

Abstract
Herein we report a proof of principle study illustrating a novel dog-human comparison strategy that addresses a central aim of cancer research, namely cancer driver–passenger distinction. We previously demonstrated that sporadic canine colorectal cancers (CRCs) share similar molecular pathogenesis mechanisms as their human counterparts. In this study, we compared the genome-wide copy number abnormalities between 29 human- and 10 canine sporadic CRCs. This led to the identification of 73 driver candidate genes (DCGs), altered in both species and with 27 from the whole genome and 46 from dog-human genomic rearrangement breakpoint (GRB) regions, as well as 38 passenger candidate genes (PCGs), altered in humans only and located in GRB regions. We noted that DCGs significantly differ from PCGs in every analysis conducted to assess their cancer relevance and biological functions. Importantly, while PCGs are not enriched in any specific functions, DCGs possess significantly enhanced functionality closely associated with cell proliferation and death regulation, as well as with epithelial cell apicobasal polarity establishment/maintenance. These observations support the notion that, in sporadic CRCs of both species, cell polarity genes not only contribute in preventing cancer cell invasion and spreading, but also likely serve as tumor suppressors by modulating cell growth. This pilot study validates our novel strategy and has uncovered four new potential cell polarity and colorectal tumor suppressor genes (RASA3, NUPL1, DENND5A, and AVL9). Expansion of this study would make more driver-passenger distinctions for cancers with large genomic amplifications or deletions, and address key questions regarding the relationship between cancer pathogenesis and epithelial cell polarity control in mammals.
Keywords: CRC driver-passenger distinction, sporadic canine CRCs, human-dog comparison, epithelial cell apicobasal polarity, genomic amplifications/deletions
INTRODUCTION
A central aim of cancer research has been to identify a handful of cancer-causative alterations (drivers) from hundreds to thousands of abnormal changes found in a cancer genome (1, 2). Discoveries of such drivers in the past have contributed to the understanding of cancer etiology and yielded prognostic markers and therapeutic intervention targets such as BCR-ABL, leading to the development of the spectacularly successful anti-leukemia drug imatinib (Gleevec) (3). As high-throughput technologies such as next-generation sequencing and high density array become routinely available, hundreds of thousands of cancer genomes have been or will be characterized and millions of cancer-related somatic mutations will be uncovered (1, 2, 4, 5). Hence, the need to efficiently distinguish drivers from passengers (incidental changes or changes occurring as a consequence of cancer) becomes increasingly pressing and has become one of the National Cancer Institute (NCI)'s Provocative Questions that need to be urgently addressed (provocativequestions.nci.nih.gov).
Drivers are typically more recurrent than passengers, and researchers have been using strategies such as increasing sample size and studying early stage cancers with advanced technologies and data analysis tools for their differentiation (1, 2, 4-6). Differing from these traditional approaches that study human cancers only, we have developed a novel human-dog comparative genomics and oncology strategy for cancer driver-passenger distinction for genes amplified or deleted in human cancers, rationalized as follows. The dog has become an increasingly important model for studying human physiology and diseases (7, 8), and sporadic canine cancer represents one of the best cancer models (9). First, these cancers are naturally-occurring and heterogeneous, unlike most genetically-modified or xenograft rodent models, and significantly, dogs share the same environment as humans and hence are exposed to the same carcinogens. Furthermore, the dog genome has been sequenced to a >7X coverage, and a relatively accurate version of its genome sequence assembly is available (10), unlike another companion animal, the cat, which was sequenced only to a 2.8X coverage (11). Critically, the dog genome is rearranged when compared to the human genome with over 300 inversions and translocations identified (10, 12), resulting in thousands of genes that are clustered in the human genome to be dispersedly located in the dog genome. We utilize these orthologous genes’ different genomic locations between the two species for driver-passenger distinction. Our hypothesis is that provided the two species share similar molecular pathways of pathogenesis for the cancer of interest, alterations that are common to both will be deemed drivers, whereas those that are found in only one species and are located in the dog-human genomic rearrangement sites will be considered as passengers.
To test this hypothesis, we conducted proof of principle studies on CRC, one of the best understood cancers for studying cancer initiation and progression (5, 13, 14). Critically, we have previously demonstrated that sporadic canine CRCs likely follow similar cancer development and progression pathways as their human counterparts (15, 16), providing the molecular justification for applying this novel cross-species comparison strategy on CRC. In this study, we compared copy number abnormality (CNAs) between human and dog sporadic CRCs (15, 17) and identified 73 driver candidate genes (DCGs) and 38 passenger candidate genes (PCGs). We noted that DCGs significantly differ from PCGs in every analysis conducted to assess their cancer relevance and biological functions. Importantly, while PCGs’ functions appear random, DCGs are significantly enriched in functions closely associated with cell proliferation/death regulation, as well as with epithelial cell apicobasal polarity establishment/maintenance. These observations emphasize the importance of cell polarity genes in CRC pathogenesis, consistent with published studies elegantly illustrating that in a Drosophila genetic model with the oncogenic RasV12 mutation, loss of cell polarity both accelerates the growth and drives the invasion of RasV12-induced benign tumors through JNK activation (18). In summary, this pilot study demonstrates that the proposed dog-human comparison strategy for cancer driver-passenger discrimination is valid. It also provides genome-wide mammalian data supporting the notion that alterations of cell polarity genes, hence loss of epithelial cell polarity, are among major drivers of CRC carcinogenesis, a critical but controversial subject in the field (19, 20).
RESULTS
CRC driver-passenger distinction via human-dog comparison
Genome-wide comparisons – 1stDCG identification
We first compared the genome-wide CNAs between 29 human sporadic CRCs (17) and 10 canine sporadic CRCs (15), with the vast majority being chromosome instability (CIN) tumors and harboring large genomic amplifications and deletions. This analysis led to the discovery of 27 genes, with each having at least one of its exons significantly altered in both species (Fig. 1), autosome-wide [sex chromosomes were excluded because the canine Y chromosome is not yet sequenced (10) and the extensive sequence duplication between X and Y chromosomes (21) complicates the analysis]. These 27 genes, which include the best known colorectal tumor suppressor APC (13) (Supplementary Table s1), are referred to as the first DCGs or 1stDCGs hereafter.
