

Abstract
Every year three million people die as a result of bacterial infections, and this number may further increase due to resistance to current antibiotics. These antibiotics target almost all essential bacterial processes, leaving only a few new targets for manipulation. The host proteome has many more potential targets for manipulation in order to control bacterial infection, as exemplified by the observation that inhibiting the host kinase Akt supports the elimination of different intracellular bacteria including Salmonella and M. tuberculosis. If host kinases are involved in the control of bacterial infections, phosphatases could be as well. Here we present an integrated small interference RNA and small molecule screen to identify host phosphatase-inhibitor combinations that control bacterial infection. We define host phosphatases inhibiting intracellular growth of Salmonella and identify corresponding inhibitors for the dual specificity phosphatases DUSP11 and 27. Pathway analysis places many kinases and phosphatases controlling bacterial infection in an integrated pathway centered around Akt. This network controls host cell metabolism, survival, and growth and bacterial survival and reflect a natural host cell response to bacterial infection. Inhibiting two enzyme classes with opposite activities–kinases and phosphatases–may be a new strategy to overcome infections by antibiotic-resistant bacteria.
Bacterial infections are responsible for the death of over three million people annually including over two million by tuberculosis, caused by Mycobacterium tuberculosis,1 and 200.000 by typhoid fever, caused by Salmonella typhi.2 Antibiotics against these bacteria can be effective in the control of infections but become gradually less effective due to the rise of (multi)drug resistance (MDR) against classical antibiotics. This problem is aggravated as the pharmaceutical industry has only few new antibiotics under development.3 The World Health Organization (WHO) and other health organizations have expressed their concern about the rise of MDR bacteria without new antibiotic developments for therapeutic alternatives. This may return society to the pre-antibiotic age where many people died of infections that are now simply treated. There is a great need for new strategies to control infections. Here we propose to target biological pathways in the host cell to control bacterial infections and provide a strategy to define host target-inhibitor combinations through an integrated chemical and genetic approach and in an unbiased fashion.
Many bacteria enter host cells and survive in phagosomes by manipulating host cells to prevent elimination.4,5 siRNA screens in Drosophila and mammalian cells have identified various biological targets and pathways in host cells controlled by S. typhimurium, M. tuberculosis, and other bacteria after intracellular infection. The feasibility of this approach was further defined by a simultaneous screen with kinase inhibitors yielding a bioactive protein kinase A (PKA) inhibitor H-89 that also inhibited intracellular growth of these bacteria as depicted in Figure 1A.6 Further studies showed that H-89 in fact inhibited the off-target kinase Akt.6S. typhimurium and M. tuberculosis activate Akt, which phosphorylates and inactivates GTPase-activating protein (GAP) AS160. As a consequence GTPase Rab14 remains active on phagosomes and recruits the scaffold Nischarin, which facilitates intracellular bacterial survival.6,7 These data imply that intracellular bacteria such as S. typhimurium and M. tuberculosis activate kinase Akt in the host cell for their own survival.6,8,9 The Akt inhibitors simply counteracted this mechanism in the host cell, effectively reducing the intracellular bacterial load. Host manipulation by small molecule inhibitors could thus represent a new class of antibiotics that are now exclusively directed against processes in their target bacteria.
Figure 1.

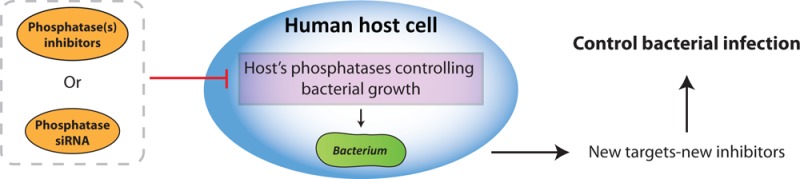
(A) The Akt protein pathway involved in Salmonella typhimurium infection. By inhibiting Akt using small molecule inhibitor H-89, intracellular growth of S. typhimurium can be blocked. (B) Outline of our approach of integrating chemical and genetic screening to define phosphatase target-inhibitor combinations in bacterial infection.
Protein kinases and protein phosphatases are basically two classes of enzymes that perform opposing chemical reactions, the phosphorylation and dephosphorylation of proteins. If kinases are involved in the control of intracellular bacterial growth, then phosphatases could be as well as these often reverse kinase-induced pathways. Over 510 kinases10 including 85 tyrosine kinases have been defined in the human genome, while only ∼150 phosphatases including 81 tyrosine phosphatases are known.11 The importance of controlling the activity of kinases in biology has long been recognized, and this has resulted in the development of several clinically approved kinase inhibitors (e.g., Imatinib) for mainly cancer treatment.12 A growing body of evidence now demonstrates that the regulation of protein and lipid dephosphorylation by phosphatases is equally important, which stimulated the development of phosphatase inhibitors.13−15 However, the development of such inhibitors is usually target-oriented, implying that first a biologically interesting phosphatase is defined before inhibitors are tested under either in vitro or cell-based conditions.16
Here we aimed at identifying phosphatase targets and corresponding small molecule inhibitors of bacterial infection in an unbiased fashion as depicted in Figure 1B. We present a strategy that integrates chemical (compound) and genetic (siRNA) inhibition screens to define host target-inhibitor combinations in controlling bacterial infections. This yielded host target-inhibitor combinations for dual specificity phosphatases (DUSPs) involved in the control in bacterial infections. The phosphatases identified were integrated in kinase networks6 that control bacterial infections on the basis of prior knowledge. Around half the phosphatases identified in our screen fitted the kinase pathways centered on the Akt pathway. The pathways controlled host cell viability, metabolism, inflammation, and phagosomal transport and were directly targeted by Salmonella effector proteins secreted into the host cell following infection. Chemical manipulation of host cell processes then counteracts the bacterial manipulation of the same processes and then support bacterial clearance in infected cells, effectively replacing antibiotics directly targeting the bacterium.
