

Abstract
In the ErbB/HER family of receptor tyrosine kinases, the deregulation of the EGFR/ErbB1/HER1, HER2/ErbB2, and HER3/ErbB3 kinases is associated with several cancers, while the HER4/ErbB4 kinase has been shown to play an anti-carcinogenic role in certain tumors. We present molecular and network models of HER4/ErbB4 activation and signaling in order to elucidate molecular mechanisms of activation and to help rationalize the effects of the clinically identified HER4 somatic mutants. Our molecular-scale simulations identify the important role played by the interactions within the juxtamembrane region during the activation process. Our results also support the hypothesis that the HER4 mutants may heterodimerize but not activate, resulting in blockage of the HER4-STAT5 differentiation pathway, in favor of the proliferative PI3K/AKT pathway. Translating our molecular simulation results into a cellular pathway model of wild type versus mutant HER4 signaling, we are able to recapitulate the major features of the PI3K/AKT and JAK/STAT activation downstream of HER4. Our model predicts that the signaling downstream of the wild type HER4 is enriched for the JAK-STAT pathway, whereas downstream of the mutant HER4 is enriched for the PI3K/AKT pathway. HER4 mutations may hence constitute a cellular shift from a program of differentiation to that of proliferation.
Keywords: Molecular Dynamics, Network Modeling, Systems Biology, Structural Biology, Receptor Tyrosine Kinase, Cellular Signaling Pathways
1. Introduction
The ErbB family of receptor tyrosine kinases (RTKs), which includes epidermal growth factor receptor (EGFR/ErbB1/HER1), ErbB2 (HER2), ErbB3 (HER3) and ErbB4 (HER4), performs a crucial role in regulating critical cellular processes including migration, differentiation, and proliferation [1–3]. The ErbB RTKs are composed of an extracellular ligand-binding domain, a transmembrane segment, an intracellular protein tyrosine kinase domain, and a C-terminal tail harboring tyrosine phosphorylation sites [4, 5]. Under physiological conditions, ligand binding promotes homo- or heterodimerization of the receptors and activation of their cytoplasmic domains [6]. Dimerization results in auto- or trans-phosphorylation of tyrosine residues in the RTK C-terminal tail segments, which serve as docking sites for signaling molecules containing SH2 or PTB domains [7, 8]. Aberrant activation of the ErbB network is frequently associated with cellular transformation and clinical malignancies such as lung, gastric, and breast cancers [9–16]. Overexpression of HER2 results in constitutive, ligand-independent activation of kinase signaling and is found in 20–30% of human breast cancers, where it is correlated with an aggressive tumor phenotype [17, 18].
Whereas deregulation of the EGFR, HER2, and HER3 kinases is associated with many types of human cancer, the HER4 kinase has recently been shown to play an anti-carcinogenic role in certain tumors, including mammary carcinomas [19, 20]. One mechanism by which HER4 is thought to impede tumor progression in mammary cells is through the activation of genes that promote cellular differentiation and inhibit proliferation, in effect steering the cell away from a program of uncontrolled growth and toward a program of differentiation [21]. HER4 is unique from the other ErbB receptors in that binding of the ligands neuregulin (NRG) and heparin-binding epidermal growth factor (HB-EGF) induces proteolytic cleavage of the 80 kDa HER4 kinase domain, termed the s80 or soluble cleavage product, and binding of s80 to the transcription factor (TF) STAT5a [22]. The s80-STAT5a complex then translocates to the nucleus to regulate expression of genes involved in mammary cell differentiation pathways, including the milk protein genes β-casein and whey acidic protein (WAP) [23–25], see Figure S1, supplementary information. Hence studies are underway to determine the molecular pathways that are stimulated by the soluble HER4 protein [20, 23, 26, 27], particularly the network of transcriptional regulatory elements that are activated upon nuclear translocation of HER4 and STAT5a [24, 25, 28, 29]. Delineation of the signaling network associated with HER4/STAT5a activity would enable the exploitation of the pathway for targeting of malignant cells. Specifically, activation of HER4 signaling in aggressive breast tumors would present a novel therapeutic approach to suppress growth of these malignancies.
Despite the unique role played by wild-type (WT) HER4 in certain tumor types, several somatic mutations in the kinase domain of HER4 were recently discovered (see Figure S2, supplementary information) in patients with breast, gastric, colorectal, and non-small cell lung cancer [30]. Tvorogov et al. [31] analyzed the basal and ligand-induced phosphorylation levels of the nine HER4 kinase domain mutants in various cell backgrounds, and found that two of the mutations, G802dup and D861Y, disrupt the catalytic activity of the HER4 kinase, whereas the remaining mutations do not appear to affect HER4 phosphorylation. The group discovered that, despite loss of kinase activity, the two mutant HER4 receptors were able to heterodimerize with HER2 and signal through the ERK and PI3K/AKT pathways. However, kinase activity of HER4 was required for ligand-induced activation of the STAT5a differentiation pathway, and as a result, the HER4 mutants were unable to activate STAT5a signaling or cell differentiation. Not only were the two mutants unable to induce cell differentiation, but when over-expressed in a breast cancer cell line, they actively suppressed the formation of differentiated acinar structures, in contrast to wild-type HER4, which induced differentiation. Thus, the HER4 mutants exhibit a selective loss-of-function phenotype, which is biased toward proliferative signaling pathways and against the cell differentiation pathway.
To explore the implications of HER4 activity for ErbB signaling and cell fate switching, a multiscale modeling approach is advantageous [32]. Multiscale computational modeling has been applied to a variety of biological systems [33–39] to help quantify the complexity inherent in intracellular signaling networks. As the biochemical processes within a cell occur on multiple spatial and temporal scales, a multiscale modeling approach is necessary to represent a hierarchy of interactions ranging from the molecular (nm, ns) to cellular signaling (μm, ms) length and time scales. Multiscale modeling provides a powerful and quantitative methodology for studying the effects of molecular perturbations, in our case, HER4 somatic mutations, on the cell phenotype, i.e., the decision between two cellular fates, proliferation and differentiation.
Given the tumor inhibitory role performed by HER4 in certain contexts and the recent interest in exploiting the HER4 pathway therapeutically, we pursue a multiscale modeling study of the HER4 kinase system at the molecular and cellular levels. A variety of modeling techniques, ranging from atomic-level molecular dynamics (MD) simulations to cellular-level modeling, are applied to investigate the WT activation mechanism of the HER4 kinase and the physiological relevance of this activity to the selection of divergent cellular signaling pathways. We begin by simulating and analyzing a molecular model of the WT HER4 homodimer in order to elucidate molecular mechanisms of activation in the WT kinase. We then apply the results of our WT simulations to help rationalize the effects of the clinically-identified HER4 somatic mutants on the cell phenotype. The results of our molecular-scale simulations support the hypothesis that the HER4 mutants may heterodimerize but not activate, resulting in blockage of the HER4-STAT5 differentiation pathway, in favor of the proliferative PI3K/AKT pathway. Our molecular simulation results of HER4 kinase activity are then translated into a cellular pathway model of WT versus mutant HER4 signaling.