Fig. 1. CRC driver-passenger distinction via human-dog comparison.

The top portion illustrates how 1stDCGs (27 total), shown in purple and with at least one of their exons significantly amplified/deleted in both species, were identified autosome-wide (see the main text and Supplementary Table s1 for details). The middle section indicates how 2ndDCGs (46 total), shown in green, and PCGs (38 total), shown in red, were identified from the human-dog GRB sites in the human genome (see the main text for criteria for DCG-PCG distinction; Supplementary Tables s1-s3 and Fig. s1 for details). The bottom area shows examples of DCG-PCG pairs identified from two GRB sites in the human genome that harbor a human-dog translocation breakpoint (left) or a human-dog inversion breakpoint (right). Genes in each example are clustered in the human genome, but are either located on different chromosomes (right), or far apart (0.2 Mb for the human versus 16.6Mb for the dog) if on the same chromosome (left), in the dog genome. In addition, genes in each example are all amplified in the human tumors; however, in the dog tumors, only DCGs [GADD45A and SLC2A1, both of which are already known cancer driver genes (22, 23)] are amplified, whereas PCGs (genes shown in red, none of which has been reported in the literature to be cancer driver genes) remain neither amplified nor deleted.
Genomic rearrangement breakpoint (GRB) regions-focused comparison – 2ndDCG and PCG identification
We then focused on the human genomic sites that harbor the human-dog GRB (a total of 324 GRBs were detected for the autosomes; see Supplementary Table s2) to identify potential driver-passenger pairs (Fig. 1), based on the following criteria. First, the greater GRB region, which includes the GRB itself and the two immediate flanking regions with each containing at least one gene, is altered in at least one human tumor. Second, at least one canine orthologous gene at one side of the GRB is altered in at least one dog tumor, while none of the canine orthologous genes at the other side of the GRB is disrupted in any of the dog tumors (Fig. 1). Once these two conditions were met, genes changed in both species were deemed drivers and those disrupted only in human tumors were considered as passengers (Fig. 1). This strategy allowed the identification of additional 46 DCGs (referred to as 2ndDCGs hereafter) and 38 PCGs from 25 GRB sites, with most genes in both groups recurrently altered in the human CRCs (Supplementary Tables s1 & s3).
Combined the two approaches described above, a total of 73 DCGs (27 1stDCG and 46 2ndDCG) and 38 PCGs were identified. We then conducted both bioinformatic and experimental analyses to evaluate their cancer relevance and the biological functions, as described below.
DCGs’ cancer relevance is strong, but PCGs’ is not
Known cancer drivers found among DCGs but not among PCGs
Besides APC, both 1stDCGs and 2ndDCGs contain known cancer genes with many already being targeted for therapeutic intervention, including the DNA damage-inducible cell-cycle regulator GADD45A (22), the constitutive glucose transporter SLC2A1 (23), the signaling molecule A3 adenosine receptor ADORA3 (24), the chromatin modifier EZH2 (6), the receptor tyrosine phosphatase PTPRD (25), and the cancer biomarker MUC16 (26). The PCGs, however, do not contain any such genes.
DCGs significantly differ from PCGs in cancer relevance examined
To assess the cancer relevance of our candidate genes, we determined their presence/absence in well-known cancer databases including Cancer Gene Census (a catalog of genes of which mutations are causally implicated in cancer) (6), KEGG cancer pathways, COSMIC (a catalog of somatic mutations of cancers) (27), and Cancer Gene Index (a database of associations between genes and cancers developed at the NCI). Then, we searched the Mouse Genome Informatics database for phenotypic changes in existing gene-knockout mouse models, including tumorigenesis and other significant alterations. It has been reported that cancer genes interact with more partners than a typical human gene (28); we thus determined the predicted protein-protein interactions and DNA-protein interactions (which imply how well a gene is transcriptionally regulated) of each gene. Finally, we also searched the PubMed cancer subset, a collection of cancer-related literatures created by the NCI and the National Library of Medicine (www.nlm.nih.gov/bsd/pubmed_subsets/cancer_strategy.html). All the information was catalogued in Supplementary Table s1, on which we performed Hotelling's T-squared tests. As shown in Fig. 2, while the difference between the two DCG groups is insignificant (p = 0.5), DCGs indeed differ notably from PCGs in these cancer-related features examined (p ≤ 0.05).
Fig. 2. DCGs significantly differ from PCGs in cancer relevance examined.

A) The top triangle shows the Hotelling's T-squared test results performed with the data documented in Supplementary Table s1 and described in the text. The numbers shown between the boxes are p-values from the tests. The table below indicates the somatic mutation status reported by a recent NGS study with 276 human CRCs (5) for each group of our candidate genes (see Supplementary Table s4 for details).
B) DCGs and PCGs responding differently in their mRNA expression changes to Taxol-treatment of HCT116 cells. The treatment of 20nM Taxol for 24 hours and 48 hours induced HCT116 cell death and cell cycle arrest (demonstrated by the multinucleate cells indicated with red arrows) (Top). The overall gene expression change at 20nM Taxol between different treatment times was indicated by the p-values, obtained from t-tests using data from Supplementary Table s5 and with “↑” representing significant increase and “↓” indicating significant decrease (middle) (see Supplementary Fig. s2 for p-values of DCGs/PCGs separated based on whether they were amplified or deleted in CRCs). The bottom shows the expression change of individual DCGs, including GADD45A and C1orf63 with similar drug-response profiles, as well as those having a slight expression decrease at 24 hours followed by a larger increase or a near complete recovery at 48 hours. Similar gene expression trends were observed for 10nM and 80nM Taxol treatment (see Supplementary Table s5).
