

Abstract
The ventrolateral thalamus (VL) is a primary relay point between the basal ganglia and the primary motor cortex (M1). Using dual probe microdialysis and locomotor behavior monitoring, we investigated the contribution of VL input into M1 during amphetamine (AMPH)-stimulated monoamine release and hyperlocomotion in rats. Tetrodotoxin (TTX) (10 uM) perfusion into the VL significantly lowered hyperactivity induced by AMPH (1 mg/kg i.p.). This behavioral response corresponded to reduced cortical glutamate and monoamine release. To determine which glutamate receptors the thalamocortical projections acted upon, we perfused either the AMPA/kainate receptor antagonist NBQX (10 μM) or the NMDA receptor antagonist (MK-801) intracortically followed by systemic AMPH. The results show that AMPA/kainate, and to a lesser extent NMDA receptors, mediated the observed effects. Since glutamate-monoamine interactions could possibly occur through local or circuit-based mechanisms, we isolated and perfused M1 tissue ex vivo to determine the extent of local glutamate-dopamine interactions. Taken together, these results demonstrate that AMPH generates hyperlocomotive states via thalamocortical signaling and that cortical AMPA receptors are an important mediator of these effects.
Keywords: dopamine, microdialysis, glutamate, amphetamine, thalamus, motor cortex
Introduction
The cortico-basal ganglia-thalamo-cortical loop is a multisynaptic circuit critical for the execution of motivational and motor commands (Alexander et al., 1986, Albin et al., 1989). Disruptions to this circuit are evident in Parkinson’s disease (PD) where striatal dopamine loss triggers an opposite dysregulation of striatofugal pathways, resulting in an overactivation of the SNr/GPi and a subsequent overinhibition of the thalamocortical relay (Gerfen et al., 1990, DeLong, 1990, Alexander and Crutcher, 1990). Conversely, L-DOPA-induced dyskinesias are thought to arise from excessive dopamine signaling and disinhibition of thalamocortical projections (Albin, 1995). The ventrolateral thalamus (VL) in particular is a primary transmitter of basal ganglia information flow into the primary motor cortex (M1; Strick, 1976). Based on its key position within the basal ganglia-thalamo-cortical loop, it is essential to describe how VL projections influence cortical neurochemistry during different states of motor activation.
Thalamocortical projections are a major source of glutamate input into cortical areas (Kharazia and Weinberg, 1994). Meanwhile, the cortical mantle is widely innervated by midbrain and brainstem monoaminergic systems including dopamine, norepinephrine (NE) and serotonin (5-HT; Brown et al., 1979, Watabe-Uchida, 2012). Although the majority of cortical glutamate-monoamine interaction studies to date have emphasized the medial prefrontal cortex, these interactions are likely conserved in M1 since it receives dopamine projections from the VTA (Hosp et al., 2011) and substantia nigra pars compacta (Watabe-Uchida, 2012), 5-HT from the raphe nucleus (Arnsten and Goldman-Rakic, 1984) and NE from the locus coeruleus (Arnsten and Goldman-Rakic, 1984, Gaspar et al., 1991). Despite the strong evidence for interactions between cortical glutamate and monoaminergic substrates (Hondo et al, 1994; Takahata and Mogghadam, 1998, 2003), the source of glutamate which takes part in those interactions has yet to be clearly defined. This is particularly relevant since glutamate can arise from both neuronal and non-neuronal sources such as astrocytes (Parpura et al., 1994).
d-Amphetamine (AMPH) potently elevates synaptic concentrations of monoamines by reversing monoamine transporters, inhibiting the intracellular vesicular monoamine transporter (VMAT; Brown et al., 2001), and increasing phasic exocytotic release (Daberkow et al., 2013), all of which results in characteristic hyperlocomotive behavior and stereotypies (Randrup and Munkvad, 1969). Thus, according to the classical model of basal ganglia function, psychostimulant-induced dopamine release at the striatal level should ultimately disinhibit thalamocortical projections, enhance cortical glutamate, and stimulate cortico-spinal projection output to facilitate movement. Indeed, previous microdialysis studies have described AMPH-induced glutamate increases in the prefrontal cortex (Reid et al., 1997, Del Arco et al., 1998).
The current study was undertaken to test the hypothesis that VL glutamatergic inputs into M1 are an essential component of AMPH-induced motor activation. To this end we unilaterally perfused the voltage-dependent Na+ channel blocker tetrodotoxin (TTX) into VL and gave AMPH systemically while monitoring locomotor behavior and thalamocortical neurochemistry. Moreover, to determine which glutamate receptors were involved in this interaction we intracortically perfused antagonists of (2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)propanoic acid (AMPA)/kainate or N-methyl-D-aspartic acid (NMDA) receptors followed by systemic AMPH. Finally, using isolated M1 slices, we were able to determine which of these neurochemical effects were due to local glutamate signaling. Since additional neurochemicals could potentially underlie behavioral responses generated by glutamate-dopamine interactions elicited by AMPH, we employed a novel benzoylation liquid chromatography-mass spectrometry (LC-MS) neurochemical monitoring system (Song et al., 2012) to examine release dynamics of glutamate, GABA, acetylcholine (ACh), dopamine, 5HT, and NE simultaneously.
Materials and Methods
Microdialysis
Adult male Sprague–Dawley rats (Harlan, Indianapolis, IN) weighing between 250 and 350 g were used for all experiments. Rats were housed in a temperature and humidity controlled room with 12 hr light/dark cycles with food and water available ad libitum. Adequate measures were taken to prevent animal pain and discomfort. All animals were treated as approved by the University of Michigan Unit for Laboratory Animal Medicine (ULAM) and in accordance with the National Institute of Health (NIH) Guidelines for the Care and Use of Laboratory Animals. Additionally, all animal experiments were conducted in accordance with Animal Research: Reporting In Vivo Experiments (ARRIVE) guidelines.
Prior to surgery, rats were anesthetized with an intraperitoneal (i.p.) injection of a ketamine (65 mg/kg) and dexdormitor (0.25 mg/kg) mixture prepared in an isotonic salt solution. Concentrically designed microdialysis probes (1 mm dialyzing length) were then implanted unilaterally into M1 and VL using a Model 963 Ultra Precise stereotaxic instrument with 0.1 mm resolution (David Kopf Instruments, Tujunga, CA). Coordinates used for M1 and the VL thalamus from bregma and dura were AP +3.2, ML ±2.8, DV −1.2 and AP −2.3, ML ±2.6, DV −6.7, respectively (Paxinos and Watson, 2007). Probes were secured to the skull by acrylic dental cement and metallic screws. Following surgery, rats were allowed to recover and experiments were run 24 hr after probe implantation. Microdialysis probes were flushed at a flow rate of 1.5 μl/min with aCSF (composition in mM: CaCl2 1.2; KCl 2.7, NaCl 148 and MgCl2 0.85) for 3 hr using a Chemyx Fusion 400 syringe pump (Chemyx, Stafford, TX). Perfusion flow rate was then reduced to 1 μl/min and samples were collected every 10 min. Baseline values were collected for 60 min and then aCSF was switched to include TTX (in VL) or glutamate receptor antagonists (in M1) for 40 min. Following this 40 min period, systemic AMPH was given. When experiments were completed, animals were euthanized and then brains were extracted and frozen at −80 C until histology.