Results and Discussion
Identifying Phosphatases Controlling Intracellular Salmonella infections
We aimed to identify phosphatases controlling intracellular bacterial infections since we already defined the opposing class of enzymes, kinases.6 Around 190 phosphatase and phosphatase-like genes encoded in the human genome were silenced with siRNAs. (Supplementary Table S1). After transfection with siRNA, the cells were grown for three days before infection with fluorescent DsRed-expressing Salmonella typhimurium17 and cultured for another 18 h before the cells were analyzed for intracellular (fluorescent Ds-Red expressing) bacteria by flow cytometry. This experiment visualized the involvement of host phosphatases in the control of intracellular growth of S. typhimurium. We identified phosphatases that when silenced accelerated or inhibited intracellular growth of S. typhimurium (Figure 2A,B). The phosphatases accelerating intracellular growth include five subunits of PPP2, a phosphatase complex that controls the activity of kinase Akt, which we had shown in a previous study limits intracellular growth of Salmonella and other intracellular bacteria.6−9 This illustrates the complementarities of phosphatase-kinase reactions in one biological process and supports our approach to control bacterial infection by targeting host proteins. Various phosphatases that strongly inhibit intracellular infection when silenced were members of the dual specificity phosphatase (DUSP) family.
Figure 2.

(A) Results from the phosphatase siRNA screen. The effect of silencing the different phosphatases (for details see Supplementary Table 1) on intracellular S. typhimurium growth is quantified by flow cytometry, and the results are expressed as a Z-score. The gray lines depict the variation in the triplicate data points. (B) The 13 phosphatases that most strongly reduced or induced intracellular growth of S. typhimurium are shown with standard deviation of triplicate measurements.
Identifying Inhibitors Controlling Intracellular Salmonella Infections
A ∼300 -compound small molecule library designed to target protein tyrosine phosphatases, including DUSPs, was screened for their effects on intracellular growth of Salmonella in the same manner as applied in the siRNA screen (Figure 3A). In the design of the inhibitors a tyrosine moiety was incorporated to target the tyrosine binding site in dual specificity and tyrosine phosphatases (highlighted in blue, Figure 3B). We identified several compounds (LH101.2, LH1.2, HA25, LH1.4, LH56.1, and LH65.3) that inhibited intracellular growth of S. typhimurium in host cells (Figure 3B). These compounds were not affecting growth of S. typhimurium or viability of host cells (Supplementary Figure S1 and S2), which suggests that the inhibitors target host processes in control of intracellular Salmonella growth.
Figure 3.

(A) Results from screening a library of compounds, aimed at inhibiting dual specificity and tyrosine phosphatases, for their effect on intracellular growth of S. typhimurium. The effect of small molecules on intracellular S. typhimurium growth is quantified by flow cytometry, and the results are expressed as a Z-score. The gray lines depict the variation in the triplicate data points. (B) Effects and structures of the most potent inhibitors of bacterial infection are depicted. Shown is Z-score and standard deviation of triplicate measurements.
Phosphatase Target-Inhibitor Combinations by Integrating Chemical and Genetic Screens
Only a limited number of phosphatases have been defined in the siRNA screen that upon silencing showed a similar affect as the small molecule inhibitors. We aimed at connecting the inhibitors with their respective targets, which can only be done in in vitro assays with purified phosphatases. We assayed phosphatase activity using 3-O-methylfluorescein phosphate (OMFP).18 The quenched OMFP substrate is hydrolyzed into fluorescent 3-O-methylfluorescein (OMF) and phosphate. We expressed and purified three DUSP family members identified in the siRNA screen as inhibiting intracellular bacterial growth when silenced: DUSP3, 11, and 27. DUSP3 (also called VHR) is reported to control ERK1 (p44MAPK3) and 2 (p38MAPK1) kinases;19 DUSP11 targets RNA (although active on tyrosine phosphate-like substrates) and is involved in cell growth control;20 and DUSP27 may be a poorly defined phosphatase with a potential role in metabolism (that could include the mTOR pathway controlled by Akt).21 We tested whether the inhibitor LH65.3 affected ERK activity in response to Salmonella infection. No effect of the compound was observed (Supplemental Figure 3). This result urged us to control the defined DUSPs again. These were identified with pools of four different siRNAs/target. Off target effects are highly unlikely when more than 2 different siRNAs/target show inhibition of intracellular bacterial growth. We have therefore deconvoluted the pool and tested the four siRNAs independently for their effect on Salmonella infection (Figure 4C). MCF7 cells were first transfected with the different siRNAs and infected 3 days later with Salmonella, and the rate of intracellular growth of Salmonella was determined 24 h later. The results are shown as a Z-score. Silencing of DUSP11 and DUSP27 with 3 or 4 different siRNAs/target confirmed inhibition of intracellular growth of Salmonella, whereas only one siRNA could be confirmed for DUSP3. This suggests that DUSP11 and DUSP27, unlike DUSP3, are involved in the control of intracellular infection by Salmonella.