2. Materials and Methods
2.1 Molecular dynamics simulations of the HER4 homodimer
The HER4 kinase homodimer system was modeled on the crystal structure of the HER4 dimer published by Qiu et al. [40] using MODELLER [41]. The receiver kinase was modeled as a HER4 monomer in the inactive conformation, and the activator kinase as a HER4 monomer in the active conformation, see Figure 1. We also constructed the mutant structures in MODELLER [42], using our equilibrated WT HER4 structure as the template. Energy minimization was performed in MODELLER to remove any unfavorable contacts, and nearest neighbor amino acids were allowed to optimize their positions.
Figure 1.

(A) Schematic of ligand-mediated dimerization of extracellular and intracellular domains in ErbB kinases. (B) Molecular model of the asymmetric dimer of the HER4 kinase domain. (C) List of bonds formed or broken during the course of the TMD simulation of HER4/ErbB4 activation. (D) Principal Component Analysis (PCA) of the HER4 dimer. Superimposing several frames from the each PCA trajectory, we depict conformational fluctuations along the first principal component. For clarity, only the key regions, namely, A-loop, P-loop, C-loop, and αC helix of the receiver kinase are depicted. That is, we take the principal component (which is a 3N dimensional vector) and only show its projection relevant a sub-dimensional vector involving the alpha-C-helix, A-loop, P-loop, N-loop atoms of the kinase undergoing activation. The structures are colored according to the RMSD, where blue regions indicate small fluctuations and red regions indicate larger fluctuations. The fluctuations are quantified according to the scale bar at the top.
Based on the asymmetric dimer interface observed in both the EGFR and HER4 crystal structures [40, 43], we constructed a HER4 wild type homodimer system in which an inactive HER4 monomer serves as the receiver kinase and an active HER4 monomer serves as the activator kinase (see Figure 1A,B). The dimer interface residues comprise portions of the C-lobe in the activator kinase and segments of the N-lobe in the receiver kinase. Recently, the juxtamembrane (JM) domain, or the 37-residue segment connecting the kinase domain to the ErbB transmembrane domain, has been shown to play an important role in the complete activation of the ErbB RTKs [44, 45]. Although the molecular mechanism underlying the activating role of the JM region is unknown, a recent crystal structure of the EGFR kinase dimer [44], which includes the complete JM domain, revealed that the JM region in the receiver kinase makes extensive contacts with the C-lobe of the activator kinase, forming a latch that ‘cradles’ the activator kinase to stabilize the asymmetric dimer interface. There have been several reported studies of the EGFR dimer [46, 47]; however, none have included the JM domain. Not only do we report the first study of the HER4 dimer, our study is also the first to include the crucial JM region.
The structures were minimized to remove unfavorable contacts and hydrogen atoms were added using the hbuild routine in CHARMM [48]. The dimer was explicitly solvated using the TIP3P model for water [49] and with the buffering distance set to 15 Å for a total system size of approximately 120,000 atoms. Sodium (Na+) and chloride (Cl−) ions were added to achieve net electroneutrality of the system and an ionic strength of 75 mM. All Na+ and Cl− ions were placed at least 8 Å away from any protein atoms and from each other. Constant pressure and temperature (NPT) simulations were performed at 300 K and 1 atm. to equilibrate the volume of the solvation box. Temperature and pressure were maintained using a Langevin piston coupling algorithm [50]. Following the NPT simulations, constant volume and temperature (NVT) simulations were performed in NAMD. Finally, a 10 nanosecond production run was completed using the same parameters as in the NVT simulations. A second dimer, in which the receiver kinase is in the active form, was constructed to serve as the target structure for the TMD simulations. This dimer was also simulated (NVT) for 10 ns. All simulations were performed in NAMD [51] using CHARMM27 force-field parameters.
2.2 Targeted molecular dynamics (TMD) simulations
TMD simulations, which were implemented in NAMD [51], were applied to accelerate the conformational change of the HER4 dimer from the inactive to the active state. The root mean square deviation (RMSD) between the current structure and the target structure was used to calculate a steering force, which was applied to every atom in the chosen molecular subset, and is calculated as follows:
RMSD(t) represents the RMSD, for a chosen molecular subset, between the current structure and the reference target structure at simulation time t. RMSD0(t) is the target RMSD value at simulation time t. The force constant, k, was deliberately chosen to be low so as not to overwhelm the thermal fluctuations, and tested for values ranging from 0.5–20 kcal/mol/Å2, and ultimately, a value of 20 was chosen; see section 2.3, below. N represents the number of atoms included in the target subset. During optimization of the TMD simulations, a variety of molecular subsets were tested, including all backbone atoms of the receiver kinase or of the catalytic site only. Ultimately, a subset, which included the heavy atoms of the catalytic site, was chosen. TMD simulations were run for 25 ns with a time step of 2 fs. The conformational transition from the initial to the target structure was determined by decreasing the value of RMSD0(t) as a function of the simulation time. The value of RMSD0(t) was linearly decreased to 0 Angstroms during the 25 ns TMD simulation. Upon reaching the target structure, MD simulations were run to allow the system to fully relax into the target conformation.
2.3 Optimization of TMD simulation restraints
As the A-loop and αC helix in the receiver kinase undergo the most pronounced conformational shifts during the transition from the inactive to the active state, we included these two catalytic sub-domains in our set of TMD restraints. Previous TMD studies of the EGFR kinase [46, 47] have focused on conformational changes in these sub-domains; however, other regions of the kinase may also be important in the activation mechanism. To incorporate a more global view of the allosteric activation mechanism, we extended our list of TMD restraints to include the entire catalytic site in the receiver kinase. To avoid over-constraining the system, we allowed the remainder of the kinase, including the JM domain, to move freely. Additional degrees of freedom were allowed in that the activator kinase was completely free to move, as it provides the activation stimulus but does not itself undergo any well-defined conformational changes.
We experimented with several different sets of restraints for the TMD simulations. The spring constant used to apply force to the dimer system was varied from k=0.5–20 kcal/mol/Å2. In addition to optimizing the force constant, we varied the set of targeted atoms. Specifically, we tested the following sets of target atoms: all backbone atoms of the receiver kinase, heavy atoms of the A-loop and αC helix, and alpha carbons, backbone atoms, or heavy atoms of the catalytic site. Restraining the heavy atoms of the catalytic site produced the most successful result in terms of achieving the target kinase conformation. We also tested the application of restraints to the JM domain, but as it is a protein terminus in our simulations, this resulted in an over constrained system. The TMD simulation time was varied from 2–25 ns. The longer the simulation and the smaller the force constant, the more accurate the results, and therefore we tested several different combinations of simulation time and force constant. We found that the minimum force constant required for our large dimer system (approximately 120,000 atoms) was k=5 kcal/mol/Å2, which, following a 10 nanosecond TMD simulation, shifted the dimer by 1 Angstrom toward the target conformation. Application of a slightly larger force constant, k=10 kcal/mol/Å2, resulted in a small improvement, shifting the dimer by about 2 Angstroms toward the active conformation. However, in running these simulations longer than 10–12 ns, we did not see an improvement in the RMSD, suggesting that, using a force constant of 5–10 kcal/mol/Å2, a prohibitively long simulation is required for such a large dimer system.