Locomotor behavior analysis
While animals were undergoing microdialysis, Logitech (Apples, Switzerland) webcams were placed above cages as previsously described (Mabrouk et al., 2011). Webcams were connected via USB port to analysis PC running Matlab 2009 (Mathworks, Natick, MA) software. Using the image acquisition toolbox in Matlab, data were collected via a custom designed motion monitoring program (Mark Dow, University of Oregon). Threshold of motion detection software was set at 150. This threshold limit was selected as not to detect small motions (such as breathing or whisker movement) but only larger motions such as walking around cage, running, rearing, etc. Data were collected every 60 seconds and then binned into 10 min intervals to correlate with neurochemistry. Data are expressed in terms of absolute locomotor activity as calculated by the detection software.
Motor cortex slice perfusions
M1 from male Sprague-Dawley rats (274–300g) was extracted, weighed and placed in chambers of a Brandel perfusion apparatus (Brandel SF-12, Gaithersburg, MD) onto Whatman GF/B filter disks. The volume of these tissue extracts was approxmiately 1 mm3. The chambers were perfused at 37 °C with oxygenated Krebs-Ringer buffer (KRB, 145 mM NaCl, 2.7mM KCl, 1.2mM KH2PO4, 1.2mM CaCl2, 1.0 mM MgCl2 mM glucose, 50 μM ascorbic acid, 24.9 mM NaHCO3, 50 μM pargyline and 50 μM tropolone) with or without 1μM NBQX or 1μM MK-801 for 60 min at a rate of 0.8 mL/min, prior to the start of collection. Fractions were collected every 1 min for 13 minutes, and a 2 min bolus of 3 μM AMPH was added to the samples at fractions 6 and 7. Based on the dead volume of the system, it was calculated that AMPH would be present in fractions 8 and 9. Eluates (0.8 mL per fraction) were collected into vials then derivatized (as described below) for dopamine analysis.
Neurotransmitter analysis using QQQ MS
ACh, glutamate, GABA, dopamine, NE, and 5HT were analyzed using a novel LC-MS method recently described by our laboratory (Song et al., 2012). For ex vivo experiments, 10 μl was withdrawn from the total sample volume and then derivatzied in the same way as dialysate samples, however, only dopamine levels were investigated. To 10 μl samples, 5 μl of 100 mM sodium tetraborate was added to the collected 10 μl dialysate sample. Then, 5 μl of 2% benzoyl chloride in acetonitrile was added to the mixture to initiate the benzyolation reaction. 5 μl of a stable 13C benzoylated isotope internal standard mixture was then added to improve quantitation. Finally, d4-ACh was added as an internal standard for ACh. A Waters nanoAcquity (Waters, Milford, MA) system automatically injected 5 μl of the sample onto a Waters HSS T3 reverse phase HPLC column (1 mm X 100 mm, 1.8 μm). Mobile phase A consisted of 10 mM ammonium formate and 0.15% formic acid. Mobile phase B was pure acetonitrile. Analytes were detected by an Agilent 6410 triple quadrupole mass spectrometer operating in positive mode using dynamic multiple reaction monitoring (MRM).
Data presentation and statistical analysis
All neurochemical data were transformed into percent baseline while behavioral data are expressed as absolute locomotor counts. Statistical analysis was performed by two-way repeated measure (RM) analysis of variance (ANOVA) using Prism 6 (Graphpad Software Inc, La Jolla, CA). In case ANOVA yielded a significant F score, post-hoc analysis was performed by contrast analysis to determine group differences. In case a significant treatment or time X treatment interaction was found, the sequentially rejective Bonferroni test was used to determine specific differences (i.e. at the single time-point level) between groups. P values <0.05 were considered to be statistically significant.
Materials
AMPH, benzoyl chloride, tetrodotoxin (TTX), NBQX, MK-801, and all LC-MS reagents were purchased from Sigma Chemical Co (St Louis, MO). Ketamine and dexdormitor were from MWI (Rochester Hills, MI). While AMPH was dissolved in isotonic saline solution, NBQX and MK-801 were dissolved in Ringer solution (described above). AMPH was administered by i.p. injections (1 mg/ml/kg) while all other drugs were perfused through the microdialysis probe (reverse dialysis).
Results
Effect of TTX perfusion in VL thalamus on AMPH-induced locomotor activity
We first wanted to determine to what extent thalamocortical projections contributed to AMPH-induced hyper locomotion. To this end, we took advantage of simultaneous locomotor behavior monitoring while perfusing TTX (10 μM) directly into the VL via reverse dialysis. Basal locomotor levels were 22.2 ± 6.3 counts/10 min (n = 13; Fig 1). RM ANOVA on locomotor activity revealed a significant effect of treatment (F1,11=37.75, p<0.0001), time (F19,209=26.35, p<0.0001) and a time X treatment interaction (F19,209=12.56, p<0.0001). Local perfusion of TTX (10 μM) had no effect on basal levels of locomotor activity. However, due to low resting activity of animal subjects, our locomotor behavior assay is limited in detecting inhibition of basal motor activity. Nevertheless, AMPH (1 mg/kg i.p.) caused a very large and reliable increase in locomotor activity (maximal ~408 locomotor counts 20 min post injection). This effect was largely attenuated by VL perfusion of TTX (Fig 1).
Figure 1.