Figure 4.

(A) Effect of identified S. typhimurium infection inhibitors on the activity of DUSP3, 11, and 27. DUSP activity (%) has been measured at an inhibitor concentration of 5 μM. Data are represented as average with standard deviation of triplicate measurements. (B) IC50 values (μM) for LH65.3 on DUSP3, 11, and 27 given as average with standard deviation of triplicate measurements. (C) Deconvolution of siRNAs for DUSP3, 11, and 27 and their effect on intracellular Salmonella growth. Shown is the Z-score for the four siRNAs tested per target. A negative Z-score indicates inhibition of intracellular growth of Salmonella.
Of note, the DUSP27 phosphatase domain produced recombinantly is highly active on our OMPF substrates. We tested the various bioactive inhibitors identified in the chemical library screen in Figure 3B for their potential to inhibit the three different DUSPs at a concentration of 5 μM in the presence of 20 μM OMFP (Figure 4A). Compound LH65.3 was able to inhibit all three DUSPs with high potency (Figure 4B) but failed to inhibit a typical tyrosine phosphatase PTP1B and lipid phosphodiesterase ENPP2 included as a control for specificity (Supplemental Figure S4). This furthermore suggests that LH65.3 inhibits intracellular S. typhimurium growth via DUSP inhibition, validating our approach.
In order to explore whether LH65.3 could be optimized further, we explored initial structure–activity relationships (SAR). We systematically synthesized a range of analogues, 27 in total. The thiazolidine-2,4-dione core was systematically equipped with different substituents through parallel synthesis. Compounds thus obtained were tested against DUSPs at a concentration of 5 μM against recombinant DUSP3, 11, and 27 (Figure 5A, percentage inhibition is shown). From these experiments, it becomes apparent that clear structure–activity relations can be observed. Moreover, selectivity for different DUSPs can be obtained. Compound 8, for example, shows a clear selectivity for DUSP3 and 11 over DUSP27. This compound was therefore resynthesized, and IC50 curves were determined side by side with LH65.3 for these three DUSPs (Figure 5B), further confirming DUSP selectivity of this LH65.3 analogue 8. Further synthesis of variants will be required to select inhibitors selective for both DUSP11 and DUSP27, the most important DUSPs in the control of intracellular bacterial infection.
Figure 5.

(A) Structure–activity analysis. Systematic variation of substituents on the thiazolidine-2,4-dione core present in LH65.3. The table shows the different substituents tested for 27 analogues. Percentage inhibition of DUSP3, 11, and 27 at a 5 μM inhibitor concentration is shown. (B) IC50 curves determined for both compound 8 and LH65.3 against these three DUSPs.
Constructing Host Cell Kinase-Phosphatase Networks in Control of Intracellular Bacterial Infections
We presented a chemical genetics screen for host kinases that when genetically silenced or chemically inhibited eliminated intracellular bacteria including Salmonella and M. tuberculosis.6 We now present a similar screen for the complementary group of enzymes, phosphatases, and identified various members that either promoted or inhibited intracellular growth of Salmonella, our model pathogen. These phosphatases likely interact with various kinase networks in the same process. To place phosphatases in the earlier identified kinase network, we introduced the various hits in network programs such as Ingenuity and STRING where connections are drawn on the basis of information from published literature. We performed a manual check and included factors not recognized by these programs: two Salmonella effectors secreted into the host cytosol for control of infection and general terms for the function of the networks (Figure 6). In addition, for some of the hits substrates are undefined but a more general function is observed (e.g., DUSP27 whose silencing is reported to affect cell viability).6,7,19,22−31 In such cases, a hit is connected not to a target protein but to a process. Finally, we included in the figure arrows and lines illustrating whether a process is activated or inhibited by an upstream candidate. Half of phosphatases identified in the siRNA screen can be placed in a large network centered around kinases Akt. Akt is activated by Salmonella effector SopB secreted in the host cell by Salmonella and promotes the intracellular survival of Salmonella (see references in Figure 6). Some of the downstream pathways have been described in more detail, such as the effect of Akt activation by SopB on AS160, Rab14, NISCH, and fusion of phagosomes with lysophagosomes where bacteria would be eliminated.6,7 The exact molecular control of other pathways is less clear but can be expected to be involved in host responses to intracellular bacterial infection. These include the control of metabolism (Salmonella requires many nutrients for propagation that will be retarded during cell starvation), cell survival, growth (probably best condition for intracellular Salmonella growth as premature death will result in detection and clearance of dead cells (+content) by macrophages), and inflammation (to control tissue responses to infection).