A force constant of k=20 kcal/mol/Å2 proved to be the minimum value required to achieve the target conformation in a reasonable simulation time, 25 ns. To gauge whether the dimer had achieved its active state, we calculated the RMSD with respect to the active dimer (target structure) and also analyzed the pattern of hydrogen bond changes during the TMD simulation. For the k=20 kcal/mol/Å2, 25 ns simulation, the final RMSD of the backbone atoms of the receiver kinase with respect to the target structure was approximately 1.2 Å, whereas the receiver had moved approximately 3.6 Å away from the starting (inactive) conformation. Due to the flexible nature of the A-loop and several other loop regions in the kinase, we did not aim to attain an RMSD of 0 Å. Rather, we focused on the key changes in the hydrogen bonding pattern and the catalytic sub-domain conformations. We also ensured that the A-loop was in its open, extended form, allowing for the binding of peptide and ATP.
2.4 Analysis of the TMD trajectories
Several types of analyses were performed on the TMD trajectories. Hydrogen bonding analysis was performed to define those bonds that broke or formed during the course of the TMD simulation. Hydrogen bonds were defined by a bond length cutoff of 3.4 Å and an angle cutoff of 150°. Bonds that fulfilled these criteria and were present in at least 60% of the trajectory were tabulated in CHARMM. Salt bridges were defined as hydrogen bonds occurring between an acidic and a basic residue and satisfying a bond length cutoff of 1.6 Å. All hydrogen bonds and salt bridges were also visualized in VMD [52] for the duration of the 10 ns simulation. Principal component analysis (PCA) of the MD trajectories was performed in CARMA [53]. During PCA, the frames of the MD simulations were aligned with respect to the backbone Cα atoms and the covariances of positional fluctuations were computed.
Ramachandran plots were constructed for key residues in order to track any changes in the dihedral angles phi and psi. The phi and psi angles were computed for the specified residues for each frame of the TMD trajectory, using VMD. The residues chosen included R841, L843, S749, and F872, as these residues undergo bonding changes during the transition to the active conformation.
RMSD values were measured for the dimer undergoing TMD, with respect to the active (target) structure and the inactive (starting) structure. The RMSD was measured with respect to the backbone atoms of the catalytic site, and also with respect to the entire receiver kinase, to gauge the progression of the TMD system along the activation pathway.
2.5 Network model of WT and mutant HER4 signaling pathways
The computational signaling model was based on modules from two published signaling models: the PI3K/AKT signaling branch was based on the model by Schoeberl et al. [54], and the JAK2/STAT5a signaling branch was based on the model by Yamada et al. [55]. Mass-action reactions describing ligand-induced ErbB receptor homo- and heterodimerization, receptor internalization and degradation, constitutive dimerization, and activation were included. The PI3K/AKT model was modified to include HER4 dimers (the original model excludes HER4, as HER4 expression levels are low in the cancer cell lines on which the model is based). Hence all four ErbB RTKs are included in the model, and allowed to dimerize in response to stimulation with the ligands neuregulin-1β (NRG-1β) and HB-EGF. The WT HER4 kinase was assumed to preferentially induce the JAK/STAT5 pathway via HER4 homodimers, whereas the mutant HER4 kinase was assumed to preferentially induce the PI3K/AKT pathway via HER4 heterodimers. Levels of phosphorylated ppAKT and pSTAT5a nuclear dimers were considered as read-outs of pathway activation for mutant and WT HER4 dimers, respectively.
The PI3K/AKT pathway has been previously well characterized and modeled, whereas the HER4-JAK-STAT pathway has not (to our knowledge) been modeled, and thus we delineate the major events here. Upon binding to NRG, HER4, which can basally associate with JAK2, as has been shown experimentally [26], homodimerizes and becomes phosphorylated. The HER4-JAK2 complex then binds and activates STAT5a, which becomes phosphorylated and dimerizes. We incorporate JAK2 into the model in this way to reflect that it is required for the HER4-mediated activation of STAT5a, though the mechanism is currently unknown. The HER4-STAT5a dimer then cleaves from the membrane and translocates to the nucleus; this event is represented in the model to reflect that STAT5a is unable to accomplish nuclear translocation without HER4 as a chaperone [23]. STAT5a and HER4 dissociate in the nucleus, where STAT5a induces expression of various genes. The only gene expression event explicitly represented in our model is the expression of SOCS, which, upon translation into SOCS protein, inhibits the JAK-STAT phosphorylation event in the cytoplasm. We note that there are a total of four negative regulators present in the JAK-STAT signaling branch, including SOCS, SHP-2, which dephosphorylates the HER4-JAK2 complex, PPX, which represents the cytoplasmic STAT5a phosphatase, and PPN, which represents the nuclear STAT5 phosphatase [55]. The network model is available in the SBML format as part of the supplementary information.
All simulations were performed in MATLAB 7.10 (MathWorks, Natick, MA). Parameter sensitivity analysis was performed by computing the normalized sensitivity, Sij, [56] defined by:
where Oi is the time-integrated (over a 4 h period) response of the ith model output (such as pAKT levels) and pj is the jth parameter (rate constant or initial condition).
3. Results
3.1 Simulation of the HER4 homodimer
To explore the molecular activation mechanism of the WT HER4 kinase, we performed atomic-level simulations of the HER4 kinase crystal structure. We ran 10 nanoseconds of molecular dynamics simulation on the HER4 dimer in the inactive state, as well as on a second HER4 dimer system representing the activated state. The purpose of these simulations was to provide a frame of reference for the analysis of bond patterns (see Figure 1C). No significant motions were revealed in either control simulation, although the active dimer was more stable than the inactive dimer. A PCA of the system trajectories revealed that the inactive dimer does move slightly toward the active conformation, in that there is a dominant shifting motion of the αC helix into the active site (Figure 1D). As in our previous simulations of the EGFR dimer [57], the dimer interface provides a stimulus to shift the inactive receiver kinase (mainly, the αC helix) away from the inactive conformation and toward the active state. The activation loop (A-loop), which is not positioned directly in the dimer interface, does not globally shift during the control simulations, and most likely requires other perturbations in order to transition to its open, extended state. Within the short time scale of our MD simulations (on the order of nanoseconds), the presence of the dimer interface is insufficient to fully activate the receiver kinase, an event which, in actuality, would occur on the order of milliseconds to seconds.
3.2 Analysis of global changes during the TMD simulation
To delineate the sequence of molecular events involved in the activation of the HER4 dimer, we undertook targeted molecular dynamics (TMD) simulations, which are used to steer a structure from one conformation to another, in our case, from the inactive to the active conformational state of the HER4 dimer (see Methods). The TMD trajectory was analyzed using several methods. First, global changes in the HER4 dimer system were assessed to determine the sequence of molecular events required to achieve the target conformation. Figure 2(A,B) tracks the RMSD of the backbone atoms of the A-loop and αC helix during the progression of the 25 ns TMD simulation. Relative to the active (target) structure, the A-loop moves from an RMSD of approximately 14 to 4 Å, and relative to the inactive (starting) structure, it moves from an RMSD of 1 to 12 Å (Figure 2B). The αC helix shifts from 4 to 2 Å away from the active structure, and from 1 to 4 Å away from the inactive structure. Interestingly, the αC helix becomes closer to the active conformation (relative to the inactive conformation) at approximately 18 ns, whereas the A-loop becomes closer to the active conformation a bit later, at 21–22 ns (Figure 2B). This order of events, i.e., the αC helix shifting before the A-loop, can partially be rationalized by the location of the αC helix within the dimer interface, such that it is expected to be directly impacted by the dimer interface stimulus. We have also observed this sequence of molecular events in our previous studies of the EGFR dimer [57]. This mechanism may be unique to the ErbB kinases in that other kinase families, such as Src, are thought to undergo the reverse order of events (A-loop moves before αC helix [58]).