DCGs significantly mutated in human CRCs but PCGs did not
We took advantage of a recent publication from The Cancer Genome Atlas, which investigated 276 human CRCs via next-generation sequencing (NGS) (5). As shown in Fig. 2 and Supplementary Table s4, both 1stDCGs and 2ndDCGs carry significantly more nonsynonymous somatic mutations than PCGs. Upon excluding missense mutations and only including nonsense mutations and fame-shift indels, DCGs become even more significant, with 14 1stDCGs (p = 0.0003) and 17 2ndDCGs (p = 0.02), but only 6 PCGs (p = 0.5), being identified. This provided additional evidence supporting the strong cancer relevance of DCGs, but not PCGs.
DCGs responded to Taxol-treatment of HCT116 cells but PCG did not
We treated HCT116, a near diploid human CRC line, with Taxol (Paclitaxel), a anticancer drug shown to stabilize microtubules, block the progression of cell cycle and trigger cancer cell death (29, 30). For PCGs, we did not observe any significant changes in gene expression on the whole at different Taxol concentrations (10nM, 20nM, and 80nM) and treatment times (24 hours and 48 hours) (Fig. 2 and Supplementary Table s5). For DCGs (especially those deleted in CRCs; see Table s5 and Fig. s2), however, we noted an initial small expression decrease after 24 hours’ treatment of most genes, for many of which this was followed by a larger expression increase, making the expression level even higher than before the drug treatment or a near complete recovery at 48 hours (Fig. 2).
We do not know if DCGs’ initial expression decrease described above is related to their enrichment in cell adhesion functions (see later sections), but both us (Fig. 2) and others (30) observed a drastic decrease in cell adhesion after 24 hours’ Taxol-treatment. The subsequent expression increase at 48 hours (Fig. 2) could possibly arise from the accelerated expression increment of cell cycle check-point genes, which arrested the cell cycle at G2/M checkpoints demonstrated by the significantly increased number of multinucleate cells observed at 48 hours as compared to 24 hours (see Fig. 2 and reference 30). Two DCGs, GADD45A and C1orf63, are especially noteworthy, with their expression level increasing along with the Taxol concentration and incubation time, reaching 18-fold for C1orf63 and 14-fold for GADD45A at 80nM for 48 hours (Fig. 2). GADD45A is known to be upregulated by stress and drug therapy, and is required at the G2/M checkpoint and arrests cell cycle progression (22). The function of C1orf63 is unknown currently; its similar Taxol response profile as GADD45A raises the possibility that C1orf63 may possess comparable functions.
In summary, while much more work is clearly needed to understand the mechanisms of the observed drug response, the study demonstrated a clear difference between DCGs and PCGs in their overall response to Taxol treatment of HCT116 cells. This supports the strong cancer relevance of DCGs (but not PCGs) concluded from the bioinformatic analyses described above.
Many DCGs contributed to cell growth, proliferation or death regulation; PCGs did not
A total of 17 DCGs (23%) but only three PCGs (8%) were annotated to participate in cell growth/death regulation, cell cycle control, and DNA repair (Supplementary Table s6). Furthermore, we have experimentally demonstrated additional 12 DCGs being critical to the proliferation/death of human CRC cells HCT116, DLD1 and SW620 (Fig. 3 and Supplementary Fig. s3). Four genes, including RASA3, DENND5A, AVL9, and NUPL1, are especially noteworthy. They are all predominantly deleted in CRCs (Table s1), and their siRNA knockdown promoted cell growth of HCT116 and other CRC cells (Fig. 3 and Fig. s3). These observations support that these genes are potential tumor suppressors of CRC. RASA3 (a RAS GTPase activator) and DENND5A (a GDP-GTP exchange factor for RAB39; see Supplementary Table s7) both participate in small GTPase signaling, AVL9 function in exocytosis in yeast (31), and NUPL1 is a nucleoporin gene (Fig. 4 and Table s1). Except for RASA3 which is known to inactivate RAS by enhancing its weak intrinsic GTPase activity (32), no any previous studies have been documented regarding their role in cancer.
Fig. 3. siRNA knockdown of DCGs promotes or inhibits the growth of HCT116 cells.

A) Cell growth promotion: The six genes shown are expressed in HCT116 and siRNA knockdown was performed, as demonstrated by the bar graphs on the top where the p-value indicates the mRNA expression difference between scramble-control (S) and knockdown (KD) of each gene. Then, the cell growth was quantified by crystal violet-staining, represented by the images in the middle, with the color intensity quantification indicated by the bar graphs below the images and the difference between scramble-control and knockdown specified by the p-values. B) Cell growth inhibition: siRNA knockdown of three genes indicated hinders cell growth. These genes and additional ones were assayed similarly in two other CRC lines SW620 and DLD1 (Supplementary Fig. s3), as well the human cervical cancer line HeLa and an immortalized primary human astrocyte (IHA).
Fig. 4. DCGs (but not PCGs) are enriched in functions associated with epithelial cell polarity establishment and maintenance, and many DCGs participate in cell proliferation/death and/or cell cycle control.

The top left images indicate that loss of epithelial cell apicobasal polarity occurs in both adenomas (tumor cells not yet penetrating the basement membrane) and adenocarcinomas (tumor cells already invading tissue layers below the epithelium). Genes shown to the right of the images are 1stDCGs (purple), 2ndDCGs (green), or PCGs (red) already known or likely functioning in epithelial cell polarity based on literature reports (see the main text), as well as gene ontology (GO) terms, INTERPRO domains, and other information provided by DAVID (33) (Supplementary Table s6). The genes with “?” were classified based on protein domain matches only. Genes shown below the images are DCGs or PCGs participating in cell proliferation/death and/or cell cycle regulations, based on annotation by DAVID (33) (Supplementary Table s6), published literature (see the main text), and experiments (Fig. 3 and Fig. s3). See Supplementary Fig. s4 for the classified functions of all DCGs and PCGs that may or may not relate to cell polarity.