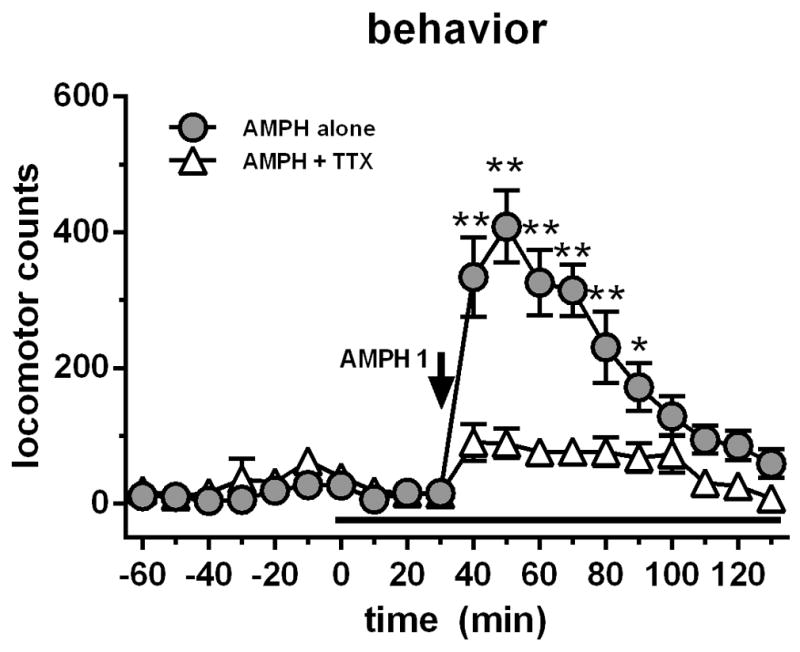
Systemic AMPH (1 mg/kg i.p.) administration caused a significant rise in locomotor activity compared to a group which received AMPH (1 mg/kg i.p.) in addition to VL perfusion of TTX (10 μM; black bar). Data are expressed as locomotor counts ± SEM.
*p<0.05, AMPH-only time point significantly different from AMPH + TTX treated group
**p<0.01, AMPH-only time point significantly different from AMPH + TTX treated group
Effect of TTX perfusion in the VL thalamus on local AMPH-induced neurochemical changes
The thalamus receives a variety of neurochemical inputs including glutamate, ACh, GABA, dopamine, 5-HT and NE from cortical, subcortical and brainstem nuclei (Sherman and Guillery, 2001). Therefore to unravel which neurochemicals might be responsible for behavioral modifications induced by AMPH, these neurochemicals were monitored simultaneously. All basal dialysate concentrations from the VL are reported in Table 1. RM ANOVA on AMPH-induced thalamic ACh release showed a significant effect of treatment (F1,13=10.06, p=0.0074), time (F19,247=2.05, p=0.0069) and a time X treatment interaction (F19,247=2.93, p<0.0001). AMPH 1 mg/kg i.p. stimulated thalamic ACh release (maximal 96% above baseline; Fig. 2A). TTX (1 μM) perfusion through the microdialysis probe prevented this effect and caused a ~30% reduction in basal values. Systemic AMPH treatment did not increase VL glutamate or GABA (Fig. 2B, 2C); however, for GABA, RM ANOVA showed an effect of TTX perfusion (F1,13=34.2, p<0.0001), of time (F19,247=1.14, p<0.0001) and a time X treatment interaction (F19,247=6.04, p<0.0001). TTX perfusion also decreased GABA levels by ~30% (Fig 2C). TTX reduced dopamine release (maximal ~60%) while AMPH stimulated it (~700% of baseline values), as expected (Fig. 2D). For dopamine, RM ANOVA showed an effect of treatment (F1,13=9.11, p=0.0008), of time (F19,247=10.22, p<0.0001) and a time X treatment interaction (F19,247=6.68, p<0.0001). Similar to dopamine, TTX reduced NE levels (maximal ~60%) while AMPH stimulated them (maximal ~560%; Fig 5E). For NE, RM ANOVA showed an effect of treatment (F1,11=15.55, p=0.0023), of time (F19,209=6.70, p<0.0001) and a time X treatment interaction (F19,209=6.41, p<0.0001). Finally, 5-HT showed a similar effect as the other monoamines with TTX reducing levels (nearly ~80%) and AMPH increasing (~300%) with respect to baseline. For 5-HT, RM ANOVA showed an effect of treatment (F1,12=27.29, p=0.0002), of time (F19,228=2.65, p=.0003) and a time X treatment interaction (F19,228=6.91, p<0.0001).
Table 1.
Basal dialysate concentrations of all neurotransmitters analyzed by LC-MS in either the primary motor cortex (M1) or the ventrolateral thalamus (VL). All concentrations are expressed as nanomolar ± SEM.
| Primary motor cortex | Ventrolateral thalamus | |||||
|---|---|---|---|---|---|---|
| Neurotransmitter | nM | SEM | n | nM | SEM | n |
| ACh | 21.2 | ±1.93 | 27 | 2.5 | ±0.28 | 25 |
| glutamate | 2459 | ±425 | 26 | 214.9 | ±24.7 | 24 |
| GABA | 28.4 | ±4.85 | 27 | 19.5 | ±2.58 | 25 |
| 5-HT | 0.60 | ±0.18 | 28 | 0.43 | ±0.16 | 26 |
| norepinephrine | 0.91 | ±0.17 | 26 | 0.74 | ±0.10 | 23 |
| dopamine | 0.24 | ±0.06 | 27 | 0.28 | ±0.09 | 25 |
Figure 2.

Systemic AMPH (1 mg/kg i.p.) administration increased thalamic ACh (A), dopamine (D), norepinephrine (E) and serotonin (F). These effects were blocked by thalamic perfusion of TTX (10 μM; black bar). Data are expressed as percent baseline ± SEM.
*p<0.05, AMPH-only time point significantly different from AMPH + TTX treated group
**p<0.01, AMPH-only time point significantly different from AMPH + TTX treated group
Figure 5.

Systemic AMPH (1 mg/kg i.p.) administration increased M1 GABA (C), dopamine (D), NE (E) and 5-HT (F). M1 perfusion of NBQX (10 μM; white triangles) blocked AMPH-induced release of GABA, dopamine, NE and 5-HT. M1 perfusion of MK-801 (10 μM) increased dopamine (D), while attenuating AMPH-stimulated dopamine and NE. Antagonist perfusion is represented by the black bar. Data are expressed as percent baseline ± SEM.
