

Abstract
Gene annotation databases (compendiums maintained by the scientific community that describe the biological functions performed by individual genes) are commonly used to evaluate the functional properties of experimentally derived gene sets. Overlap statistics, such as Fishers Exact test (FET), are often employed to assess these associations, but don't account for non-uniformity in the number of genes annotated to individual functions or the number of functions associated with individual genes. We find FET is strongly biased toward over-estimating overlap significance if a gene set has an unusually high number of annotations. To correct for these biases, we develop Annotation Enrichment Analysis (AEA), which properly accounts for the non-uniformity of annotations. We show that AEA is able to identify biologically meaningful functional enrichments that are obscured by numerous false-positive enrichment scores in FET, and we therefore suggest it be used to more accurately assess the biological properties of gene sets.
Evaluating the functional properties of gene sets is a routine step in understanding high-throughput biological data1,2 and is commonly used both to verify that the genes implicated in a biological experiment are functionally relevant1 and to discover unexpected shared functions between those genes3,4. Many functional annotation databases have been developed in order to classify genes according their various roles in the cell5,6,7,8,9. Among these, the Gene Ontology (GO)10,11 is one of the most widely used by many functional enrichment tools (for example1,2,12,13,14) and is highly regarded both for its comprehensiveness and its unified approach for annotating genes in different species to the same basic set of underlying functions10.
It has recently been observed that many classification databases, including the Gene Ontology, exhibit a heavy-tailed distribution in the number of genes annotated to individual categories15. However, there has been little investigation into how these underlying annotation properties may influence the results of functional analysis techniques. In this work we find that traditional functional enrichment approaches spuriously identify significant associations between functional terms in GO and random gene sets, if the number of annotations made to genes in the gene set is high. We also investigate the properties of curated experimentally-derived gene signatures, i.e. sets of genes whose combined expressed patterns are associated with specific biological conditions, and find that many contain a disproportionate number of highly annotated genes. Furthermore, traditional overlap statistics report significant associations between these signatures and randomly constructed collections of functional terms. Consequently, we propose a scheme, called Annotation Enrichment Analysis (AEA), that evaluates the overlap in annotations between a set of genes and the set of terms belonging to a branch of the GO hierarchy, using a randomization protocol to build a null model. By looking at annotation overlap instead of gene overlap, our approach takes into account the annotation properties of the Gene Ontology. It effectively eliminates biases due to database construction and highlights relevant biological functions in experimentally-defined gene signatures. We also provide a simple analytic approximation to AEA (which we call AEA-A, for Annotation Enrichment Analysis Approximation) that is able to partially compensate for the biases we find using traditional approaches. Implementations of both AEA and AEA-A are provided at http://www.networks.umd.edu.
In this study, we primarily focus on Gene Ontology annotations associated with human genes. The Gene Ontology10 takes the form of a directed acyclic graph (DAG) in which “child” functional categories (“terms”) are subclassified under one or more other, more general categories, called “parent” terms. “Branches” in the Gene Ontology can therefore be defined as sets of terms that contain a parent term and all of its progeny. Note that these branches contain overlapping sets of terms since each term can be a descendant of multiple ancestors at each level of the DAG. Using this structure, individual genes are annotated to various functional categories. These annotations are transitive up the hierarchy such that a parent term will take on all the gene annotations associated with any of its progeny16. Consequently, terms with many progeny often contain many gene annotations whereas terms with few progeny generally have fewer associated genes. “Biological Process,” “Molecular Function,” and “Cellular Component” are the three most general terms in GO, defining three independent branches such that every other term can only belong to one of these three categories. As a consequence all genes in GO are annotated to at least one, and often all three, of these categories.
The most widely used statistics for evaluating which functional categories are enriched in a set of genes are based on gene counts and include Fisher's Exact Test, the binomial test, and the chi-squared test17. Although these statistics vary in exact implementation, they all rely on the same basic underlying assumption that all genes have an equal probability of being selected under the null hypothesis. Of these tests, Fisher's Exact Test (FET) is the most common statistic and is used by many of the most popular functional enrichment tools (see Table 2 in18), and therefore we choose it to represent a “typical” evaluation of gene set functional enrichment. FET estimates enrichment by evaluating the overlap between genes in a given experimental gene set with genes annotated to a GO term. Genes in the experimentally-derived gene set are assumed to have an equal likelihood of being identified, consistent with the null model of FET. By mathematical construction FET also assumes that the genes annotated to a functional term are equally likely to be identified (see Equation 3 in the Methods section); however, because some genes are annotated to many functional terms while others are only annotated to a few, it follows that genes do not have an equal likelihood of being identified in the context of gene functional annotations, inconsistent with FET's null model. We investigate how this false assumption might alter predictions made in the context of functional enrichment analysis.