In summary, 28 out of 73 total (38%) DCGs are closely associated with cell proliferation and death regulation, cell cycle control, and/or DNA repair (Figs. 3 and 4; Table s6), as known or putative cancer genes. For PCGs, however, only three out of 38 total (<10%) are involved in such processes (ASAH2B, CTGF, and SERBP1), some of which may be false negatives that could be resolved by increasing sample size.
DCGs (but not PCGs) significantly enriched in functions closely associated with epithelial cell apicobasal polarity establishment and maintenance
1stDCGs enriched in cell adhesion/motility functions and 2ndDCG enriched in vesicle-trafficking and ion-transport functions, whereas PCGs not enriched in any specific functions
We applied the frequently-used DAVID Gene Functional Classification Tools (33) on the candidate genes and observed notably more functional groups being over-represented in DCGs than in PCGs. These include cell adhesion (p = 0.02), adherent junctions (p = 0.04), cell motility (p = 0.05), and cell morphogenesis (p = 0.05) for 1stDCGs, as well as vesicle-trafficking (p = 0.03) and ion transport (p = 0.05) for 2ndDCGs (Supplementary Table s8). For PCGs, however, no any significant enrichment was found, with C2H2-zinc finger (ZNF) genes being the most enriched group (p = 0.14) (Table s8).
Because 2ndDCGs and PCGs were identified from the 324 GRB sites rather than autosome-wide (Fig. 1), we performed the same enrichment analysis using the 4,401 genes encoded in 1Mb sequences surrounding each GRB (0.5Mb each of upstream and downstream of the GRB center) as the background, instead of the entire autosome gene set as above. The analysis revealed ion homeostasis/transport and vesicle-trafficking remaining significant (p ≤ 0.05) for 2ndDCGs as above (Table s8). For PCGs, however, still no significant enrichment was observed, and C2H2-ZNFs (p = 0.23) were replaced by nuclear lumen-associated genes as the most enriched, which stay nevertheless insignificant (p = 0.17) (Table s8).
GRB sites enriched in C2H2-ZNFs and G-protein coupled receptors (GPCRs)
The following analysis explained why up to five C2H2-ZNFs are among PCGs (Table s1). Compared to the whole autosome set, the 324 GBR sites, which were used to identify PCGs and 2ndDCGs (Fig. 1), are significantly enriched with C2H2-ZNF genes (p ≤ 0.0001; Supplementary Table s9). However, when comparing the 25 GBR sites, where PCGs and 2ndDCGs locate, against the entire 324 GBR set, the difference is insignificant (p > 0.3). Hence, consistent with published studies (34), the GRB sites harbor significantly more C2H2-ZNFs than other genomic regions [for which a possible explanation could be that, like segmental duplications that are also enriched at the GRB sites (12), C2H2-ZNF-formed gene clusters could mediate genomic rearrangements, which in turn expand this gene family via duplications and deletions]. Thus, the five C2H2-ZNFs in PCGs were pulled out by random chance. Similar conclusions were reached for GPCRs and olfactory receptors (ORs) (Tables s1 and s9). C2H2-ZNFs and GPCRs are the only gene families of which multiple members were found among PCGs.
Many DCGs likely function in cell polarity, while PCGs do not
DCGs’ significant enrichment in cell adhesion and trafficking functions described above indicate that many DCGs could possibly be cell polarity genes, rationalized as follows. Colon epithelial cells are asymmetric, exhibiting apical and basal polarity (Fig. 4). This allows them to form permeability barriers between two compartments in the body and to vectorially transport ions and solutes between the compartments. According to published literature (35), development of epithelial cell polarity requires cell-cell and cell-substratum adhesions achieved by genes functioning in various adhesion complexes, cell junctions, and cytoskeleton organization. In addition, the polarized plasma membrane, which is divided into functionally and structurally distinct apical and basolateral domains, is established and maintained by polarized intracellular protein sorting, trafficking and targeting involving a variety of genes and organelles including trans-Golgi network and endosomes.
We found that up to 78% of 1stDCGs and 72% of 2ndDCGs, but only 26% of PCGs, might participate in these processes of establishing and maintaining epithelial cell polarity. Specifically, as many as 56% of 1stDCGs (15 genes out of 27 total, with 12 certain and three likely) and 39% of 2ndDCGs (18 genes out of 46 total, with 13 certain and five likely), but only 13% of PCGs (five genes out of 38 total, with three certain and two likely), possess functionality in cell-cell or cell-substratum adhesions, as listed in Fig. 4 and Table s6. For polarized ion-transport and trafficking, we noted up to 33% of 2ndDCGs (15 genes with 13 certain and two likely) and 22% of 1stDCGs (six genes with four certain and two likely), but only 13% of PCGs (five genes) having or likely having such functions (Fig. 4).
Significantly, a substantial portion of DCGs are already known to be located in different portions of the polarized plasma membrane (Fig. 4 and Table s6); however, we did not find such information for any of the PCGs. Furthermore, a total of eight 1stDCGs and six 2ndDCGs are already-known epithelial cell polarity genes (Fig. 4 and Table s6). Many function in cell adhesion, including protocadherin PCDH9; the multifunctional CRC tumor suppressor APC; membrane-associated guanylate kinase MPP7 and DLGAP2 (DLG-associated protein 2) (36); basement membrane genes LAMA1 and FREM2; cytoskeleton genes ACTC1 and KIFAP3 (37); and small GTPase signaling molecules RAP1A and ARHGEF7 (38). Others function in polarized ion and small molecule transporting, including potassium/chloride cotransporter SLC12A6 (39), glucose/cation symporter SLC2A1, apical membrane-located v-type proton ATPase subunit ATP6V1B2 (40), and phosphodiesterase ENPP1 (40). Furthermore, 1stDCGs contain three additional putative polarity genes: 1) EFNB2, a member of Eph/Ephrin signaling pathway which regulates the mesenchymal-epithelial transition (MET) (41); 2) EYA2, a tyrosine phosphatase that dephosphorylates histone H2AX and involves in epithelial-mesenchymal transition (EMT) (42); and 3) MYCBP2, a MYC-binding protein whose functions include cytoskeleton organization (37). Similarly, 2ndDCGs also harbor five such genes: 1) AVL9, an exocytosis gene in yeast (31); 2) CHRNA7, a cholinergic receptor whose functions include regulating calcium homeostasis (43); 3) EPDR1, an ependymin-related gene that is involved a cell-substratum adhesion; 4) MLANA, a palmitoylated integral membrane protein likely involving in sorting and degradation of melanosome proteins (44); and 5) SFRP4, a WNT-signaling gene. In summary, as many as 41% of 1stDCGs and 24% of 2ndDCGs are already-known or likely polarity genes. For PCGs, only three (8%) are possibly such genes [CTGF, SEC24C, TRIM62 (45)].