Figure 2.

Transition of the HER4 homodimer system during the TMD simulation. The RMSD is plotted for (A) all backbone atoms of the receiver kinase, and (B) the backbone atoms of the A-loop and αC helix, with respect to the active (target) structure (blue) and with respect to the inactive (starting) structure (red). Results are shown for the simulation in which t=25ns and k=20 kcal/mol/Å2. (C) Bond distance for the E743-R841 salt bridge during the TMD simulation. Top panel displays the bond distance in the inactive dimer (10ns MD control simulation), in which the bond spontaneously breaks and forms during the course of the simulation. Bottom panel displays the bond distance in the 25ns TMD simulation; at t=18ns, the bond breaks. (D) Ramachandran plots for key residues during the 25ns TMD simulation: (i) R841, (ii) L843, (iii) S749, and (iv) F872. Each data point marks a time point in the TMD simulation; this way, any flipping of the dihedral angles can be detected.
3.3 Analysis of local interactions during the TMD simulation
In order to translate the global pattern of sub-domain motion that we observed for the HER4 TMD system into changes in specific, or local, interactions, we next analyzed the TMD trajectory for hydrogen bonds and salt bridges which may have broken or formed during the conformational transition to the active state (see Methods). As the pattern of hydrogen bonds maintaining the inactive state differs significantly from the bond pattern in the active state, we expect to observe these changes during the course of the TMD. Figure 1C summarizes all bonds, which have either broken or formed during the transition from the inactive to the active conformation. The E743-K726 salt bridge, which is conserved among the active ErbB kinases and is required for the coordination of the α and β phosphates of ATP, represents one of the key bonds which must form during kinase activation. In the inactive state, there are two residues which sequester E743 and K726, therefore autoinhibiting kinase activation by preventing formation of the key salt bridge. These two sequestering bonds are D836-K726 and R841-E743, which must break during the activation process. Indeed, in our TMD simulation, these two bonds have broken, freeing the E743 and K726 residues to form the crucial salt bridge (Figure 1C and Figure 2C). Figure 2C tracks the change in bond length for the R841-E743 sequestering bond during the course of the TMD simulation. In comparison to the 10 ns MD control simulation, in which the R841-E743 bond spontaneously forms and breaks during the course of the simulation, the bond successfully breaks (at t=18 ns) during the 25 ns TMD simulation. The Ramachandran plot in Figure 2D(i) tracks the change in the backbone dihedral angles for the R841 residue (see Methods), revealing that the angle phi flips from a small negative value to a large positive value during the conformational transition, facilitating breakage of the R841-E743 bond. Other interactions which are severed during the TMD simulation include E730-K856, which allows the E739-K856 bond (between the A-loop and αC helix) to form, and the E847-R870 bond in the A-loop.
Another important interaction is L843-R813, which does not appear in the inactive dimer, but forms during the TMD simulation. This bond couples the A-loop and C-loop in the active state, and helps to maintain the A-loop in its open, extended form. In fact, several bonds which bridge the A-loop and C-loop occur in the active kinase, and can be considered ‘fastening bonds’ in that they stabilize the A-loop in its activated conformation [59, 60]. Several of these interactions were observed to form during our TMD simulation. Figure 2D(ii) depicts the Ramachandran plot, which tracks any changes in the backbone dihedral angles for the L843 residue. It is apparent from the plot that the angle phi flips from a small negative value to a large positive value during the course of the TMD simulation, facilitating the formation of the L843-R813 fastening bond.
In addition to the bonding pattern in the receiver kinase changing in response to the TMD restraints, several inter-kinase bonds (i.e., bonds between the receiver and activator kinases) also broke during the 25 ns TMD simulation. Specifically, the D742-K917 bond, which occurs between the αC helix of the receiver kinase and the C-lobe of the activator kinase, is significantly weakened. The S764-E896 bond fully breaks during the TMD simulation. Such inter-dimer bonds have not been explored in previous TMD simulations of the EGFR kinase, yet may be worthwhile to investigate further, as previously-overlooked interactions between regions of the receiver and activator monomers may play a key role in kinase activation.
The bonding pattern in the JM region of the receiver kinase also shifts during the course of the TMD simulation. The following bonds change: N681-S749 is weakened, E690-S764 forms, and K689-E715 and E692-W712 break. One bond, L685-Q903, forms between the JM domain of the receiver kinase and the C-lobe of the activator kinase. In addition, Figure 2D(iii) displays the Ramachandran plot for S749, revealing that the phi angle flips from a negative to a positive value. Cross-referencing these JM residues with the list of JM amino acids shown to abolish EGFR phosphorylation in the scanning alanine mutagenesis assay performed by Red Brewer et al. [44], we discovered significant overlap. In particular, N681 (N676 in EGFR) and L685 (L680 in EGFR) both participate in JM interactions that are altered during our HER4 TMD simulation, and mutation of the analogous residues in EGFR was shown to abrogate kinase activity [44]. Thus our results for the HER4 dimer are in agreement with the experimental results, and highlight from a structural perspective the importance of the JM region as a key component of the activation mechanism in the ErbB kinases (at least, in EGFR and HER4).
3.4 Rationalizing the molecular effects of the HER4 somatic mutations
As discussed in the Introduction, two of the nine clinically identified HER4 somatic mutants (see Figure S2) exhibit a selective loss-of-function phenotype in terms of the downstream signaling pathways that are preferentially activated. To provide insight into the molecular mechanisms by which the HER4 mutants exert their preferential signaling effects, we assessed the mutants D861Y (biochemical numbering is D836 and will be used henceforth) and G802dup (biochemical numbering is G777) in the framework of our WT HER4 simulations. The D836Y mutation is located in the DFG motif, a highly conserved amino acid segment which is positioned at the N-terminal end of the A-loop in many eukaryotic kinases, and is critical for efficient phosphotransfer. Based on our WT HER4 simulations and our mutant D836Y structure, it is apparent that in the WT kinase, D836 forms a crucial set of interactions with N823 and D818 (C-loop), the catalytic aspartic acid which deprotonates the hydroxyl group of the substrate tyrosine residue. The bulky tyrosine side chain in the D836Y mutant appears to disrupt these interactions, which are key in orienting the peptide substrate for phosphoryl transfer; thus the catalytic activity of the kinase is predicted to be abrogated. Furthermore, in our TMD simulation of the WT HER4 dimer, D836 sequesters the key activating salt bridge by bonding with K726, an interaction which is released in the active kinase. Thus D836 serves important roles in both the inactive and active kinase conformations.
G777 forms part of the ATP-binding pocket in the HER4 kinase. Our structural mutant model revealed that the G777dup mutation perturbed the relative orientation of the energy-minimized neighboring residues, including H776, C778, and L779. Furthermore, other neighboring residues which were perturbed played key roles in our TMD simulation of the WT kinase. In particular, L773, which contributes to the ATP-binding pocket and is displaced in the G777dup mutant, bonds with P722 during the TMD simulation of the WT HER4 kinase. Although ATP is not explicitly included in our models, the disruption of the ATP-binding pocket introduced by the G777dup mutation is expected to significantly impair the proper docking of ATP into the catalytic site. Thus our simulations help to predict, at molecular resolution, the specific structural perturbations which are likely to be introduced by the D836Y and G777dup mutants.