Figure 6.

An integrated host kinase-phosphatase network in control of bacterial infections. The hits from two siRNA screens for host kinases and phosphatases, as involved in intracellular growth of S. typhimurium, are placed in a connecting interactome network. The network is generated on the basis of published literature, and activation or inactivation of the various proteins in the network by their upstream regulators is indicated. Salmonella effector proteins secreted in the host cytosol can control host Akt and are included. These effectors modify the host for their survival. In addition, general terms for the relevance of the various pathways are included. Small encircled numbers at the various connections refer to refs (6, 7, 19, and 22−31).
Two chemical genetics screens yield kinases and phosphatases of which a major portion can be placed in only a limited number of integrated signaling networks controlling processes that bacteria such as Salmonella also want to control. In general terms, these are intracellular transport, cell viability, and nutrient state. These processes all promote fast growth without prior elimination of Salmonella. This also may explain why Salmonella has invested in generating an exquisite Type III secretion system and effector proteins to control these host processes. Chemical counterattack may then be an attractive way to control these infections.
Our chemical genetics screens provide such new small chemical entities, identified in an almost unbiased manner. Our chemical genetics approach also identifies the corresponding targets, thus providing the material for improving the drugs to eliminate intracellular bacteria by inhibiting host pathways activated by bacteria to create optimal growth conditions in cells. We identified an inhibitor that appears to inhibit three DUSPs, two of which are relevant in host responses to bacterial infections. This new chemical entity may find application in the more detailed studies on the function of these DUSPs, as this enzyme class is rather poorly understood. Further optimization of the inhibitor structures is then a prerequisite.
Our approach to target the phosphatases of host cells rather than targeting the bacteria itself for controlling bacterial infection has several implications. First, the number of targets in the bacteria itself for selective inhibition of bacterial growth has been estimated to be very limited.32 The host proteome may contain a wealth of new targets to control intracellular growth of bacteria such as Salmonella and M. tuberculosis. We and others demonstrated that kinases can be used to control bacterial growth,6,8,9 and here we add the complementary group of enzymes, phosphatases. Second, whereas it is easy for bacteria to become drug-resistant,33 it might be more difficult, maybe impossible, for bacteria to manipulate the host in this endeavor. Drug resistance for inhibitors targeting host cell processes is expected to be a minor issue. Third, as Salmonella and other bacteria survive in host cells by manipulating host cell cell biology, this screen unravels new biology. Here we integrated host phosphatases into a host kinase network that both control the survival of intracellular bacteria such as Salmonella. The pathway centers around Akt that is manipulated by effector proteins of Salmonella for the good of its own survival.6,22 Akt is controlled by a series of phosphatases acting up- and downstream (for example, phosphatase PPAPDC3 controls Akt substrate kinase mTOR that is involved in metabolism regulation (Figure 6)). Many phosphatase hits control Akt as this is a central molecule in host cell responses to bacterial infections. Akt controls cell survival but also bacterial survival by preventing maturation of phagosomes into phagolysosomes where they would have been degraded.6,7 Many hits from the two complementary screens could be placed in one network involving kinases and phosphatases known to control processes relevant for bacterial survival or elimination within host cells. Fourth, inhibitors targeting host processes to control bacterial infection may be developed for the treatment of other diseases and now find broader use. For example, kinase inhibitors targeting the host kinase Akt to control bacterial infections6,8,9 are now being tested in phase II/III trials as anticancer drugs.34
To conclude, we have demonstrated the feasibility of a new approach to generate phosphatase target-inhibitor combinations by integrating genetic and chemical inhibition screens (Figure 1B). This approach allows the discovery of novel antibacterial compounds acting in an unexplored manner by supporting host cells to control bacterial infection.
Methods
Synthesis of the Inhibitor Library
Inhibitors were synthesized as previously described with minor adjustments to the protocol.35−37
HPLC–MS and 1H and 13C NMR spectra for LH65.3 can be found in the Supporting Information.