Deleted (in CRC) DCGs’ expressions increase significantly in a cell culture system where cell-cell/cell-ECM (extracellular matrix) adhesions are establishing
The above analysis is corroborated by mRNA expression changes of DCGs and PCGs in an in vitro human embryonic stem cell (hESC) differentiation system, hESC WA09 → ISL1+ nascent mesoderm, during which cell-cell and cell-ECM adhesions are being built. Among 265 most significantly upregulated genes during this differentiation revealed by microarray analyses, 96 of them function in cell adhesion, ECM building, and/or are secreted (Supplementary Table s10), making focal adhesion and ECM-receptor interaction the most significantly enriched pathways among these upregulated genes (FDR < 0.2). As summarized in Fig. 5, we observed a significant expression increase on the whole for DCGs that were predominantly deleted in CRCs (Table s1), especially for those from 1stDCGs where the overall expression increased by two-fold (Supplementary Table s11). Meanwhile, the expression of PCGs barely changed on average. This result is consistent with the cell adhesion function listed for many of the deleted DCGs in Fig. 4.
Fig. 5. Expression changes of DCGs and PCGs in an in vitro hESC differentiation system.
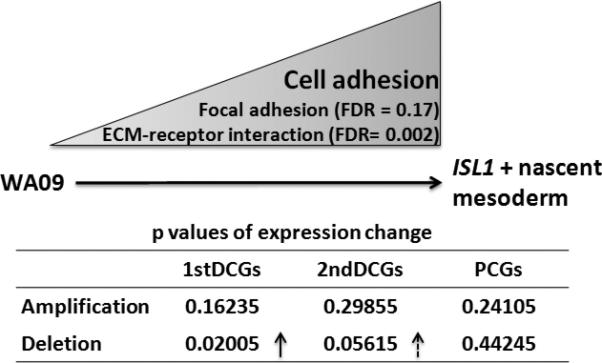
During this differentiation cell-cell/cell-ECM adhesions are establishing, demonstrated by microarray expression studies (see the main text). The expression level of individual genes of 1stDCGs, 2ndDCGs, and PCGs in hESC WA09 and its differentiated ISL1+ nascent mesoderm was determined by qRT-PCR (Supplementary Table s11). T-tests were then performed to determine the significance of the expression difference for the genes of each group, which were further separated depending upon if a gene was predominantly amplified (indicated by “Amplification” in the image) or deleted (indicated by “Deletion”) in CRCs examined (Supplementary Tables s1 & s11), between the two cell types. “↑” indicates a significant increase in gene expression.
Although both bioinformatic and experimental analyses described above supports the likelihood that DCGs participate in cell polarity building and maintenance whereas PCGs do not, we acknowledge that for numerous DCGs (Fig. 4), it remains to be experimentally determined whether they are polarity genes or not.
Other DCG-PCG differences in functional groups
Besides those described above, we also noted several other functional groups being more prominent in DCGs than in PCGs (Table s7). These include: 1) ubiquitin-dependent protein degradation genes (six for DCGs versus none for PCGs); 2) redox genes (five for DCGs versus none for PCGs); 3) calcium-binding/dependent genes (10 for DCGs versus one for PCGs); and 4) developmental genes (13 for DCGs versus three for PCGs).
DISCUSSION
Our novel dog-human comparison strategy for cancer diver-passenger discrimination is valid
This pilot project provides a proof of principle study successfully demonstrating the efficiency of our approach on CRCs with large genomic amplifications and deletions. First, the identified DCGs significantly differ from the PCGs in every analysis performed, including cancer relevance based on existing cancer databases and literature, significant somatic mutations in CRCs discovered by NGS (5), and response to the anticancer drug Taxol (Fig. 2). Importantly, both bioinformatic and experimental analyses revealed that, while PCGs’ functions appear random, DCGs hold significantly enhanced functionalities in cell proliferation and death regulation, and cell cycle controls, as well as in cell adhesion, polarized trafficking and other cell polarity-related features that are critical in maintaining colon epithelium integrity (Figs. 2-5) (see below). Alteration of these functions promotes tumor cell growth, invasion and migration. Hence, DCGs displayed strong cancer relevance but PCGs did not, supporting that our approach is valid.
Polarity genes are cancer driver genes?
Loss of cell polarity is a hallmark of cancers that originate from epithelial cells (19). Although its role as a driver or a passenger of cancer is still under debate at present (19), cell polarity has been proposed to be critical in cancer initiation by controlling asymmetric division of cancer stem cells, as well as in cancer progression by preventing cancer cell invasion and spreading (20). This has become increasingly appreciated in recent years as more studies demonstrate the importance of the EMT in cancer (46). Critically, loss of cell polarity has been shown to be cancer-driving in Drosophila genetic models (18).