We also examined the two mutations in the context of the HER4 dimer interface. Our WT simulations confirmed that the mutations are unlikely to disrupt dimerization of HER4 directly, due to their position relative to the simulated dimer interface. The message supported by our simulations is that the mutants abolish kinase activity but not dimerization of HER4, a hypothesis which is underscored by the experiments and modeling of Tvorogov et al. [31]. Although the effects of these mutants may be predicted through analysis of the crystal structure of the HER4 homodimer, this gives only a static picture of the impact of such perturbations. Our HER4 simulations provide a dynamic picture of the crucial roles played by D836 and G777 in the WT kinase activation mechanism.
3.5 Branched signaling model of WT versus mutant HER4 pathways
Due to the disrupted kinase activity of the HER4 mutants, they are unable to activate STAT5a, which requires HER4 kinase activity. Thus, in the inactivating HER4 mutant systems, there is a bias toward the cellular proliferative signaling pathways, which are induced by HER4 heterodimers. In order to capture this divergence in downstream signaling pathways between the WT and mutant HER4 kinases, we constructed a branched HER4 signaling model to explore under what conditions each respective pathway is selected (Figure 3). The purpose of our model is to explore two signaling branches of the HER4 signaling model. In our network model, the PI3K/AKT cascade is stimulated by ErbB heterodimers with mutant HER4 as well as WT HER4 homodimers. Whereas, the JAK2/STAT5a cascade is only induced by WT HER4 homodimers. Figure 4A displays time course plots for several model species, including phosphorylated HER4-JAK2 dimers, cytoplasmic STAT5a, phosphorylated nuclear STAT5a dimers, and cytoplasmic SOCS mRNA, in response to stimulation with 5 nM NRG. It is apparent that levels of pHER4-JAK2 peak at approximately 30 minutes post-ligand stimulation, and levels of phosphorylated nuclear STAT5a dimer peak at approximately 1 hour post-stimulation. Due to the time lag during which STAT5a translocates to the nucleus and induces expression of SOCS mRNA, levels of cytoplasmic mRNA do not peak until approximately 2 hours post-induction. The damped oscillations in the temporal responses in ppAKT and pSTATn are due to the negative feedback loops featuring the respective phosphatase molecules.
Figure 3.

Schematic of the HER4 signaling model, highlighting the two divergent pathways: PI3K/AKT and JAK2/STAT5a. HER3 is used to illustrate the activation of the PI3K/AKT pathway, but other ErbB dimer combinations may also activate PI3K/AKT. Only HER4 homodimers can activate the JAK2/STAT5a pathway.
Figure 4.
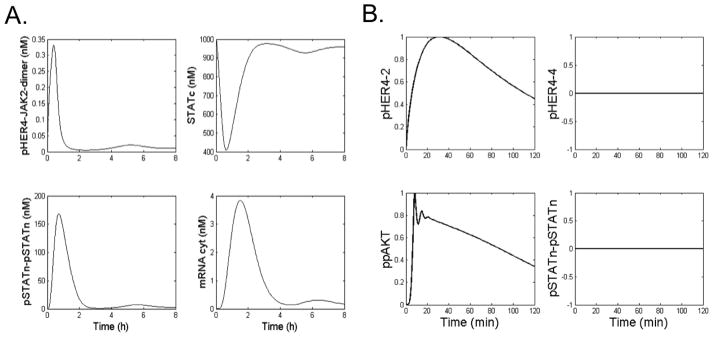
(A) Time courses for key species in the WT HER4-JAK-STAT signaling model, including phosphorylated HER4-JAK2 dimer (pHER4-JAK2-dimer), cytoplasmic STAT5 (STATc), phosphorylated nuclear STAT5a dimers (pStatn-pStatn), and cytoplasmic SOCS mRNA, in response to stimulation with 5 nM NRG. (B) Time courses for key species in the mutant HER4-JAK-STAT model, including phosphorylated HER4-2 dimers (pHER4-2), phosphorylated HER4 homodimers (pHER4-4), doubly phosphorylated AKT (ppAKT,) and phosphorylated nuclear STAT5a dimers.
In response to an in silico cell expressing WT HER4, the activation of HER4 homodimers and the JAK2/STAT5a signaling branch is increased. Thus the JAK2/STAT5a cascade becomes the predominant signaling pathway. In direct contrast, in response to expression of mutant HER4, signaling through phosphorylated HER4 homodimers is essentially turned off, and PI3K/AKT is the predominant pathway induced by activated ErbB dimers (Figure 4B). The model recapitulates the major features of PI3K/AKT and JAK/STAT signaling dynamics, including the importance of the STAT5 nuclear phosphatase in governing both peak and steady state levels of nuclear pSTAT dimer. Our model is also the first (to our knowledge) to explicitly incorporate the HER4-JAK-STAT signaling branch. Specifically, the mutants signal through the proliferative ERK and PI3K/AKT pathways but not through the STAT5a differentiation pathway.
3.6 Parameter sensitivity analysis of the HER4 signaling model
To identify the key proteins that direct signaling in our branched signaling model, parameter sensitivity analysis was performed with respect to phosphorylated AKT (ppAKT) and nuclear phosphorylated STAT5a (pSTAT5a). Figure S3(A), supplementary material, displays the normalized sensitivity of ppAKT to various species in the model, and it is apparent that HER2, HER3, and HER4 represent the most sensitive species in the signaling network, as they can all equally heterodimerize to activate the AKT pathway. EGFR is not a strong determinant of the extent of AKT phosphorylation, as expected from the weak ability of NRG-1β to elicit EGFR dimers. PTEN (the PIP3 phosphatase), PP2A, and the ErbB phosphatase exhibited a negative sensitivity in the analysis, as these phosphatases negatively regulate the signaling network through dephosphorylation of key molecular species. JAK2 also exhibited a negative sensitivity, suggesting that an increase in JAK2 expression would dampen AKT signaling. This result can be rationalized in that JAK2 competes with the AKT pathway (specifically, with PI3K) for binding to HER4 kinase.
Figure S3(B) displays the normalized sensitivity of phosphorylated nuclear STAT homodimers (pSTATn_pSTATn) to the model species. STATc (cytoplasmic STAT5a) and JAK2 expectedly represent the most sensitive species. EGFR, HER2, and HER3 are not sensitive, as only HER4 can stimulate the JAK2/STAT5a pathway. Interestingly, HER4 is not a very sensitive species, as it is in excess compared to JAK2 levels (i.e., JAK2 is the limiting species and thus exhibits high sensitivity). However, if we decrease the initial concentration of HER4 such that it is below that of JAK2, the HER4 sensitivity increases to a normalized value of 0.59 (Figure S3(C)), while the JAK2 sensitivity decreases considerably, as it is no longer the limiting species. Thus the ratio of HER4:JAK2 levels, which varies among different cell types, is crucial, as is the ratio of HER4:PI3K.
4. Discussion and Conclusion
In this work, two modeling techniques, namely, atomic-level MD simulations and network modeling simulations, were applied to investigate the WT activation mechanism of the HER4 kinase and the physiological relevance of this activity to the selection of divergent cellular signaling pathways. A molecular model of the WT HER4 homodimer was first simulated in order to elucidate molecular mechanisms of activation in the WT kinase. We then applied the results of our WT simulations to help rationalize the effects of the clinically identified HER4 somatic mutants on the cell phenotype.