Phosphatase siRNA Screen
Gene silencing was performed in 96-well plates with a human breast cancer (MCF-7) and glioblastoma (A-172) cell line. Cells were seeded at a density of 5000 cells per well and reverse transfected with DharmaFECT transfection reagent no. 4 and 50 nM siRNA (Human siGenome SMARTpool phosphatase library, Dharmacon). Two days after transfection, cells were infected with Salmonella typhimurium expressing DsRed,17 which was performed based on the infection protocol described by Steele-Mortimer et al. with minor changes.38 In short, 2 days before infection a bacterial culture was streaked from the frozen stock on LB agar plates. The next day, an overnight bacterial culture was prepared by inoculating 5 mL of LB medium with one colony from the agar plate. The overnight culture was incubated at 310 K and 200 rpm for 16–20 h and then diluted 1:33 in fresh, prewarmed (310 K) ampicillin (100 μg mL–1) containing LB medium for a further incubation of 3.5 h. The bacterial culture (4 mL) was transferred to a 15-mL Falcon tube and pelleted by centrifugation at 4000 rpm for 10 min at RT. The pellet was washed once with DMEM/FCS (8%) and resuspended in DMEM/8% FCS (310 K). The cells were infected at an MOI of 50. After 20 min, the cells were washed 4 times with DMEM/8% FCS containing 100 μg mL–1 gentamycin and further cultured for 60 min in DMEM/FCS medium with 100 μg mL–1 gentamicin to kill remaining extracellular bacteria. For the remaining infection period, the antibiotic concentration was lowered to 10 μg mL–1. After overnight infection cells were washed once with PBS, and 30 μL of trypsin/EDTA was added for 5 min followed by addition of 30 μL of PBS/1% BSA. The sample was fixed by addition of 60 μL of PBS with 7% (v/v) formalin
Samples were analyzed by flow cytometry (BD FACSArray) for DsRed fluorescence as marker for Salmonella infection and proliferation. The data were normalized (cellHTS2, Bioconductor) and transformed into Z-scores.39
Small Molecule Screen
The infection of mammalian cells (MCF-7 or A172) with S. typhimurium expressing DsRed17 was performed based on the infection protocol described above.38 In order to infect the cells with the MOI 20 for 30 min, the bacterial culture was diluted according to the following assumption: OD595 of 1 ≈ 1.3 × 109 CFU mL–1. To infect cells with bacteria, the cell culture medium was aspirated, and 100 μL of bacteria in DMEM/FCS was added to the wells. Plates were centrifuged at 1000 rpm at RT for 5 min and incubated at 310 K with 5% CO2 in a humidified cell culture incubator to allow invasion for 1 h. The cells were washed 4 times with DMEM/FCS containing 100 μg mL–1 gentamycin and incubated for another 1 h in DMEM/FSC containing 100 μg mL–1 gentamycin. For the remaining infection period, the medium was replaced with DMEM/FCS containing 10 μg mL–1 gentamicin and compound as indicated. Compounds were tested at 10 μM in triplicate.
After overnight infection, cells were washed once with PBS and incubated in 30 μL of trypsin/EDTA for 5–10 min to resuspend cells. Subsequently, 30 μL of PBS/1% BSA to quench trypsin, and 60 μL of PBS/formalin (7%) was added to fix cells and bacteria. After at least a 2 h incubation, fixed cells were analyzed by flow cytometry, as previously described.6
Effect of Compounds on S. typhimurium Growth in LB Medium
An overnight bacterial culture was prepared by inoculating 5 mL of ampicillin (100 μg mL–1) containing LB medium with one colony from an agar plate. The overnight culture was incubated at 310 K and 200 rpm for 16–20 h and then diluted 1:33 in fresh ampicillin (100 μg mL–1) containing LB medium. In a 96-well plate, 100 μL of the bacteria-containing medium was pipetted to LB medium (100 μL) containing 20 μM of inhibitor. Bacterial growth was monitored by measuring the optical density at 595 nm (OD595).
Cell toxicity
Cell toxicity was determined using the CellTiter-Blue assay. Cells (1.5 × 105 mL–1) were incubated in presence or absence of inhibitors for 24 h. Phenylarsine oxide (PAO) was used as a control for cell death. After 19 h of incubation, 20 μL of resazurin (0.125 mg mL–1) was added for 5 h. Fluorescence was measured at λex/λem = 544/560 nm.
DUSP Activity Assay.18
Measuring DUSP activity using 3-O-methylfluorescein phosphate (OMFP) as a substrate40 was performed as follows. In a black flat-bottom 96-well plate, 0.9 μL of DMSO containing inhibitor was added to 45 μL of recombinant DUSP (∼0.1 U) in a Tris-HCl buffer (9 mM Tris-HCl, 11 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.01% Triton X-100, pH 7.4). Finally, 45 μL of 40 μM OMFP in the same Tris-HCl buffer was added to each well using a multichannel pipet, and instantaneously fluorescence was measured at RT (λex/λem = 485/520 nm). The above-described mixture with DMSO alone was used as a 100% activity control. OMFP without DUSP was taken as control for autohydrolysis of OMFP. For each inhibitor, the percentage inhibition (PI) was determined at a final inhibitor concentration of 5 μM. IC50 values of inhibitors have been determined in an inhibitor concentration range of 0.01–6 μM. Data were analyzed using Graphpad Prism software.