While further research is clearly needed, this study also underscores the importance of cell polarity in CRC pathogenesis. First, nearly half of our DCGs function in cell-cell adhesion or cell-substratum adhesion (Fig. 4). Disruption of these genes would free tumor cells to invade tissue layers below the basement membrane and spread to other places (Fig. 4), which in fact occurred in all human CRCs [classified as T1, T2, T3 or T4 tumors (17)] and most of the canine CRCs [many are invasive adenocarcinomas (15)] investigated in this study. Hence, alterations of these genes with cell polarity-related functions are indeed cancer drivers in this regard. Second, many DCGs (but not PCGs) contributes to cell proliferation and death regulation (Figs. 3 and 4). Hence, this pilot study indicates that, in sporadic CRCs of both species, genes closely associated with epithelial cell polarity establishment/maintenance not only play a critical role in preventing cancer cell invasion and spreading, but also likely serve as tumor suppressors by modulating cell growth. This is consistent with the conclusion from Drosophila genetic model studies (18), and supports the notion that alterations of polarity genes and loss of cell polarity are a cancer driver (20), which can be further tested by future experimental validation of those DCGs with putative polarity and cancer-driving roles (Fig. 4).
Unlike previously published papers that are limited to the mere identification of genes recurrently amplified/deleted in CRCs, this human-dog comparison approach allowed us to further classify these genes as either cancer driver or passenger candidates, effectively narrowing down the targets for downstream functional validation. Expanding this pilot study to a larger scale would likely make many more driver-passenger distinctions and address key questions regarding cancer pathogenesis and epithelial cell polarity control. Meanwhile, we caution this pilot study demonstrating only that the proposed dog-human comparison for driver-passenger discrimination is valid for amplified/deleted genes in cancers with large genomic amplifications or deletions, e.g., CRCs with CIN (5, 13). For genes that are mutated via base substitutions or small indels [e.g., in CRCs with microsatellite instability (5, 13)] or abnormal epigenetic modifications (47), it remains to be tested whether this approach is effective or not.
MATERIALS AND METHODS
All analyses were performed with the canFam version 2.0 and the human genome NCBI build 36.1 (hg18). CNAs in both human (17) and dog (15) CRCs were identified as described previously (15). Amplified/deleted genes were identified using the RefGene annotation for the human and the xenoRefGene annotation for the dog, downloaded from the University of California Santa Cruz (UCSC) genome site (www.genome.ucsc.edu). Genes recurrently disrupted in both species were identified with the GISTIC algorithm (48). The 324 GRBs for the autosomes (Table s2) were identified by integrating our human/dog genomic synteny/rearrangement data (12) and the UCSC hg18-canfam2 net alignment data with manual curation.
The Cancer Gene Census (6), containing 410 putative driver genes with 19 explicitly stated to be CRC-related when downloaded, and COSMIC (27) data v48 (containing 4,602 mutated genes when downloaded) were both from www.sanger.ac.uk, while the Cancer Gene Index was from cabig.nci.nih.gov/inventory/data-resources/cancer-gene-index. The human cancer pathways included hsa05200 to hsa05223 of the KEGG Release 54.0 obtained from www.genome.jp/kegg/. Substantial phenotypic change information (e.g., tumorigenesis, lethality, defect, and sterility) of existing gene-knockout mouse models was obtained from MGI phenotype database v4.35 from www.informatics.jax.org. Protein-protein interactions were determined using the I2D database v1.9 at ophid.utoronto.ca/, while DNA-protein interactions were determined by counting the transcription factor binding sites in DAVID v6.7 (david.abcc.ncifcrf.gov/) (33). Gene functional classification was investigated using DAVID v6.7 (33), the Pfam database (pfam.sanger.ac.uk), and literature reports.
HCT116 cells and other CRC lines, kindly provided by Dr. Bert Vogelstein of Johns Hopkins University, were cultured and authenticated following the instruction provided by ATCC. Taxol (Paclitaxel) was purchased from Sigma-Aldrich (Product No. T7402). The expression levels of DCGs and PCGs in HCT116 cells, WA09 and ISL1+ nascent mesoderm cells were determined by quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR) as described previously (49). The primers were listed in Supplementary Table s12, and GAPDH was used as the normalization gene. siRNA primers (Supplementary Table s13) were designed using Silencer® siRNA Construction Kit Template Design Tool and siRNA Target Finder (Ambion). Then, siRNA and scramble-control RNA were synthesized with Silencer® siRNA Construction Kit (cat no. AM1620) from Ambion. Transfection was performed with siPORT NeoFX Transfection Agent (cat no. AM4510) from Ambion. Cell growth and crystal violet-staining were performed as described (50). Images were quantified using ImagJ (rsbweb.nih.gov/ij/) and t-tests were performed between scramble-control and knockdown. WA09 hESCs were maintained in StemPro defined media (Invitrogen). Differentiation to ISL1+ nascent mesoderm was achieved by supplementation of defined media with Wnt3a (25 ng/ml) and BMP4 (50 ng/ml) for 4 days. Microarray experiments were then performed with Affymetrix Human Gene 1.0 ST arrays at the Emory Biomarker Service Center.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Ms. Huan Xiong for her work; Dr. Bert Vogelstein for providing various CRC lines and Dr. Timothy A. Chan of Memorial Sloan-Kettering Cancer Center for providing the IHA cells; and Drs. Dong M Shin, J David Puett, Claiborne Glover, Georgia Chen, and Lisa J Stubb for their help and advice on this study.