*p<0.05, AMPH alone group significantly different from NBQX + AMPH group
**p<0.01, AMPH alone group significantly different from NBQX + AMPH group
#p<0.05, AMPH alone group significantly different from MK801 + AMPH group
##p<0.01, AMPH alone group significantly different from MK801 + AMPH group
Effect of TTX perfusion in the VL thalamus on AMPH-induced neurochemical changes in the M1 motor cortex
The M1 receives a dense glutamatergic projection from the thalamus (Herkenham, 1980, Grofova et al., 1974, McFarland and Haber, 2002, Hooks et al., 2013). In order to determine the contribution of this circuit to AMPH-stimulated hyperactivation and neurochemical changes, we performed simultaneous dual probe microdialysis in M1 and VL. All basal dialysate concentrations from the M1 are reported in Table 1. Neither thalamic TTX perfusion nor systemic AMPH treatment affected ACh values in M1. AMPH however stimulated the release of cortical glutamate (maximal ~210%) while VL TTX perfusion prevented this effect (Fig 3B). For glutamate, RM ANOVA did not show a significant effect of treatment (F1,11=4.42, p=0.0594), but an effect of time (F19,209=4.23, p<0.0001) and a significant time X treatment interaction (F19,209=2.55, p<0.0001). AMPH 1 mg/kg i.p. modestly stimulated cortical GABA release (maximal ~30% above baseline) and TTX (1 μM) perfusion through the microdialysis probe in the VL prevented this effect (Fig 3C). For GABA, RM ANOVA showed an effect of treatment (F1,13=6.27, p=0.02), but not time nor a time X treatment interaction. As expected, AMPH stimulated the release of dopamine (maximal ~410%) at the level of M1 while simultaneous perfusion of TTX (1 μM) into the VL largely attenuated this effect (Fig. 3D). For dopamine, RM ANOVA showed an effect of treatment (F1,13=8.48, p=0.012), of time (F19,247=12.80, p<0.0001) and a time X treatment interaction (F19,247=2.61, p=0.0004). Similar to dopamine, AMPH stimulated the release of NE (maximal ~550%) at the level of the motor cortex while simultaneous perfusion of TTX (1 μM) into the VL largely attenuated this effect (Fig. 3E). For NE, RM ANOVA showed an effect of treatment (F1,11=8.96, p=0.012), of time (F19,209=9.26, p<0.0001) and a time X treatment interaction (F19,209=3.42, p<0.0001). Finally, 5-HT showed a similar, but more modest effect compared to the other monoamines with AMPH stimulating cortical release (maximal ~275%) and VL TTX perfusion attenuating these effects (Fig. 3F). For 5-HT, RM ANOVA showed an effect of treatment (F1,12=6.59, p=0.024), of time (F19,228=1.91, p=.014) and a time X treatment interaction (F19,228=1.87, p=0.018).
Figure 3.

Systemic AMPH (1 mg/kg i.p.) administration increased cortical glutamate (B), dopamine (D), norepinephrine (E), and serotonin (F). These effects were attenuated by thalamic perfusion of TTX (10 μM; black bar). Data are expressed as percent baseline ± SEM.
*p<0.05, AMPH-only time point significantly different from AMPH + TTX treated group
**p<0.01, AMPH-only time point significantly different from AMPH + TTX treated group
Effect of ionotropic glutamate receptor antagonist perfusion in M1 motor cortex on AMPH-induced locomotor activity
Based on our finding that VL activity strongly affected AMPH-induced hyperlocomotion and neurochemistry, we wanted to determine if these effect were mediated through ionotropic glutamate receptors in M1. To this end, we perfused either AMPA (NBQX) or NMDA (MK-801) receptor antagonists in M1 and then treated subjects with AMPH (1 mg/kg i.p.). Basal locomotor levels were 29.2 ± 10.6 counts/10 min (n = 18; Fig 4). RM ANOVA on locomotor activity did not reveal a significant effect of treatment, but an effect of time (F19,285=29.72, p<0.0001) and a time X treatment interaction (F38,285=2.30, p<0.0001). Cortical perfusion of either NBQX (10 μM) or MK-801 (10 μM) had no effect on basal levels of locomotor activity while AMPH stimulated it (maximal ~400 counts). The effect of AMPH-induced hyperactivation was partially attenuated by local M1 cortex perfusion of NBQX, but not MK-801 (Fig. 4).
Figure 4.
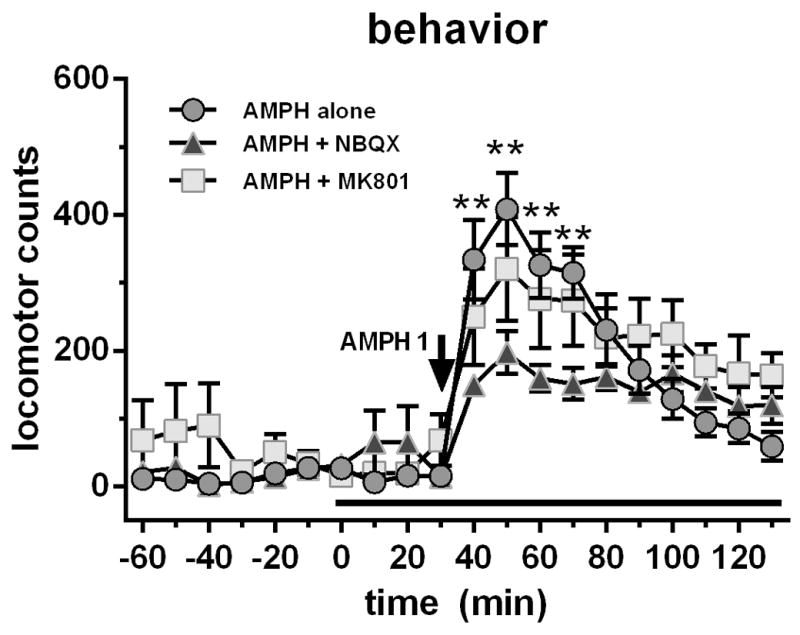
Systemic AMPH (1 mg/kg i.p.) administration caused a significant rise in locomotor activity. Perfusion of the AMPA receptor antagonist NBQX (10 μM) in M1 motor cortex attenuated these effects while perfusion of the NMDA receptor antagonist (10 μM) was without effect. Data are expressed as locomotor counts ± SEM.
**p<0.01, AMPH alone group significantly different from NBQX + AMPH group
Effect of ionotropic glutamate receptor antagonist perfusion in M1 motor cortex on local AMPH-induced neurochemical changes
No difference was seen on ACh or glutamate levels during antagonist perfusion or AMPH treatments (although a trend for AMPH to increase glutamate was observed; Fig. 5B). AMPH 1 mg/kg i.p. modestly stimulated cortical GABA release (maximal ~45% above baseline) and NBQX, and to a lesser extent MK-801, prevented this increase (Fig. 5C). For GABA, RM ANOVA showed an effect of treatment (F2,16=12.85, p=0.0005), not time, but a time X treatment interaction (F38,304=1.45, p=0.048). MK-801 (10 μM) perfusion into M1 enhanced basal dopamine levels. As expected, AMPH stimulated the release of dopamine while local perfusion of NBQX (10 μM), and to a lesser extent MK-801 (10 μM) attenuated this effect (Fig. 5D). For dopamine, RM ANOVA showed an effect of treatment (F2,17=4.41, p=0.029), of time (F19,323=15.03, p<0.0001) and a time X treatment interaction (F38,323=2.99, p<0.0001). Neither antagonist had an effect on basal cortical NE release. Similar to dopamine, AMPH stimulated the release of cortical NE, but differently, NBQX and MK-801 entirely prevented this effect (Fig. 5E). For NE, RM ANOVA showed an effect of treatment (F2,16=6.99, p=0.007), of time (F19,304=8.92, p<0.0001) and a time X treatment interaction (F38,304=3.63, p<0.0001). Finally, 5-HT showed a similar effect as the other monoamines with AMPH stimulating cortical release, and NBQX perfusion in M1 strongly attenuated these effects while the effect of MK-801 was far less pronounced. For 5-HT, RM ANOVA showed an effect of treatment (F2,16=4.83, p=0.023), of time (F19,304=2.08, p=.006) and a time X treatment interaction (F38,304=1.73, p=0.007).