Cloning, Expression, and Purification of DUSPs
PCR fragments of phosphatase domains of DUSP3, 11, and 27 containing a His-tag sequence have been cloned into a pETNKI-His-3c-LIC-kan vector41 and were sequence verified. The resulting constructs have been transformed into BL21 (DE3) cells. Transformed cells were grown overnight in LB medium (2 mL) containing kanamycin (30 μg mL–1) and were subsequently inoculated in LB medium (1 L) with kanamycin (30 μg mL–1) until an OD595 of 0.6 was reached. Protein expression was induced by IPTG (0.5 mM) overnight at 293 K. Cells were spun down (3,000g, 15 min, 277 K), and the resulting cell pellet was resuspended in lysis buffer (40 mM Tris, 200 mM NaCl, 5 mM β-mercaptoethanol, 5 mM imidazole, pH 8.0). After sonication, cell debris and insoluble proteins were removed by centrifugation (14,000g, 30 min, 277 K), and the soluble fraction was incubated with Talon beads and washed lysis buffer. Protein was eluted with elution buffer (40 mM Tris-HCl, 200 mM NaCl, 5 mM β-mercaptoethanol, 300 mM imidazole, pH 8.0), and elution fractions were analyzed by SDS-PAGE for protein content. DUSP-containing fractions were pooled and diluted with 3 volumes of 40 mM Tris-HCl, 5 mM β-mercaptoethanol pH 8.0 to decrease the NaCl concentration of the protein solution to 50 mM. The DUSP proteins were further purified by resource Q anion-exchange chromatography and eluted from the column in a linear gradient from 50 to 500 mM NaCl in 40 mM Tris-HCl, 5 mM β-mercaptoethanol, pH 8.0. A final purification step of all DUSPs included a size exclusion chromatography (S75 10/60 column) using 40 mM Tris, 100 mM NaCl, 5 mM β-mercaptoethanol, pH 8.0 as running buffer. Fractions containing DUSP protein were pooled and concentrated using a 10 kDa cutoff Centriprep column and stored at 193 K until further usage.
Pathway Analyses
The hits defined in the siRNA screens for host kinases6 and phosphatases were loaded into Ingenuity PathwayFinder to generate networks on the basis of published literature. The major pathways were around Akt and p38ERK1/2 and were further developed using Ingenuity as well as STRING, and the pathways were further improved by manual analyses of literature connected to the various steps in the network (references indicated in Figure 6). Sometimes (as for DUSP27) the target kinase(s) is unknown but the process affected is described. In such cases, the target phosphatase was linked to this process. In the network, we include a general description of biological processes to illustrate how the various kinases, phosphatases, and Salmonella effectors may all contribute in the manipulation of limited cell biology for (control of) intracellular bacterial propagation.
Acknowledgments
In memory of L. J. D. Hendrickx 06-01-1986/28-05-2010. We want to thank D. Bumann for supplying us with S. typhimurium expressing DsRed. This work was supported by grants from The Netherlands Organization for Scientific Research (NWO), the Dutch Cancer Society (KWF), an ERC Advanced Grant, and The Netherlands Proteomics Centre supported by The Netherlands Genomics Initiative.
Glossary
Abbreviations
- CFU
colony forming units
- DUSP
dual specificity phosphatase
- GAP
GTPase-activating protein
- FACS
fluorescence-activated cell sorter
- MDR
multidrug-resistant
- OD
optical density
- PKA
protein kinase A
Supporting Information Available
Information about the effect of compounds on S. typhimurium growth in LB medium and on cell viability of cells, list of screened phosphatase and phosphatase-like siRNAs, effect of compounds on Phospho-p44/42 MAPK levels, enzyme selectivity of LH65.3, and details on the synthesis of the inhibitor library including spectral data for LH65.3. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
¶ These author contributed equally to this work.
The authors declare no competing financial interest.