The WT activation pathway for the HER4 kinase was first analyzed through simulations of a HER4 homodimer system. The simulations highlighted key molecular interactions which are predicted to be important in the HER4 activation mechanism. We discovered that the order of events, in terms of motions of the individual catalytic sub-domains, differed from those of other kinase families such as Src, a distinction which may partially be attributed to the fact that phosphorylation of the A-loop tyrosine residue is not required for ErbB kinase activation. In addition to delineating global motions and hydrogen bonding events taking place within the activated kinase, we also identified several inter-kinase bonds (i.e., bonds between the receiver and activator kinases) which broke during the TMD simulation. Moreover, the bonding pattern in the JM region of the receiver kinase shifted during the course of the TMD simulation, a result which is in agreement with previous experimental studies demonstrating that the JM domain is a crucial contributor to the kinase allosteric activation mechanism [44, 45]. These inter-kinase and JM domain bonds have not been explored in previous simulations of the EGFR kinase, yet our results indicate that they warrant further investigation, in order to piece together the complete activation pathway of the ErbB kinases.
We applied the results of our WT analysis to rationalize the effects of two clinically reported HER4 somatic mutations, D836Y and G777dup. Specific residues perturbed by the mutations were cross-referenced with bonds that broke or formed during our TMD simulations of the WT HER4 kinase, underscoring the importance of D836 and G777 in the activation of the WT kinase. Our results confirmed the effects of the mutants on disruption of the catalytic site and impairment of kinase activity. However, our WT simulations confirmed dynamically and allosterically that the mutations are unlikely to disrupt dimerization of HER4 directly, due to their position relative to the simulated dimer interface. This observation of impaired activation but not dimerization in the mutant systems underscores the difference between the wild type and mutant systems at the network level. It is noteworthy that the HER4 mutants are unique among the ErbB kinases in that similar mutations have been reported in EGFR and HER2, yet these mutations confer a gain-of-function phenotype [61, 62]. Specifically, tumors expressing these mutant EGFR and HER2 RTKs demonstrate increased ErbB phosphorylation and kinase activity. In addition, these gain-of-function mutations in EGFR and HER2 confer sensitivity of tumors to specific kinase inhibitors [10, 63–65], suggesting that these tumors have become addicted to signaling through the mutant ErbB kinases. By contrast, similar mutations in HER4, which we have analyzed in this work, result in a selective loss-of-function phenotype by abolishing HER4 kinase activity but not dimerization. These HER4 mutants do not demonstrate sensitivity to specific kinase inhibitors [31], as do the analogous EGFR and HER2 mutants.
As HER4 may represent the only member of the ErbB family of RTKs to play an anti-cancer role in certain tumor contexts, it is crucial to quantitatively evaluate the signaling dynamics of the HER4 pathway in the cell environment. Thus we translated the results of our structural studies of the WT HER4 kinase, which corroborated that the HER4 somatic mutants abrogate the ability of HER4 to activate but not dimerize, into a cell signaling model of HER4 activity, which branches into two different pathways: the JAK2/STAT5a network (induced by WT HER4) and the PI3K/AKT cascade (induced by mutant HER4). We find that HER4 homodimers predominantly induce the JAK2/STAT5 pathway, although in an in vivo context, it is likely that a combination of hormones and ligands, including prolactin and hydrocortisone, contributes to the preferential stimulation of HER4 homodimers rather than other ErbB dimer combinations, in response to ligands such as NRG (which can also bind to HER3). Our model is able to recapitulate the major features of the PI3K/AKT and JAK/STAT pathways, yet is the first, to our knowledge, to explicitly incorporate the HER4-JAK-STAT signaling axis as well as the unique events occurring in the HER4-STAT5a pathway, including translocation of the activated kinase into the nucleus to effect gene transcription.
Additional experiments in the HER4-stimulated mammary epithelial cells are necessary in order to delineate the role of crosstalk between the HER4 pathway and other signaling cascades, including the glucocorticoid receptor and prolactin pathways, in promoting the HER4-induced differentiation signal, emphasizing that these synergistic networks must be taken into account when considering therapeutic modulation of the HER4 pathway. We suggest that HER4-mediated STAT5a activation may constitute a cellular shift from a program of proliferation to a program of differentiation: precisely the same shift that can potentially be modulated to bias tumor cells toward the HER4 differentiation pathway for therapeutic purposes.
Based on our hypotheses regarding the HER4 mutants, parallels can be drawn to themes which emerged from our previous multiscale modeling studies of the HER3 kinase [33, 60, 66, 67]. In particular, we have shown through our multiscale studies of the HER3 and HER4 kinases that ErbB kinases devoid of intrinsic kinase activity are not necessarily benign in terms of their role in cell signaling and induction of cancer. Indeed, although the kinase activity of the ErbB RTKs plays a crucial function in cell signaling, there are other, equally important roles performed by the ErbB kinases, which allow for participation in signaling despite lack of catalytic activity. In our previous study of the HER3 kinase, we found that, although HER3 exhibits relatively weak catalytic activity in comparison to the other ErbB family members, it plays a key role in the development of drug resistance to certain kinase inhibitors, due to its ability to synergize with other signaling processes in the cell and amplify its weak signal. In the case of HER4, the kinase-dead HER4 somatic mutants retain the ability to induce proliferative signaling, due to their ability to dimerize with other ErbB RTKs. Indeed, the various activities of the ErbB kinases complement each other to produce a holistically functioning signaling unit, compensating for any deficiencies in the individual ErbB proteins. In tumor cells predisposed to uncontrolled signaling, this otherwise advantageous evolutionary characteristic of the ErbB system can have deleterious consequences, and must be considered when designing therapeutics targeted to the ErbB family.
Supplementary Material
Acknowledgments
This work was supported in part by National Science Foundation Grants CBET-0853389, CBET-0853539, CBET-1133267, and CBET-1244507. The research leading to these results has received funding from the European Commission Grant FP7-ICT-2011-9-600841. Computational resources were provided in part by the National Partnership for Advanced Computational Infrastructure under Grant No. MCB060006 from XSEDE. S.E.T. was supported by the National Institutes of Health under Ruth L. Kirschstein National Research Service Award 2T32HL007954 from the NIHNHLBI, a National Science Foundation Graduate Research Fellowship, and a Graduate Assistantship in Areas of National Need (GAANN) from the Department of Education.
Abbreviations
- MD
Molecular Dynamics
- WT
wild type
- RTK
Receptor Tyrosine Kinase
- A-loop
Activation Loop
- TMD
Targeted Molecular Dynamics
- JM
Juxta Membrane
- RMSD
Root Mean-Squared Deviation
- PCA
Principal Component Analysis
- TF
Transcription Factor
Footnotes
Conflict of interest statement
The authors have no conflict of interest to declare.