Effect of ionotropic glutamate receptor antagonist perfusion in M1 motor cortex on AMPH-induced neurochemical changes in M1 motor cortex slice preparations
Since our results pointed towards a complex neurochemical interaction which could be due to either local or circuit-driven processes, we isolated M1 ex vivo to examine the contribution of local interactions. We chose to focus on dopamine release in response to local AMPH suprafusion of M1 tissue with or without concomitant suprafusion of ionotropic glutamate receptor antagonists. As expected, AMPH (3 μM) stimulated dopamine release from M1 slices (maximal 183% above baseline; Fig 6). Perfusion of either NBQX (1 μM) or MK-801 (1 μM) into the M1 cortex attenuated the dopamine releasing effects of AMPH in M1 slices (Fig 6). For dopamine, RM ANOVA did not show an effect of treatment, but did show an effect of time (F12,156=14.42, p<0.0001) and a time X treatment interaction (F24,156=2.44, p=0.0006).
Figure 6.
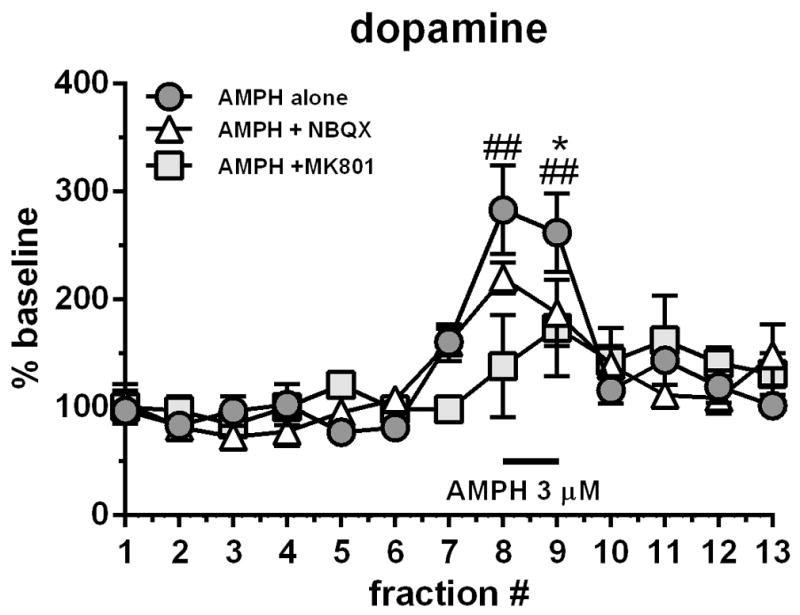
Isolated ex vivo M1 slices perfused with AMPH (3 μM; black bar) caused a 3-fold increase in dopamine levels. Co-perfusion of MK-801 (1 μM) and NBQX (1 μM) attenuated this increase. Data are expressed as percent baseline ± SEM.
*p<0.05, NBQX-treated group significantly different from AMPH alone group
##p<0.01, MK-801-treated group significantly different from AMPH alone group
Discussion
The present study describes a mechanism by which systemic AMPH regulates cortical neurochemistry and motor function, through thalamocortical glutamate projections. As expected, systemic AMPH treatment stimulated hyperlocomtion which corresponded to cortical monoamine and glutamate release. These effects were largely attenuated by concomitant TTX perfusion in the ipsilateral VL or intracortical glutamate antagonist perfusion, suggesting that VL glutamate input into M1 contributes to AMPH-induced movement. Considering the larger inhibitory effect of NBQX with respect to MK-801, glutamate influence appears to be mediated by AMPA receptors and to a lesser extent, NMDA receptors.
AMPH-induced behavior and thalamocortical neurochemistry are sensitive to VL thalamus perfusion of TTX
Based on its key position between the basal ganglia and the cortex, we asked how heightened monoamine levels (via systemic AMPH treatment) affected thalamic neurochemistry and to what extent this nucleus drives cortical neurotransmission and movement. These results demonstrate that AMPH-mediated movement is processed extrastriatally through thalamocortical glutamate transmission.
TTX is a selective voltage-dependent sodium channel blocker, and is commonly used to disclose effects solely due to neuronal activity. TTX perfusion effectively “shuts down” the targeted nucleus by preventing action potentials and both tonic and phasic impulse-dependent release events (Narahashi, 1974, Westerink et al., 1987). We did not observe any changes in resting locomotor activity during TTX perfusion in VL, but did see a robust attenuation of AMPH-induced hyperactivity, suggesting that AMPH-induced hyperactivation is derived from thalamic activation. As expected and previously reported outside the thalamus, local TTX perfusion in the VL significantly reduced levels of all extracellular monoamines (Westerink et al., 1987, Benwell et al., 1993), GABA and ACh (Osborne et al., 1990, Williams et al, 1994, Douglas et al., 2001). On the other hand, TTX perfusion did not affect glutamate levels; this is likely due to the ability of non-neuronally derived glutamate sources such as astrocytes to maintain homeostatic extracellular levels of this transmitter (Timmerman and Westerink, 1997). AMPH stimulated the release of all three monoamines measured (dopamine, 5HT and NE) both in the VL and in M1 while thalamic TTX perfusion nearly abolished these effects locally in VL. This suggests that AMPH-induced release of monoamines is largely dependent on normal neuronal firing (i.e. impulse dependent release) of monoamine efferents projecting to the thalamus. These findings are in line with recent works which showed that systemic AMPH treatment in behaving or anesthetized animals produced a phasic, exocytotic-driven release of dopamine at the striatal level (Daberkow et al., 2013, Avelar et al., 2013).
