated subpopulation may consist of multiple different glycoforms, as in the case of CREB (Rexach et al., 2012).
O-GlcNAcylation stoichiometries must be carefully calculated to obtain accurate, reproducible results. To determine the stoichiometry of a specific glycosylated subpopulation, the ratio of the intensity of that mass-shifted band to the intensity of all the protein bands (shifted and non-shifted) is taken, accounting for background signal in the control lane. Specifically, in the experimental lane, the intensity of the non-shifted band (en) and each mass-shifted band should be individually quantified (es1, es2, …). In the control lane, the intensity of the background signal corresponding to each of the mass-shifted bands should be individually quantified (cs1, cs2, …). The following formula can then be used to calculate the total protein glycosylation stoichiometry:
Alternatively, the glycosylation stoichiometry of a specific glycosylated subpopulation si can be calculated as:
When quantifying immunoblots, care should be taken to use a quantitative immunoblotting system (e.g. an Odyssey system) or ECL film exposures in which the intensity of each band is within the linear range.
Time considerations
The entire labeling experiment can be completed in two to three days. Both the ketogalactose labeling step and the aminooxy PEG labeling steps require approximately 90 minutes of hands-on time. Cell lysis, protein concentration measurements, SDS-PAGE, and immunoblotting procedures can take variable amounts of hands-on time, depending on individual approaches to each step.
Troubleshooting
| Problem | Possible Cause | Solution |
|---|---|---|
| No mass tag shift is detected even though other O-GlcNAc detection methods suggest that the protein is glycosylated, and a control O-GlcNAc protein (e.g. Nup62 or Sp1) in the lysate is showing the anticipated mass shift. | The protein glycosylation stoichiometry level is below the detection limit of the antibody. | Increase the amount of the protein of interest on the SDS-PAGE gel, either by labeling a greater amount of lysate or by enriching for the protein through subcellular fractionation or immunoprecipitation. |
| No or low levels of glycosylation are detected on positive control proteins (e.g. Nup62 or Sp1), despite verified reagents and good quality sample. | The mass-tagged proteins are difficult to transfer to the immnoblotting membrane and have failed to fully transfer. | Increase the transfer time by 33%. |
| The labeling reaction did not proceed quantitatively. | Increase enzyme and substrate concentrations and/or reaction times. | |
| The glycosylated protein fraction was not adequately separated from the non-glycosylated protein fraction. | The protein was not sufficiently resolved. | Run the protein of interest further through the gel to allow for adequate subpopulation separation. Consider a gradient gel or altering the acrylamide percentage. |
| The size of the mass tag chosen was too small. | Choose a larger size mass tag (e.g. 5-kDa instead of 2-kDa). We usually use a 2-kDa PEG mass tag for proteins smaller than 50-kDa and a 5-kDa PEG mass tag for proteins larger than 50-kDa. | |
| Multiple bands are seen in both the control and experimental lanes of the immunoblot. | The antibody chosen for analyzing the protein of interest identifies multiple proteins and is obscuring the mass shift. | Immunoblot with a different antibody against the protein of interest. |
| The protein is modified by other post-translational modifications (i.e. ubiquitinylation) that cause a shift in the protein’s apparent molecular weight. | Pre-treat the samples to remove these other post-translational modifications. |
Acknowledgments
We thank P. Qasba (National Cancer Institute at Frederick) for helpful discussions on GalT and generously providing the Y298L GalT construct. This work was supported by the National Institutes of Health (R01 GM084724 to L.C.H.-W., F31 NS056525 to J.E.R., and 5T32 GM07737 to P.M.C.).