Author Status
# Deceased.
Supplementary Material
References
- Harries A. D.; Dye C. (2006) Tuberculosis. Ann. Trop. Med. Parasitol. 100, 415–431. [DOI] [PubMed] [Google Scholar]
- Buckle G. C.; Walker C. L.; Black R. E. (2012) Typhoid fever and paratyphoid fever: Systematic review to estimate global morbidity and mortality for 2010. J. Global Health 2, 10401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H. (2009) Multidrug resistance in bacteria. Annu. Rev. Biochem. 78, 119–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meresse S.; Steele-Mortimer O.; Moreno E.; Desjardins M.; Finlay B.; Gorvel J. P. (1999) Controlling the maturation of pathogen-containing vacuoles: a matter of life and death. Nat. Cell Biol. 1, E183–E188. [DOI] [PubMed] [Google Scholar]
- Vergne I.; Chua J.; Singh S. B.; Deretic V. (2004) Cell biology of mycobacterium tuberculosis phagosome. Annu. Rev. Cell Dev. Biol. 20, 367–394. [DOI] [PubMed] [Google Scholar]
- Kuijl C.; Savage N.; Marsman M.; Tuin A.; Janssen L.; Egan D.; Ketema M.; van den Nieuwendijk R.; van den Eeden S.; Geluk A.; Poot A.; van der Marel G.; Beijersbergen R.; Overkleeft H.; Ottenhoff T.; Neefjes J. (2007) Intracellular bacterial growth is controlled by a kinase network around PKB/AKT1. Nature 450, 725–730. [DOI] [PubMed] [Google Scholar]
- Kuijl C.; Pilli M.; Alahari S. K.; Janssen H.; Khoo P. S.; Ervin K. E.; Calero M.; Jonnalagadda S.; Scheller R. H.; Neefjes J.; Junutula J. R. (2013) Rac and Rab GTPases dual effector Nischarin regulates vesicle maturation to facilitate survival of intracellular bacteria. EMBO J. 32, 713–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D.; Nath L.; Kamal M. A.; Varshney A.; Jain A.; Singh S.; Rao K. V. (2010) Genome-wide analysis of the host intracellular network that regulates survival of Mycobacterium tuberculosis. Cell 140, 731–743. [DOI] [PubMed] [Google Scholar]
- Thornbrough J. M.; Hundley T.; Valdivia R.; Worley M. J. (2012) Human genome-wide RNAi screen for host factors that modulate intracellular Salmonella growth. PLoS One 7, e38097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G.; Whyte D. B.; Martinez R.; Hunter T.; Sudarsanam S. (2002) The protein kinase complement of the human genome. Science 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- Alonso A.; Sasin J.; Bottini N.; Friedberg I.; Friedberg I.; Osterman A.; Godzik A.; Hunter T.; Dixon J.; Mustelin T. (2004) Protein tyrosine phosphatases in the human genome. Cell 117, 699–711. [DOI] [PubMed] [Google Scholar]
- Druker B. J.; Tamura S.; Buchdunger E.; Ohno S.; Segal G. M.; Fanning S.; Zimmermann J.; Lydon N. B. (1996) Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 2, 561–566. [DOI] [PubMed] [Google Scholar]
- Tonks N.; Neel B. (2001) Combinatorial control of the specificity of protein tyrosine phosphatases. Curr. Opin. Cell Biol. 13, 182–195. [DOI] [PubMed] [Google Scholar]
- Schneider R.; Beumer C.; Simard J. R.; Grutter C.; Rauh D. (2013) Selective detection of allosteric phosphatase inhibitors. J. Am. Chem. Soc. 135, 6838–6841. [DOI] [PubMed] [Google Scholar]
- Vintonyak V. V.; Waldmann H.; Rauh D. (2011) Using small molecules to target protein phosphatases. Bioorg. Med. Chem. 19, 2145–2155. [DOI] [PubMed] [Google Scholar]
- Stanford S. M.; Panchal R. G.; Walker L. M.; Wu D. J.; Falk M. D.; Mitra S.; Damle S. S.; Ruble D.; Kaltcheva T.; Zhang S.; Zhang Z. Y.; Bavari S.; Barrios A. M.; Bottini N. (2012) High-throughput screen using a single-cell tyrosine phosphatase assay reveals biologically active inhibitors of tyrosine phosphatase CD45. Proc. Natl. Acad. Sci. U.S.A. 109, 13972–13977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sörensen M.; Lippuner C.; Kaiser T.; Mißlitz A.; Aebischer T.; Bumann D. (2003) Rapidly maturing red fluorescent protein variants with strongly enhanced brightness in bacteria. FEBS Lett. 552(2–3), 110–114. [DOI] [PubMed] [Google Scholar]
- Tierno M.; Johnston P.; Foster C.; Skoko J.; Shinde S.; Shun T.; Lazo J. (2007) Development and optimization of high-throughput in vitro protein phosphatase screening assays. Nat. Protoc. 2, 1134–1144. [DOI] [PubMed] [Google Scholar]
- Todd J. L.; Tanner K. G.; Denu J. M. (1999) Extracellular regulated kinases (ERK) 1 and ERK2 are authentic substrates for the dual-specificity protein-tyrosine phosphatase VHR. A novel role in down-regulating the ERK pathway. J. Biol. Chem. 274, 13271–13280. [DOI] [PubMed] [Google Scholar]
- Caprara G.; Zamponi R.; Melixetian M.; Helin K. (2009) Isolation and characterization of DUSP11, a novel p53 target gene. J. Cell Mol. Med. 13, 2158–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg I.; Nika K.; Tautz L.; Saito K.; Cerignoli F.; Friedberg I.; Godzik A.; Mustelin T. (2007) Identification and characterization of DUSP27, a novel dual-specific protein phosphatase. FEBS Lett. 581, 2527–2533. [DOI] [PubMed] [Google Scholar]