Additionally, systemic AMPH stimulated the release of ACh in the VL to a similar degree as previously reported in the mediodorsal thalamus (Rada et al., 2007). This release was TTX-dependent like that observed for monoamine release in this area. The role of thalamic ACh is generally associated with wakefulness (Williams et al., 1994) and these results are in line with the known wake-promoting effects of AMPH (Engber et al., 1998). The precise mechanism by which ACh is released by AMPH in this area remains a matter of speculation. However, others have shown in hippocampal and striatal areas that AMPH, indirectly acting through DA D1-like receptors elevates extracellular levels (Imperato et al., 1993). Since DA in the current experiment was strongly reduced by TTX perfusion it suggests that DA transmission positively modulates VL ACh.
A main finding of the current study was that inhibition of the VL via TTX perfusion significantly attenuated cortical monoamine release. Although this was initially quite surprising, it suggests that glutamate potently modulates cortical monoamine signaling. Indeed, glutamate antagonists delivered directly to the cortex also attenuated AMPH-induced monoamine levels. This type of glutamate modulation has been shown for dopamine in both cortical and subcortical brain structures (Shimizu et al., 1990, Wang 1991, Takahata and Moghaddam, 1998). Our findings show for the first time that VL glutamate input is responsible for this interaction in M1 and that psychostimulant-evoked monoamine release and movement can be attenuated by cortical glutamate receptor blockade. It is interesting to note that others have shown AMPH-mediated motor behaviors to be strongly attenuated by systemic administration of glutamate antagonists (Karler et al, 1991, Kelley and Throne, 1992, Karler et al 1995). Our findings point to inhibition of thalamocortical glutamate signaling as the primary mediator of those effects. We also extend the findings of glutamate control over cortical dopamine release to both 5-HT and NE in M1.
Cortical perfusion of glutamate antagonists generates a similar behavioral and neurochemical profile as thalamic TTX perfusion
M1 is strongly innervated by glutamatergic projections originating in the VL and VA (for review see Parent and Hazrati, 1995). Here we show that these projections are necessary for AMPH-stimulated hyperactivity and cortical monoamine release. We sought to determine which glutamate receptor subtypes might be responsible for these effects. NBQX perfusion in M1 attenuated AMPH-stimulated hyperactivity while MK-801 produced only a trend to decrease hyperactivity. This was a clue that TTX-induced effects were mediated via cortical AMPA/kainate receptors. Our neurochemical findings however suggested a more complex interaction than a simple glutamate-AMPA receptor mediation of monoamine function. Perfusion of either antagonist did not reduce basal glutamate levels or significantly prevent the rise in cortical glutamate release following AMPH treatment, suggesting that these receptors are not positioned on thalamocortical terminals.
MK-801 perfusion in itself enhanced dopamine levels but did not affect 5-HT or NE. This is in line with previous works in the mPFC which showed that local MK-801 (10 μM) perfusion enhanced cortical dopamine 2-fold (Hondo et al., 1994) and a more recent work has described cortical activation by NMDA receptor blockade as occurring through the inhibition of cortical GABAergic interneurons (Homayoun and Moghaddam, 2007). In the current study MK-801 did not significantly reduce GABA levels in itself (although a trend was observed) but did prevent AMPH-induced GABA release, suggesting that NMDA rececptor blockade modulates stimulated, but not basal GABA. However, this reduction in stimulated GABA corresponded to reductions in AMPH-induced dopamine and NE release; therefore, apart from increasing tonic dopamine levels through cortical disinhibition, MK-801 may also inhibit phasic dopamine release via receptor blockade at dopamine terminals. In fact, there is strong evidence for NMDA receptor localization to dopamine terminals (Roberts and Sharif 1978, Krebs et al., 1991, Desce et al., 1992). Therefore, the effects of MK-801 occur through a local cortical mechanism since our ex vivo experiments showed that MK-801 perfusion increased dopamine in isolated M1 tissue (data not shown) but also partially prevented AMPH-stimulated dopamine release, similar to our in vivo results. It is interesting to note that while MK-801 did not greatly affect AMPH-stimulated 5-HT release, it completely abolished AMPH-stimulated NE release. This strongly suggests that cortical NMDA receptors have diverse pre- and postsynaptic localization on different cell types to selectively modulate cortical neurochemistry.
In contrast to MK-801, NBQX perfusion had no effect on basal dopamine levels and almost completely blocked AMPH-induced dopamine, NE and 5-HT and GABA release. Therefore, similar to our reported behavioral data, AMPA receptor blockade was efficient at preventing AMPH-induced effects. Since NBQX itself did not increase dopamine levels, we can conclude that disinhibition of mesocortical neurons was not involved. However, there is also evidence for presynaptic localization of AMPA receptors (Schenk and Matteoli, 2004) which may contribute to the observed effect. This is in line with a previous work that showed that cortical AMPA receptor blockade effectively attenuated cortical dopamine release induced by a mild stressor (Takahata and Moghaddamn, 1998). Therefore if all monoamine terminals expressed AMPA receptors, NBQX might block phasic release in response to AMPH treatment. However, this idea is not entirely supported by our ex vivo data which showed only a partial attenuation of dopamine release by NBQX. One possibility is that AMPA receptors located on cortical pyramidal neurons projecting to the midbrain and brain stem are inhibited by NBQX, thus causing less glutamatergic drive of AMPH-stimulated mesocortical, raphe-cortical and coeruleus-cortical release. This view is in line with other work demonstrating that AMPH stimulates phasic release (Daberkow et al., 2013); therefore, reducing cell excitability with glutamate receptor antagonists would inhibit release events. These data support the idea that AMPA receptor antagonism might be a reasonable target for diseases in which overactivation of thalamocortical projections results in pathological movement. In fact the use of AMPA receptor antagonists has previously been shown to improve symptoms of MPTP-treated monkeys rendered dyskinetic by chronic L-DOPA treatment (Konitsiotis et al., 2000). Our results suggest that cortical AMPA receptors may be responsible for those effects.
Conclusions
Taken together, VL glutamate input acting on NMDA and AMPA receptors positioned on different populations of M1 elements (i.e. GABergic interneurons, presynaptic dopamine neurons and postsynaptic glutamate neurons) modulate basal and evoked cortical monoamine release dependent on the degree of activation. AMPH-stimulated monoamine release in M1 and hyperlocomotor activity appears to depend on the integrity of this thalamocortical signalling. In addition, these results also suggest that AMPH stimulates release via a TTX-independent mechanism.
Acknowledgments
We would like to thank Prof. Michele Morari for the critical reading of the manuscript. This work was supported by the National Institute on Drug Abuse T32 training grant (DA07268) to the Biology of Drug Abuse program at the University of Michigan (O.S.M.) and R37 EB003320 to R.T.K., R01 DA11697 (M.E.G.) and the National Institute for General Medicine T32 GM007767 (S.M.). Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number 2UL1TR000433. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The authors of this article declare that they do not have any conflicts of interest.