References
- 1.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 2.Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 3.Sweeney C, Fambrough D, Huard C, Diamonti AJ, et al. Growth factor-specific signaling pathway stimulation and gene expression mediated by ErbB receptors. J Biol Chem. 2001;276:22685–22698. doi: 10.1074/jbc.M100602200. [DOI] [PubMed] [Google Scholar]
- 4.Schlessinger J. Cell Signaling by Receptor Tyrosine Kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 5.Hubbard SR, Till JH. Protein Tyrosine Kinase Structure and Function. Annual Review of Biochemistry. 2000;69:373–398. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- 6.Linggi B, Carpenter G. ErbB receptors: new insights on mechanisms and biology. Trends Cell Biol. 2006;16:649–656. doi: 10.1016/j.tcb.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 7.Schulze WX, Deng L, Mann M. Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol Syst Biol. 2005;1:2005 0008. doi: 10.1038/msb4100012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Birtwistle MR, Hatakeyama M, Yumoto N, Ogunnaike BA, et al. Ligand-dependent responses of the ErbB signaling network: experimental and modeling analyses. Mol Syst Biol. 2007;3:144. doi: 10.1038/msb4100188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi SH, Mendrola JM, Lemmon MA. EGF-independent activation of cell-surface EGF receptors harboring mutations found in gefitinib-sensitive lung cancer. Oncogene. 2007;26:1567–1576. doi: 10.1038/sj.onc.1209957. [DOI] [PubMed] [Google Scholar]
- 10.Ji H, Li D, Chen L, Shimamura T, et al. The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell. 2006;9:485–495. doi: 10.1016/j.ccr.2006.04.022. [DOI] [PubMed] [Google Scholar]
- 11.Lee JW, Soung YH, Kim SY, Nam SW, et al. ERBB2 kinase domain mutation in the lung squamous cell carcinoma. Cancer Lett. 2006;237:89–94. doi: 10.1016/j.canlet.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 12.Minami Y, Shimamura T, Shah K, LaFramboise T, et al. The major lung cancer-derived mutants of ERBB2 are oncogenic and are associated with sensitivity to the irreversible EGFR/ERBB2 inhibitor HKI-272. Oncogene. 2007;26:5023–5027. doi: 10.1038/sj.onc.1210292. [DOI] [PubMed] [Google Scholar]
- 13.Shigematsu H, Takahashi T, Nomura M, Majmudar K, et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005;65:1642–1646. doi: 10.1158/0008-5472.CAN-04-4235. [DOI] [PubMed] [Google Scholar]
- 14.Willmore-Payne C, Holden JA, Layfield LJ. Detection of epidermal growth factor receptor and human epidermal growth factor receptor 2 activating mutations in lung adenocarcinoma by high-resolution melting amplicon analysis: correlation with gene copy number, protein expression, and hormone receptor expression. Hum Pathol. 2006;37:755–763. doi: 10.1016/j.humpath.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 15.Alimandi M, Romano A, Curia MC, Muraro R, et al. Cooperative signaling of ErbB3 and ErbB2 in neoplastic transformation and human mammary carcinomas. Oncogene. 1995;10:1813–1821. [PubMed] [Google Scholar]
- 16.Da Silva L, Simpson P, Smart C, Cocciardi S, et al. HER3 and downstream pathways are involved in colonization of brain metastases from breast cancer. Breast Cancer Research. 2010;12:R46. doi: 10.1186/bcr2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang SE, Narasanna A, Perez-Torres M, Xiang B, et al. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell. 2006;10:25–38. doi: 10.1016/j.ccr.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 18.Anglesio MS, Arnold JM, George J, Tinker AV, et al. Mutation of ERBB2 Provides a Novel Alternative Mechanism for the Ubiquitous Activation of RAS-MAPK in Ovarian Serous Low Malignant Potential Tumors. Molecular Cancer Research. 2008;6:1678–1690. doi: 10.1158/1541-7786.MCR-08-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feng SM, Sartor CI, Hunter D, Zhou H, et al. The HER4 cytoplasmic domain, but not its C terminus, inhibits mammary cell proliferation. Mol Endocrinol. 2007;21:1861–1876. doi: 10.1210/me.2006-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muraoka-Cook RS, Feng SM, Strunk KE, Earp HS. 3rd, ErbB4/HER4: role in mammary gland development, differentiation and growth inhibition. J Mammary Gland Biol Neoplasia. 2008;13:235–246. doi: 10.1007/s10911-008-9080-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tidcombe H, Jackson-Fisher A, Mathers K, Stern DF, et al. Neural and mammary gland defects in ErbB4 knockout mice genetically rescued from embryonic lethality. Proceedings of the National Academy of Sciences. 2003;100:8281–8286. doi: 10.1073/pnas.1436402100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clark DE, Williams CC, Duplessis TT, Moring KL, et al. ERBB4/HER4 potentiates STAT5A transcriptional activity by regulating novel STAT5A serine phosphorylation events. J Biol Chem. 2005;280:24175–24180. doi: 10.1074/jbc.M414044200. [DOI] [PubMed] [Google Scholar]
- 23.Williams CC, Allison JG, Vidal GA, Burow ME, et al. The ERBB4/HER4 receptor tyrosine kinase regulates gene expression by functioning as a STAT5A nuclear chaperone. J Cell Biol. 2004;167:469–478. doi: 10.1083/jcb.200403155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kabotyanski EB, Huetter M, Xian W, Rijnkels M, Rosen JM. Integration of prolactin and glucocorticoid signaling at the beta-casein promoter and enhancer by ordered recruitment of specific transcription factors and chromatin modifiers. Mol Endocrinol. 2006;20:2355–2368. doi: 10.1210/me.2006-0160. [DOI] [PubMed] [Google Scholar]
- 25.Kabotyanski EB, Rijnkels M, Freeman-Zadrowski C, Buser AC, et al. Lactogenic hormonal induction of long distance interactions between beta-casein gene regulatory elements. J Biol Chem. 2009;284:22815–22824. doi: 10.1074/jbc.M109.032490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muraoka-Cook RS, Sandahl M, Hunter D, Miraglia L, Earp HS. 3rd, Prolactin and ErbB4/HER4 signaling interact via Janus kinase 2 to induce mammary epithelial cell gene expression differentiation. Mol Endocrinol. 2008;22:2307–2321. doi: 10.1210/me.2008-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muraoka-Cook RS, Sandahl M, Husted C, Hunter D, et al. The intracellular domain of ErbB4 induces differentiation of mammary epithelial cells. Mol Biol Cell. 2006;17:4118–4129. doi: 10.1091/mbc.E06-02-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amin DN, Perkins AS, Stern DF. Gene expression profiling of ErbB receptor and ligand-dependent transcription. Oncogene. 2004;23:1428–1438. doi: 10.1038/sj.onc.1207257. [DOI] [PubMed] [Google Scholar]
- 29.Amin DN, Tuck D, Stern DF. Neuregulin-regulated gene expression in mammary carcinoma cells. Exp Cell Res. 2005;309:12–23. doi: 10.1016/j.yexcr.2005.04.034. [DOI] [PubMed] [Google Scholar]
- 30.Soung YH, Lee JW, Kim SY, Wang YP, et al. Somatic mutations of the ERBB4 kinase domain in human cancers. International Journal of Cancer. 2006;118:1426–1429. doi: 10.1002/ijc.21507. [DOI] [PubMed] [Google Scholar]
- 31.Tvorogov D, Sundvall M, Kurppa K, Hollmen M, et al. Somatic Mutations of ErbB4. Journal of Biological Chemistry. 2009;284:5582–5591. doi: 10.1074/jbc.M805438200. [DOI] [PubMed] [Google Scholar]