Since functional enrichment analysis often involves comparing a gene set to all the terms in GO, multiple-hypothesis corrections are generally applied to the results of these statistical tests18. These corrections decrease the value at which a comparison between a gene set and a GO term should be considered significant. Commonly used multiple-hypothesis corrections include the Bonferroni, Benjamini and the False Discovery Rate. Of these, the Bonferroni is the most conservative and adjusts the value at which a test is considered “significant” by the number of tests made19. The False Discovery Rate (FDR) adjusts the value at which a test is considered “significant” based on the rank of the predicted level of significance20,21. It provides approximately the same correction as the Bonferroni for the most significantly-ranked p-values but will not adjust tests that are the least-significant. It is important to note that although these corrections will change the critical value of individual tests, they do not affect the rank ordering of the results.
Results
Annotation properties of the gene ontology
To start our analysis we downloaded information regarding gene-term annotations for human genes from the Gene Ontology website (geneontology.org) and used this data to construct a gene-term bipartite graph, represented as an nG × nT adjacency matrix, where nG is the total number of genes and nT is the total number of terms listed in the annotation file. In this matrix a value of one indicates a known connection between the corresponding gene and term, and a value of zero indicates that the gene is not associated with that term. In this bipartite graph many terms are only associated with a small handful of genes, while some terms are associated with many genes. A histogram of the “degree”, kt, of terms (the number of genes annotated to individual terms) reveals a heavy-tailed relationship (Figure 1(a)). In contrast, a histogram of the “degree”, kg, of genes (the number of terms to which individual genes are annotated) shows that although some genes have many more annotations than others, the distribution is not as heavy-tailed as the term degree distribution (Figure 1(b)). We note that the annotation properties of the Gene Ontology are often shared by other databases (see Supplemental Figure S1), and therefore, we believe that the methods we develop below, although tested using the Gene Ontology, could be applied to functional enrichment analysis using other functional classifications.
Figure 1. The cumulative degree distributions of (a) terms and (b) genes in human GO annotations.
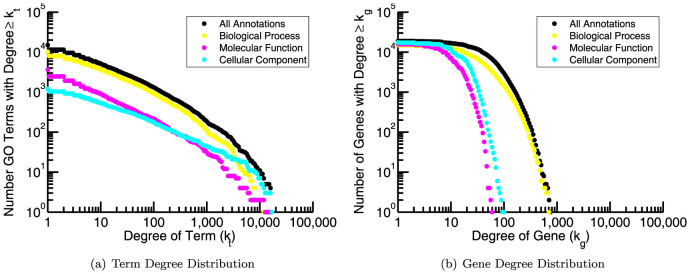
“Biological Process” terms make up the majority of terms and annotations. The average number of “Biological Process” terms to which an individual gene is annotated is 43.2 while the average number of annotations made to an individual term is 64.4.
We also point out that the “Biological Process” ontology contains a significant fraction of the total annotations. Although all three ontologies are used in functional enrichment analysis, many studies using GO focus on this ontology, both for its size and because its members describe dynamical processes performed by the cell. We do the same in the following analysis. The total number of annotations made to the “Biological Process” ontology is 656783, originating from 15213 genes to 10192 terms. Consequently, the average number of annotations made by an individual gene to this ontology is 43.2 and the average number of annotations made to an individual term is 64.4. These values will be useful to keep in mind, especially as we investigate the annotation properties of gene signatures and of the terms for which they are enriched.
Annotation properties influence the results of functional enrichment analysis
One of our goals is to determine the effect of annotation database properties on functional enrichment analysis. To do this, we first created 200 random gene sets with Ng members each, but in which we controlled the total number of annotations (Mg) made by the member genes (for more details see Methods). In practice, experimentally-derived sets of genes can range from only a handful (≈ 10) to a few thousand members. In this analysis we chose Ng = 200 since this represents a “typical” gene set size.