References
- Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;10:366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- Albin RL. The pathophysiology of chorea/ballism and Parkinsonism. Parkinsonism Relat Disord. 1995;1:3–11. doi: 10.1016/1353-8020(95)00011-t. [DOI] [PubMed] [Google Scholar]
- Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci. 1986;9:357–381. doi: 10.1146/annurev.ne.09.030186.002041. [DOI] [PubMed] [Google Scholar]
- Alexander GE, Crutcher MD. Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci. 1990;13:266–271. doi: 10.1016/0166-2236(90)90107-l. [DOI] [PubMed] [Google Scholar]
- Arnsten AF, Goldman-Rakic PS. Selective prefrontal cortical projections to the region of the locus coeruleus and raphe nuclei in the rhesus monkey. Brain Res. 1984;306:9–18. doi: 10.1016/0006-8993(84)90351-2. [DOI] [PubMed] [Google Scholar]
- Avelar AJ, Juliano SA, Garris PA. Amphetamine augments vesicular dopamine release in the dorsal and ventral striatum through different mechanisms. J Neurochem. 2013;125:373–385. doi: 10.1111/jnc.12197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benwell ME, Balfour DJ, Lucchi HM. Influence of tetrodotoxin and calcium on changes in extracellular dopamine levels evoked by systemic nicotine. Psychopharmacology. 1993;112:467–474. doi: 10.1007/BF02244896. [DOI] [PubMed] [Google Scholar]
- Brown RM, Crane AM, Goldman PS. Regional distribution of monoamines in the cerebral cortex and subcortical structures of the rhesus monkey: concentrations and in vivo synthesis rates. Brain Res. 1979;168:133–150. doi: 10.1016/0006-8993(79)90132-x. [DOI] [PubMed] [Google Scholar]
- Brown JM, Hanson GR, Fleckenstein AE. Regulation of the vesicular monoamine transporter-2: a novel mechanism for cocaine and other psychostimulants. J Pharmacol Exp Ther. 2001;296:762–767. [PubMed] [Google Scholar]
- Daberkow DP, Brown HD, Bunner KD, Kraniotis SA, Doellman MA, Ragozzino ME, Garris PA, Roitman MF. Amphetamine paradoxically augments exocytotic dopamine release and phasic dopamine signals. J Neurosci. 2013;33:452–463. doi: 10.1523/JNEUROSCI.2136-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Arco A, Martínez R, Mora F. Amphetamine increases extracellular concentrations of glutamate in the prefrontal cortex of the awake rat: a microdialysis study. Neurochem Res. 1998;23:1153–1158. doi: 10.1023/a:1020769816332. [DOI] [PubMed] [Google Scholar]
- DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13:281–285. doi: 10.1016/0166-2236(90)90110-v. [DOI] [PubMed] [Google Scholar]
- Desce JM, Godeheu G, Galli T, Artaud F, Chéramy A, Glowinski J. L-glutamate-evoked release of dopamine from synaptosomes of the rat striatum: involvement of AMPA and N-methyl-D-aspartate receptors. Neuroscience. 1992;47:333–339. doi: 10.1016/0306-4522(92)90249-2. [DOI] [PubMed] [Google Scholar]
- Douglas CL, Baghdoyan HA, Lydic R. M2 muscarinic autoreceptors modulate acetylcholine release in prefrontal cortex of C57BL/6J mouse. J Pharmacol Exp Ther. 2001;299:960–966. [PubMed] [Google Scholar]
- Engber TM, Dennis SA, Jones BE, Miller MS, Contreras PC. Brain regional substrates for the actions of the novel wake-promoting agent modafinil in the rat: comparison with amphetamine. Neuroscience. 1998;87:905–911. doi: 10.1016/s0306-4522(98)00015-3. [DOI] [PubMed] [Google Scholar]
- Gaspar P, Duyckaerts C, Alvarez C, Javoy-Agid F, Berger B. Alterations of dopaminergic and noradrenergic innervations in motor cortex in Parkinson’s disease. Ann Neurol. 1991;30:365–374. doi: 10.1002/ana.410300308. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ, Jr, Sibley DR. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250:1429–1432. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- Grofová I, Rinvik E. Light and electron microscopical studies of the normal structure and main afferent connections of the nuclei ventralis lateralis and ventralis anterior thalami of the cat. Confin Neurol. 1974;36:256–272. doi: 10.1159/000102800. [DOI] [PubMed] [Google Scholar]
- Herkenham M. Laminar organization of thalamic projections to the rat neocortex. Science. 1980;4430:532–535. doi: 10.1126/science.7352263. [DOI] [PubMed] [Google Scholar]
- Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27:11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondo H, Yonezawa Y, Nakahara T, Nakamura K, Hirano M, Uchimura H, Tashiro N. Effect of phencyclidine on dopamine release in the rat prefrontal cortex; an in vivo microdialysis study. Brain Res. 1994;633:337–342. doi: 10.1016/0006-8993(94)91558-x. [DOI] [PubMed] [Google Scholar]
- Hooks BM, Mao T, Gutnisky DA, Yamawaki N, Svoboda K, Shepherd GM. Organization of cortical and thalamic input to pyramidal neurons in mouse motor cortex. J Neurosci. 2013;33:748–760. doi: 10.1523/JNEUROSCI.4338-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosp JA, Pekanovic A, Rioult-Pedotti MS, Luft AR. Dopaminergic projections from midbrain to primary motor cortex mediate motor skill learning. J Neurosci. 2011;31:2481–2487. doi: 10.1523/JNEUROSCI.5411-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imperato A, Obinu MC, Gessa GL. Effects of cocaine and amphetamine on acetylcholine release in the hippocampus and caudate nucleus. Eur J Pharmacol. 1993;238:377–381. doi: 10.1016/0014-2999(93)90869-j. [DOI] [PubMed] [Google Scholar]
- Karler R, Calder LD, Turkanis SA. DNQX blockade of amphetamine behavioral sensitization. Brain Res. 1991;552:295–300. doi: 10.1016/0006-8993(91)90095-d. [DOI] [PubMed] [Google Scholar]
- Karler R, Calder LD, Thai LH, Bedingfield JB. The dopaminergic, glutamatergic, GABAergic bases for the action of amphetamine and cocaine. Brain Res. 1995;671:100–104. doi: 10.1016/0006-8993(94)01334-e. [DOI] [PubMed] [Google Scholar]
- Kelley AE, Throne LC. NMDA receptors mediate the behavioral effects of amphetamine infused into the nucleus accumbens. Brain Res Bull. 1992;29:247–254. doi: 10.1016/0361-9230(92)90034-u. [DOI] [PubMed] [Google Scholar]