- 32.Telesco SE, Radhakrishnan R. Structural systems biology and multiscale signaling models. Ann Biomed Eng. 2012;40:2295–2306. doi: 10.1007/s10439-012-0576-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Telesco SE, Shih AJ, Jia F, Radhakrishnan R. A multiscale modeling approach to investigate molecular mechanisms of pseudokinase activation and drug resistance in the HER3/ErbB3 receptor tyrosine kinase signaling network. Molecular Biosystems. 2011;7:2066–2080. doi: 10.1039/c0mb00345j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fedosov DA, Caswell B, Karniadakis GE. A multiscale red blood cell model with accurate mechanics, rheology, and dynamics. Biophys J. 2010;98:2215–2225. doi: 10.1016/j.bpj.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y, Purvis J, Shih A, Weinstein J, et al. A multiscale computational approach to dissect early events in the Erb family receptor mediated activation, differential signaling, and relevance to oncogenic transformations. Ann Biomed Eng. 2007;35:1012–1025. doi: 10.1007/s10439-006-9251-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rausenberger J, Hussong A, Kircher S, Kirchenbauer D, et al. An integrative model for phytochrome B mediated photomorphogenesis: from protein dynamics to physiology. PLoS One. 2010;5:e10721. doi: 10.1371/journal.pone.0010721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shih AJ, Purvis J, Radhakrishnan R. Molecular systems biology of ErbB1 signaling: Bridging the gap through multiscale modeling and high-performance computing. Mol Biosyst. 2008;4:1151–1159. doi: 10.1039/b803806f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vasalou C, Henson MA. A multiscale model to investigate circadian rhythmicity of pacemaker neurons in the suprachiasmatic nucleus. PLoS Comput Biol. 2010;6:e1000706. doi: 10.1371/journal.pcbi.1000706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Purvis J, Liu Y, Ilango V, Radhakrishnan R. Proc Foundations in Systems Biology II. IRB Verlag; Stuttgart: 2007. Efficacy of tyrosine kinase inhibitors in the mutants of the epidermal growth factor receptor: A multiscale molecular/systems model for phosphorylation and inhibition; pp. 289–294. [Google Scholar]
- 40.Qiu C, Tarrant MK, Choi SH, Sathyamurthy A, et al. Mechanism of activation and inhibition of the HER4/ErbB4 kinase. Structure. 2008;16:460–467. doi: 10.1016/j.str.2007.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fiser A, Sali A. Modeller: generation and refinement of homology-based protein structure models. Methods in Enzymology. 2003;374:461–491. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- 42.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 43.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 44.Red Brewer M, Choi SH, Alvarado D, Moravcevic K, et al. The Juxtamembrane Region of the EGF Receptor Functions as an Activation Domain. Molecular Cell. 2009;34:641–651. doi: 10.1016/j.molcel.2009.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thiel KW, Carpenter G. Epidermal growth factor receptor juxtamembrane region regulates allosteric tyrosine kinase activation. Proceedings of the National Academy of Sciences. 2007;104:19238–19243. doi: 10.1073/pnas.0703854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dixit A, Verkhivker GM. Hierarchical Modeling of Activation Mechanisms in the ABL and EGFR Kinase Domains: Thermodynamic and Mechanistic Catalysts of Kinase Activation by Cancer Mutations. PLoS Comput Biol. 2009;5:e1000487. doi: 10.1371/journal.pcbi.1000487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Papakyriakou A, Vourloumis D, Tzortzatou-Stathopoulou F, Karpusas M. Conformational dynamics of the EGFR kinase domain reveals structural features involved in activation. Proteins: Structure, Function, and Bioinformatics. 2009;76:375–386. doi: 10.1002/prot.22353. [DOI] [PubMed] [Google Scholar]
- 48.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. Journal of Physical Chemistry B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 49.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics. 1983;79:926–935. [Google Scholar]
- 50.Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation: The Langevin piston method. J Chem Phys. 1995;103:4613–4621. [Google Scholar]
- 51.Phillips JC, Braun R, Wang W, Gumbart J, et al. Scalable molecular dynamics with NAMD. Journal of Computational Chemistry. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 53.Glykos NM. Software news and updates. Carma: a molecular dynamics analysis program. J Comput Chem. 2006;27:1765–1768. doi: 10.1002/jcc.20482. [DOI] [PubMed] [Google Scholar]
- 54.Schoeberl B, Pace EA, Fitzgerald JB, Harms BD, et al. Therapeutically targeting ErbB3: a key node in ligand-induced activation of the ErbB receptor-PI3K axis. Sci Signal. 2009;2:ra31. doi: 10.1126/scisignal.2000352. [DOI] [PubMed] [Google Scholar]
- 55.Yamada S, Shiono S, Joo A, Yoshimura A. Control mechanism of JAK/STAT signal transduction pathway. FEBS Lett. 2003;534:190–196. doi: 10.1016/s0014-5793(02)03842-5. [DOI] [PubMed] [Google Scholar]
- 56.Zi Z, Zheng Y, Rundell AE, Klipp E. SBML-SAT: a systems biology markup language (SBML) based sensitivity analysis tool. BMC Bioinformatics. 2008;9:342. doi: 10.1186/1471-2105-9-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shih AJ, Telesco SE, Choi SH, Lemmon MA, Radhakrishnan R. Molecular dynamics analysis of conserved hydrophobic and hydrophilic bond interaction networks in ErbB family kinases. Biochemical Journal. 2011;436:241–251. doi: 10.1042/BJ20101791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang S, Banavali NK, Roux Bt. Mapping the conformational transition in Src activation by cumulating the information from multiple molecular dynamics trajectories. Proceedings of the National Academy of Sciences. 2009;106:3776–3781. doi: 10.1073/pnas.0808261106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Telesco SE, Radhakrishnan R. Atomistic insights into regulatory mechanisms of the HER2 tyrosine kinase domain: a molecular dynamics study. Biophys J. 2009;96:2321–2334. doi: 10.1016/j.bpj.2008.12.3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Telesco SE, Shih AJ, Liu Y, Radhakrishnan R. Molecular Simulation of Structure, Dynamics, and Function in the HER2 Receptor Tyrosine Kinase and Relevance to Cancer Mutations. Cancer Research Journal. 2011;4:1–35. [PMC free article] [PubMed] [Google Scholar]
- 61.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. New England Journal of Medicine. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 62.Paez JG, Janne PA, Lee JC, Tracy S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 63.Iyevleva AG, Novik AV, Moiseyenko VM, Imyanitov EN. EGFR mutation in kidney carcinoma confers sensitivity to gefitinib treatment: A case report. Urologic Oncology: Seminars and Original Investigations. 2009;27:548–550. doi: 10.1016/j.urolonc.2008.03.022. [DOI] [PubMed] [Google Scholar]
- 64.Mulloy R, Ferrand A, Kim Y, Sordella R, et al. Epidermal growth factor receptor mutants from human lung cancers exhibit enhanced catalytic activity and increased sensitivity to gefitinib. Cancer Research. 2007;67:2325–2330. doi: 10.1158/0008-5472.CAN-06-4293. [DOI] [PubMed] [Google Scholar]
- 65.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-Sensitizing EGFR Mutations in Lung Cancer Activate Anti-Apoptotic Pathways. Science. 2004;305:1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 66.Shi F, Telesco SE, Liu Y, Radhakrishnan R, Lemmon MA. ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc Natl Acad Sci U S A. 2010;107:7692–7697. doi: 10.1073/pnas.1002753107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shih AJ, Telesco SE, Radhakrishnan R. Analysis of somatic mutations in cancer: molecular mechanisms of activation in the ErbB family of receptor tyrosine kinases. Cancers. 2011;3:1195–1231. doi: 10.3390/cancers3011195. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.