- Odendall C.; Rolhion N.; Forster A.; Poh J.; Lamont D. J.; Liu M.; Freemont P. S.; Catling A. D.; Holden D. W. (2012) The Salmonella kinase SteC targets the MAP kinase MEK to regulate the host actin cytoskeleton. Cell Host Microbe 12, 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson R. M. (1998) Tuberculosis: old problems and new approaches. Proc. Natl. Acad. Sci. U.S.A 95, 13352–13354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A.; Bonni A.; Zigmond M. J.; Lin M. Z.; Juo P.; Hu L. S.; Anderson M. J.; Arden K. C.; Blenis J.; Greenberg M. E. (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868. [DOI] [PubMed] [Google Scholar]
- Ugi S.; Imamura T.; Maegawa H.; Egawa K.; Yoshizaki T.; Shi K.; Obata T.; Ebina Y.; Kashiwagi A.; Olefsky J. M. (2004) Protein phosphatase 2A negatively regulates insulin’s metabolic signaling pathway by inhibiting Akt (protein kinase B) activity in 3T3-L1 adipocytes. Mol. Cell. Biol. 24, 8778–8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thong F. S.; Bilan P. J.; Klip A. (2007) The Rab GTPase-activating protein AS160 integrates Akt, protein kinase C, and AMP-activated protein kinase signals regulating GLUT4 traffic. Diabetes 56, 414–423. [DOI] [PubMed] [Google Scholar]
- Seely B. L.; Staubs P. A.; Reichart D. R.; Berhanu P.; Milarski K. L.; Saltiel A. R.; Kusari J.; Olefsky J. M. (1996) Protein tyrosine phosphatase 1B interacts with the activated insulin receptor. Diabetes 45, 1379–1385. [DOI] [PubMed] [Google Scholar]
- Li L.; Ljungman M.; Dixon J. E. (2000) The human Cdc14 phosphatases interact with and dephosphorylate the tumor suppressor protein p53. J. Biol. Chem. 275, 2410–2414. [DOI] [PubMed] [Google Scholar]
- Galaktionov K.; Beach D. (1991) Specific activation of cdc25 tyrosine phosphatases by B-type cyclins: evidence for multiple roles of mitotic cyclins. Cell 67, 1181–1194. [DOI] [PubMed] [Google Scholar]
- Guo L.; Martens C.; Bruno D.; Porcella S. F.; Yamane H.; Caucheteux S. M.; Zhu J.; Paul W. E. (2013) Lipid phosphatases identified by screening a mouse phosphatase shRNA library regulate T-cell differentiation and Protein kinase B AKT signaling. Proc. Natl. Acad. Sci. U.S.A. 110, E1849–E1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GeneCARDS.
- Cudic M.; Otvos L. Jr. (2002) Intracellular targets of antibacterial peptides. Curr. Drug Targets 3(2), 101–106. [DOI] [PubMed] [Google Scholar]
- Lynch M. (2010) Evolution of the mutation rate. Trends Genet. 26, 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granville C. A.; Memmott R. M.; Gills J. J.; Dennis P. A. (2006) Handicapping the race to develop inhibitors of the phosphoinositide 3-kinase/Akt/mammalian target of rapamycin pathway. Clin. Cancer Res. 12, 679–689. [DOI] [PubMed] [Google Scholar]
- Albers H.; van Meeteren L.; Egan D.; van Tilburg E.; Moolenaar W.; Ovaa H. (2010) Discovery and optimization of boronic acid based inhibitors of autotaxin. J. Med. Chem. 53, 4958–4967. [DOI] [PubMed] [Google Scholar]
- Albers H.; Hendrickx L.; van Tol R.; Hausmann J.; Perrakis A.; Ovaa H. (2011) Structure-based design of novel boronic acid-based inhibitors of autotaxin. J. Med. Chem. 54, 4619–4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers H. M.; Dong A.; van Meeteren L. A.; Egan D. A.; Sunkara M.; van Tilburg E. W.; Schuurman K.; van Tellingen O.; Morris A. J.; Smyth S. S.; Moolenaar W. H.; Ovaa H. (2010) Boronic acid-based inhibitor of autotaxin reveals rapid turnover of LPA in the circulation. Proc. Natl. Acad. Sci. U.S.A. 107, 7257–7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele-Mortimer O.; Méresse S.; Gorvel J.; Toh B.; Finlay B. B. (1999) Biogenesis of Salmonella typhimurium-containing vacuoles in epithelial cells involves interactions with the early endocytic pathway. Cell. Microbiol. 1, 33–49. [DOI] [PubMed] [Google Scholar]
- Boutros M.; Bras L.; Huber W. (2006) Analysis of cell-based RNAi screens. Genome Biol. 7, R66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K.; Southwick E.; Kerns J.; Rosi K.; Carr B.; Wilcox C.; Lazo J. (2000) Cdc25 inhibition and cell cycle arrest by a synthetic thioalkyl vitamin K analogue. Cancer Res. 60, 1317–1325. [PubMed] [Google Scholar]
- Luna-Vargas M.; Christodoulou E.; Alfieri A.; van Dijk W.; Stadnik M.; Hibbert R.; Sahtoe D.; Clerici M.; Marco V.; Littler D.; Celie P.; Sixma T.; Perrakis A. (2011) Enabling high-throughput ligation-independent cloning and protein expression for the family of ubiquitin specific proteases. J. Struct. Biol. 175, 113–119. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.