Financial support: This work was funded by the American Cancer Society and the Georgia Cancer Coalition (PI: S. Zhao), as well as NCI P50 CA128613 (PI: Dr. Dong M Shin) and GM085354 (PI: Dr. Stephen Dalton).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
REFERENCES
- 1.Haber DA, Settleman J. Cancer: drivers and passengers. Nature. 2007;446(7132):145–6. doi: 10.1038/446145a. Epub 2007/03/09. [DOI] [PubMed] [Google Scholar]
- 2.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458(7239):719–24. doi: 10.1038/nature07943. Epub 2009/04/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sawyers CL. Chronic myeloid leukemia. The New England journal of medicine. 1999;340(17):1330–40. doi: 10.1056/NEJM199904293401706. Epub 1999/04/29. [DOI] [PubMed] [Google Scholar]
- 4.Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabe RR, et al. International network of cancer genome projects. Nature. 2010;464(7291):993–8. doi: 10.1038/nature08987. Epub 2010/04/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–7. doi: 10.1038/nature11252. Epub 2012/07/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, et al. A census of human cancer genes. Nature reviews Cancer. 2004;4(3):177–83. doi: 10.1038/nrc1299. Epub 2004/03/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boyko AR. The domestic dog: man's best friend in the genomic era. Genome Biol. 2011;12(2):216. doi: 10.1186/gb-2011-12-2-216. Epub 2011/02/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neff MW, Rine J. A fetching model organism. Cell. 2006;124(2):229–31. doi: 10.1016/j.cell.2006.01.008. Epub 2006/01/28. [DOI] [PubMed] [Google Scholar]
- 9.Rowell JL, McCarthy DO, Alvarez CE. Dog models of naturally occurring cancer. Trends in molecular medicine. 2011;17(7):380–8. doi: 10.1016/j.molmed.2011.02.004. Epub 2011/03/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438(7069):803–19. doi: 10.1038/nature04338. Epub 2005/12/13. [DOI] [PubMed] [Google Scholar]
- 11.Pontius JU, Mullikin JC, Smith DR, Lindblad-Toh K, Gnerre S, Clamp M, et al. Initial sequence and comparative analysis of the cat genome. Genome research. 2007;17(11):1675–89. doi: 10.1101/gr.6380007. Epub 2007/11/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji X, Zhao S. DA and Xiao-two giant and composite LTR-retrotransposon-like elements identified in the human genome. Genomics. 2008;91(3):249–58. doi: 10.1016/j.ygeno.2007.10.014. Epub 2007/12/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87(2):159–70. doi: 10.1016/s0092-8674(00)81333-1. Epub 1996/10/18. [DOI] [PubMed] [Google Scholar]
- 14.Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268–74. doi: 10.1126/science.1133427. Epub 2006/09/09. [DOI] [PubMed] [Google Scholar]
- 15.Tang J, Le S, Sun L, Yan X, Zhang M, Macleod J, et al. Copy number abnormalities in sporadic canine colorectal cancers. Genome research. 2010;20(3):341–50. doi: 10.1101/gr.092726.109. Epub 2010/01/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Youmans L, Taylor C, Shin E, Harrell A, Ellis AE, Seguin B, et al. Frequent alteration of the tumor suppressor gene APC in sporadic canine colorectal cancers. PloS ONE. doi: 10.1371/journal.pone.0050813. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Camps J, Grade M, Nguyen QT, Hormann P, Becker S, Hummon AB, et al. Chromosomal breakpoints in primary colon cancer cluster at sites of structural variants in the genome. Cancer research. 2008;68(5):1284–95. doi: 10.1158/0008-5472.CAN-07-2864. Epub 2008/03/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Igaki T, Pagliarini RA, Xu T. Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Current biology : CB. 2006;16(11):1139–46. doi: 10.1016/j.cub.2006.04.042. Epub 2006/06/07. [DOI] [PubMed] [Google Scholar]
- 19.Royer C, Lu X. Epithelial cell polarity: a major gatekeeper against cancer? Cell death and differentiation. 2011;18(9):1470–7. doi: 10.1038/cdd.2011.60. Epub 2011/05/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee M, Vasioukhin V. Cell polarity and cancer--cell and tissue polarity as a non-canonical tumor suppressor. Journal of cell science. 2008;121(Pt 8):1141–50. doi: 10.1242/jcs.016634. Epub 2008/04/05. [DOI] [PubMed] [Google Scholar]
- 21.Skaletsky H, Kuroda-Kawaguchi T, Minx PJ, Cordum HS, Hillier L, Brown LG, et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature. 2003;423(6942):825–37. doi: 10.1038/nature01722. Epub 2003/06/20. [DOI] [PubMed] [Google Scholar]
- 22.Reinhardt HC, Hasskamp P, Schmedding I, Morandell S, van Vugt MA, Wang X, et al. DNA damage activates a spatially distinct late cytoplasmic cell-cycle checkpoint network controlled by MK2-mediated RNA stabilization. Molecular cell. 2010;40(1):34–49. doi: 10.1016/j.molcel.2010.09.018. Epub 2010/10/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amann T, Hellerbrand C. GLUT1 as a therapeutic target in hepatocellular carcinoma. Expert opinion on therapeutic targets. 2009;13(12):1411–27. doi: 10.1517/14728220903307509. Epub 2009/10/31. [DOI] [PubMed] [Google Scholar]
- 24.Jacobson KA, Klutz AM, Tosh DK, Ivanov AA, Preti D, Baraldi PG. Medicinal chemistry of the A3 adenosine receptor: agonists, antagonists, and receptor engineering. Handbook of experimental pharmacology. 2009(193):123–59. doi: 10.1007/978-3-540-89615-9_5. Epub 2009/07/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Veeriah S, Brennan C, Meng S, Singh B, Fagin JA, Solit DB, et al. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(23):9435–40. doi: 10.1073/pnas.0900571106. Epub 2009/05/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bouanene H, Miled A. Conflicting views on the molecular structure of the cancer antigen CA125/MUC16. Disease markers. 2010;28(6):385–94. doi: 10.3233/DMA-2010-0719. Epub 2010/08/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic acids research. 2011;39(Database issue):D945–50. doi: 10.1093/nar/gkq929. Epub 2010/10/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin J, Gan CM, Zhang X, Jones S, Sjoblom T, Wood LD, et al. A multidimensional analysis of genes mutated in breast and colorectal cancers. Genome research. 2007;17(9):1304–18. doi: 10.1101/gr.6431107. Epub 2007/08/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nature reviews Cancer. 2004;4(4):253–65. doi: 10.1038/nrc1317. Epub 2004/04/02. [DOI] [PubMed] [Google Scholar]