Literature Cited
- Clark PM, Dweck JF, Mason DE, Hart CR, Buck SB, Peters EC, Agnew BJ, Hsieh-Wilson LC. Direct in-gel fluorescence detection and cellular imaging of O-GlcNAc-modified proteins. J Am Chem Soc. 2008;130:11576–11577. doi: 10.1021/ja8030467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart GW, Housley MP, Slawson C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 2007;446:1017–1022. doi: 10.1038/nature05815. [DOI] [PubMed] [Google Scholar]
- Khidekel N, Arndt S, Lamarre-Vincent N, Lippert A, Poulin-Kerstien KG, Ramakrishnan B, Qasba PK, Hsieh-Wilson LC. A chemoenzymatic approach toward the rapid and sensitive detection of O-GlcNAc posttranslational modifications. J Am Chem Soc. 2003;125:16162–16163. doi: 10.1021/ja038545r. [DOI] [PubMed] [Google Scholar]
- Khidekel N, Ficarro SB, Clark PM, Bryan MC, Swaney DL, Rexach JE, Sun YE, Coon JJ, Peters EC, Hsieh-Wilson LC. Probing the dynamics of O-GlcNAc glycosylation in the brain using quantitative proteomics. Nat Chem Biol. 2007;3:339–348. doi: 10.1038/nchembio881. [DOI] [PubMed] [Google Scholar]
- Khidekel N, Ficarro SB, Peters EC, Hsieh-Wilson LC. Exploring the O-GlcNAc proteome: direct identification of O-GlcNAc-modified proteins from the brain. Proc Natl Acad Sci U S A. 2004;101:13132–13137. doi: 10.1073/pnas.0403471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love DC, Hanover JA. The hexosamine signaling pathway: deciphering the “O-GlcNAc code”. Sci STKE. 2005:re13. doi: 10.1126/stke.3122005re13. [DOI] [PubMed] [Google Scholar]
- Matsuura A, Ito M, Sakaidani Y, Kondo T, Murakami K, Furukawa K, Nadano D, Matsuda T, Okajima T. O-linked N-acetylglucosamine is present on the extracellular domain of notch receptors. J Biol Chem. 2008;283:35486–35495. doi: 10.1074/jbc.M806202200. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan B, Qasba PK. Structure-based design of beta 1,4-galactosyltransferase I (beta 4Gal-T1) with equally efficient N-acetylgalactosaminyltransferase activity: point mutation broadens beta 4Gal-T1 donor specificity. J Biol Chem. 2002;277:20833–20839. doi: 10.1074/jbc.M111183200. [DOI] [PubMed] [Google Scholar]
- Rexach JE, Clark PM, Hsieh-Wilson LC. Chemical approaches to understanding O-GlcNAc glycosylation in the brain. Nat Chem Biol. 2008;4:97–106. doi: 10.1038/nchembio.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rexach JE, Clark PM, Mason DE, Neve RL, Peters EC, Hsieh-Wilson LC. Dynamic O-GlcNAc modification regulates CREB-mediated gene expression and memory formation. Nat Chem Biol. 2012;8:253–261. doi: 10.1038/nchembio.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rexach JE, Rogers CJ, Yu SH, Tao J, Sun YE, Hsieh-Wilson LC. Quantification of O-glycosylation stoichiometry and dynamics using resolvable mass tags. Nat Chem Biol. 2010;6:645–651. doi: 10.1038/nchembio.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai HC, Khidekel N, Ficarro SB, Peters EC, Hsieh-Wilson LC. Parallel identification of O-GlcNAc-modified proteins from cell lysates. J Am Chem Soc. 2004;126:10500–10501. doi: 10.1021/ja047872b. [DOI] [PubMed] [Google Scholar]
- Thannhauser TW, Konishi Y, Scheraga HA. Sensitive quantitative analysis of disulfide bonds in polypeptides and proteins. Anal Biochem. 1984;138:181–188. doi: 10.1016/0003-2697(84)90786-3. [DOI] [PubMed] [Google Scholar]
- Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA, 3rd, Peters EC, Driggers EM, Hsieh-Wilson LC. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science. 2012;337:975–980. doi: 10.1126/science.1222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachara NE. Detecting the “O-GlcNAc-ome”; detection, purification, and analysis of O-GlcNAc modified proteins. Methods Mol Biol. 2009;534:251–279. doi: 10.1007/978-1-59745-022-5_19. [DOI] [PubMed] [Google Scholar]