
Figure 2.
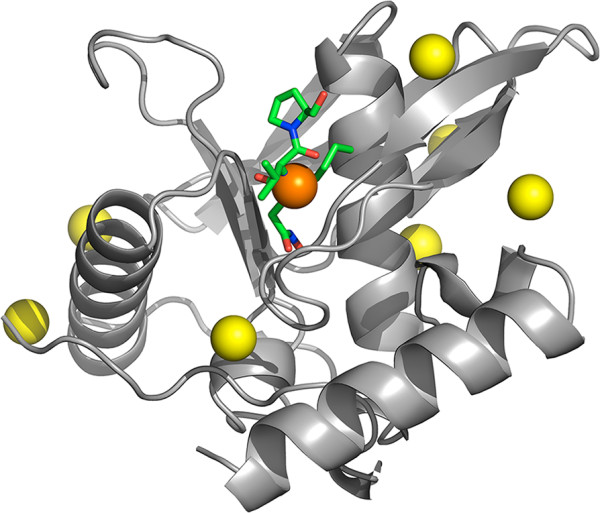
Peptide deformylase, automatic structural modeling and pocket analysis. The spheres displayed on the homology model of Pseudomonas aeruginosa peptide deformylase [Swiss-Prot:Q9I7A8] (based on template [PDB:1N5N]) represent putative ligand-binding sites as predicted by the automatic pocket analysis. The orange sphere marks the only cavity predicted to significantly affect overall protein flexibility. To illustrate the relevance of this prediction, we show the location of the antibiotic ligand (in ‘sticks’ representation) after superimposing the homology model to the known structure of the antibiotic-bound protein [PDB:1LRY] (RMSD 0.5 Å). The position of antibiotic Actinonin matches precisely the cavity marked by the procedure. The same cavity is also estimated to be very well conserved at the structural level (100% presence in the protein family).