- 30.Llovera L, Mansilla S, Portugal J. Apoptotic-like death occurs through a caspase-independent route in colon carcinoma cells undergoing mitotic catastrophe. Cancer letters. 2012;326(1):114–21. doi: 10.1016/j.canlet.2012.08.001. Epub 2012/08/14. [DOI] [PubMed] [Google Scholar]
- 31.Harsay E, Schekman R. Avl9p, a member of a novel protein superfamily, functions in the late secretory pathway. Molecular biology of the cell. 2007;18(4):1203–19. doi: 10.1091/mbc.E06-11-1035. Epub 2007/01/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nafisi H, Banihashemi B, Daigle M, Albert PR. GAP1(IP4BP)/RASA3 mediates Galphai-induced inhibition of mitogen-activated protein kinase. The Journal of biological chemistry. 2008;283(51):35908–17. doi: 10.1074/jbc.M803622200. Epub 2008/10/28. [DOI] [PubMed] [Google Scholar]
- 33.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. Epub 2009/01/10. [DOI] [PubMed] [Google Scholar]
- 34.Dehal P, Predki P, Olsen AS, Kobayashi A, Folta P, Lucas S, et al. Human chromosome 19 and related regions in mouse: conservative and lineage-specific evolution. Science. 2001;293(5527):104–11. doi: 10.1126/science.1060310. Epub 2001/07/07. [DOI] [PubMed] [Google Scholar]
- 35.Nelson WJ. Adaptation of core mechanisms to generate cell polarity. Nature. 2003;422(6933):766–74. doi: 10.1038/nature01602. Epub 2003/04/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bryant DM, Mostov KE. From cells to organs: building polarized tissue. Nature reviews Molecular cell biology. 2008;9(11):887–901. doi: 10.1038/nrm2523. Epub 2008/10/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li R, Gundersen GG. Beyond polymer polarity: how the cytoskeleton builds a polarized cell. Nature reviews Molecular cell biology. 2008;9(11):860–73. doi: 10.1038/nrm2522. Epub 2008/10/24. [DOI] [PubMed] [Google Scholar]
- 38.Iden S, Collard JG. Crosstalk between small GTPases and polarity proteins in cell polarization. Nature reviews Molecular cell biology. 2008;9(11):846–59. doi: 10.1038/nrm2521. Epub 2008/10/24. [DOI] [PubMed] [Google Scholar]
- 39.Bachmann O, Juric M, Seidler U, Manns MP, Yu H. Basolateral ion transporters involved in colonic epithelial electrolyte absorption, anion secretion and cellular homeostasis. Acta Physiol (Oxf) 2011;201(1):33–46. doi: 10.1111/j.1748-1716.2010.02153.x. Epub 2010/06/10. [DOI] [PubMed] [Google Scholar]
- 40.Belleannee C, Da Silva N, Shum WW, Brown D, Breton S. Role of purinergic signaling pathways in V-ATPase recruitment to apical membrane of acidifying epididymal clear cells. American journal of physiology Cell physiology. 2010;298(4):C817–30. doi: 10.1152/ajpcell.00460.2009. Epub 2010/01/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barrios A, Poole RJ, Durbin L, Brennan C, Holder N, Wilson SW. Eph/Ephrin signaling regulates the mesenchymal-to-epithelial transition of the paraxial mesoderm during somite morphogenesis. Current biology : CB. 2003;13(18):1571–82. doi: 10.1016/j.cub.2003.08.030. Epub 2003/09/19. [DOI] [PubMed] [Google Scholar]
- 42.Farabaugh SM, Micalizzi DS, Jedlicka P, Zhao R, Ford HL. Eya2 is required to mediate the pro-metastatic functions of Six1 via the induction of TGF-beta signaling, epithelialmesenchymal transition, and cancer stem cell properties. Oncogene. 2011 doi: 10.1038/onc.2011.259. Epub 2011/06/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krais AM, Hautefeuille AH, Cros MP, Krutovskikh V, Tournier JM, Birembaut P, et al. CHRNA5 as negative regulator of nicotine signaling in normal and cancer bronchial cells: effects on motility, migration and p63 expression. Carcinogenesis. 2011;32(9):1388–95. doi: 10.1093/carcin/bgr090. Epub 2011/05/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levy F, Muehlethaler K, Salvi S, Peitrequin AL, Lindholm CK, Cerottini JC, et al. Ubiquitylation of a melanosomal protein by HECT-E3 ligases serves as sorting signal for lysosomal degradation. Molecular biology of the cell. 2005;16(4):1777–87. doi: 10.1091/mbc.E04-09-0803. Epub 2005/02/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lott ST, Chen N, Chandler DS, Yang Q, Wang L, Rodriguez M, et al. DEAR1 is a dominant regulator of acinar morphogenesis and an independent predictor of local recurrence-free survival in early-onset breast cancer. PLoS medicine. 2009;6(5):e1000068. doi: 10.1371/journal.pmed.1000068. Epub 2009/06/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. The Journal of clinical investigation. 2009;119(6):1420–8. doi: 10.1172/JCI39104. Epub 2009/06/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin B, Yao B, Li JL, Fields CR, Delmas AL, Liu C, et al. DNMT1 and DNMT3B modulate distinct polycomb-mediated histone modifications in colon cancer. Cancer research. 2009;69(18):7412–21. doi: 10.1158/0008-5472.CAN-09-0116. Epub 2009/09/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(50):20007–12. doi: 10.1073/pnas.0710052104. Epub 2007/12/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ji X, Tang J, Halberg R, Busam D, Ferriera S, Pena MM, et al. Distinguishing between cancer driver and passenger gene alteration candidates via cross-species comparison: a pilot study. BMC cancer. 2010;10:426. doi: 10.1186/1471-2407-10-426. Epub 2010/08/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beck CR, Collier P, Macfarlane C, Malig M, Kidd JM, Eichler EE, et al. LINE-1 retrotransposition activity in human genomes. Cell. 2010;141(7):1159–70. doi: 10.1016/j.cell.2010.05.021. Epub 2010/07/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.