- Kharazia VN, Weinberg RJ. Glutamate in thalamic fibers terminating in layer IV of primary sensory cortex. J Neurosci. 1994;14:6021–6032. doi: 10.1523/JNEUROSCI.14-10-06021.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konitsiotis S, Blanchet PJ, Verhagen L, Lamers E, Chase TN. AMPA receptor blockade improves levodopa-induced dyskinesia in MPTP monkeys. Neurology. 2000;54:1589–1595. doi: 10.1212/wnl.54.8.1589. [DOI] [PubMed] [Google Scholar]
- Krebs MO, Trovero F, Desban M, Gauchy C, Glowinski J, Kemel ML. Distinct presynaptic regulation of dopamine release through NMDA receptors in striosome- and matrix-enriched areas of the rat striatum. J Neurosci. 1991;11:1256–1262. doi: 10.1523/JNEUROSCI.11-05-01256.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabrouk OS, Li Q, Song P, Kennedy RT. Microdialysis and mass spectrometric monitoring of dopamine and enkephalins in the globus pallidus reveal reciprocal interactions that regulate movement. J Neurochem. 2011;118:24–33. doi: 10.1111/j.1471-4159.2011.07293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland NR, Haber SN. Thalamic relay nuclei of the basal ganglia form both reciprocal and nonreciprocal cortical connections, linking multiple frontal cortical areas. J Neurosci. 2002;18:8117–8132. doi: 10.1523/JNEUROSCI.22-18-08117.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narahashi T. Chemicals as tools in the study of excitable membranes. Physiol Rev. 1974;54:813–889. doi: 10.1152/physrev.1974.54.4.813. [DOI] [PubMed] [Google Scholar]
- Osborne PG, O’Connor WT, Drew KL, Ungerstedt U. An in vivo microdialysis characterization of extracellular dopamine and GABA in dorsolateral striatum of awake freely moving and halothane anaesthetised rats. J Neurosci Methods. 1990;34:99–105. doi: 10.1016/0165-0270(90)90047-j. [DOI] [PubMed] [Google Scholar]
- Parent A, Hazrati LN. Functional anatomy of the basal ganglia. I. The cortico-basal ganglia-thalamo-cortical loop. Brain Res Brain Res Rev. 1995;20:91–127. doi: 10.1016/0165-0173(94)00007-c. [DOI] [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;6483:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 6. Academic Press; 2007. [DOI] [PubMed] [Google Scholar]
- Rada P, Hernandez L, Hoebel BG. Feeding and systemic D-amphetamine increase extracellular acetylcholine in the medial thalamus: a possible reward enabling function. Neurosci Lett. 2007;416:184–187. doi: 10.1016/j.neulet.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Randrup A, Munkvad I. Pharmacological studies on the brain mechanisms underlying two forms of behavioral excitation: stereotyped hyperactivity and “rage”. Ann N Y Acad Sci. 1969;159:928–938. doi: 10.1111/j.1749-6632.1969.tb12989.x. [DOI] [PubMed] [Google Scholar]
- Reid MS, Hsu K, Jr, Berger SP. Cocaine and amphetamine preferentially stimulate glutamate release in the limbic system: studies on the involvement of dopamine. Synapse. 1997;27:95–105. doi: 10.1002/(SICI)1098-2396(199710)27:2<95::AID-SYN1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Roberts PJ, Sharif NA. Effects of l-glutamate and related amino acids upon the release of [3H]dopamine from rat striatal slices. Brain Res. 1978;157:391–395. doi: 10.1016/0006-8993(78)90048-3. [DOI] [PubMed] [Google Scholar]
- Schenk U, Matteoli M. Presynaptic AMPA receptors: more than just ion channels? Biol Cell. 2004;96:257–260. doi: 10.1016/j.biolcel.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Sherman SM, Guillery RW. Exploring the Thalamus. 1. Academic Press; 2001. [Google Scholar]
- Shimizu N, Duan SM, Hori T, Oomura Y. Glutamate modulates dopamine release in the striatum as measured by brain microdialysis. Brain Res Bull. 1990;25:99–102. doi: 10.1016/0361-9230(90)90258-2. [DOI] [PubMed] [Google Scholar]
- Song P, Mabrouk OS, Hershey ND, Kennedy RT. In vivo neurochemical monitoring using benzoyl chloride derivatization and liquid chromatography-mass spectrometry. Anal Chem. 2012;84:412–419. doi: 10.1021/ac202794q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strick PL. Anatomical analysis of ventrolateral thalamic input to primate motor cortex. J Neurophysiol. 1976;39:1020–1031. doi: 10.1152/jn.1976.39.5.1020. [DOI] [PubMed] [Google Scholar]
- Takahata R, Moghaddam B. Glutamatergic regulation of basal and stimulus-activated dopamine release in the prefrontal cortex. J Neurochem. 1998;71:1443–1449. doi: 10.1046/j.1471-4159.1998.71041443.x. [DOI] [PubMed] [Google Scholar]
- Takahata R, Moghaddam B. Activation of glutamate neurotransmission in the prefrontal cortex sustains the motoric and dopaminergic effects of phencyclidine. Neuropsychopharmacology. 2003;28:1117–1124. doi: 10.1038/sj.npp.1300127. [DOI] [PubMed] [Google Scholar]
- Timmerman W, Westerink BH. Brain microdialysis of GABA and glutamate: what does it signify? Synapse. 1997;27:242–261. doi: 10.1002/(SICI)1098-2396(199711)27:3<242::AID-SYN9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Wang JK. Presynaptic glutamate receptors modulate dopamine release from striatal synaptosomes. J Neurochem. 1991;57:819–822. doi: 10.1111/j.1471-4159.1991.tb08224.x. [DOI] [PubMed] [Google Scholar]
- Watabe-Uchida M, Zhu L, Ogawa SK, Vamanrao A, Uchida N. Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron. 2012;74:858–873. doi: 10.1016/j.neuron.2012.03.017. [DOI] [PubMed] [Google Scholar]
- Westerink BH, Tuntler J, Damsma G, Rollema H, de Vries JB. The use of tetrodotoxin for the characterization of drug-enhanced dopamine release in conscious rats studied by brain dialysis. Naunyn Schmiedebergs Arch Pharmacol. 1987;336:502–507. doi: 10.1007/BF00169306. [DOI] [PubMed] [Google Scholar]
- Williams JA, Comisarow J, Day J, Fibiger HC, Reiner PB. State-dependent release of acetylcholine in rat thalamus measured by in vivo microdialysis. J Neurosci. 1994;14:5236–5242. doi: 10.1523/JNEUROSCI.14-09-05236.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]