As an initial test, we used FET to determine the enrichment of all 10192 GO terms from the “Biological Process” ontology in each randomly constructed gene set. Figure 2(a) shows the results for the subset of terms that have 200 or more unique gene annotations, ordered based on their total number of unique gene annotations. The trend is striking. Even though they have the same number of members, gene sets with a higher number of annotations are more enriched in GO terms compared to gene sets with a lower number of annotations. Although we expect a minimum p-value across all tests of approximately 10−4, instead we observe that random gene sets with the fewest annotations have a minimum p-value around 10−3, while random gene sets with the highest annotation levels have a minimum p-value close to 10−6 (Supplemental Figure S2(a)). For high degree gene sets, we also observe that high degree GO branches tend to be more significantly enriched (i.e., have lower p-values) than low degree branches. We point out that although multiple-hypothesis corrections will sufficiently raise a p-value such that either very few or no false positives occur, these biases cannot be overcome in this manner (see Supplemental Figure S2(b)).
Figure 2.

(a) The enrichment (measured by p-value) of 200 randomly generated gene sets in GO branches. The branches are ordered based on how many genes are annotated to the parent term (kt) and the gene sets are ordered based on the total the number of annotations (Mg) made by the 200 genes in that set. Although we tested enrichment for all branches, for simplicity we only visualize the subset of branches with 200 or more unique gene annotations. There is an obvious bias toward significant enrichment between high degree gene-set/term pairs using Fisher's Exact Test (FET). (b) A plot of the p-values predicted by FET as a function of rank for five of the random gene sets shows that FET is both overly conservative for low degree gene sets and anti-conservative for high degree gene sets. (c)–(d) Analogous plots to (a) and (b) illustrating that this observed annotation bias can be correctly mitigated by using Annotation Enrichment Analysis (AEA).
In order to better interpret these results, for five of our random gene sets (〈k〉 ≈ {21, 32, 43, 54, 65}), we directly compared the distributions of the p-values predicted by FET to the expected distribution (evenly distributed values from zero to one). Figure 2(b) plots, in rank order, the p-values calculated for these random gene sets for the set of terms that contain at least one gene annotation from a member of the given random gene set. The deep dip below the diagonal for the the more highly-annotated gene sets demonstrates that FET is anti-conservative for these gene sets; in addition, FET also appears to be overly conservative for gene sets with a lower overall annotation level. Plots using all terms are shown in Supplemental Figure S3(a).
Annotation enrichment analysis corrects for annotation bias
Clearly annotation properties of both gene sets and functional categories can influence the results of functional enrichment analysis. In order to mitigate these effects, we suggest that instead of evaluating the overlap between genes, as is traditionally done in functional enrichment analysis, one instead considers the overlap between the annotations made to a gene set and a branch of terms in the Gene Ontology. To accurately capture the significance of annotation overlap we develop a randomization scheme that preserves the transitive annotation features of the GO DAG while calculating the probability of obtaining a certain number of annotations between a gene set and a GO branch. We call this approach Annotation Enrichment Analysis (AEA) and illustrate it in Figure 3.
Figure 3. An outline of how Annotation Enrichment Analysis (AEA) calculates the significance of association between a given gene set and the collection of terms that belong to a branch in the GO hierarchy.

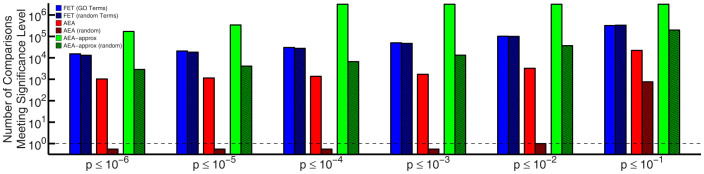
In this randomization is it useful to think of the Gene Ontology as a bipartite graph (see above). We begin by determining Mg, the total number of annotations to a gene set, Mt, the total number of annotations to the terms in a GO branch, and Mgt, the number of annotations stretching between this gene set and branch. We then determine a distribution for the expected number of co-annotations. To do this we, simultaneously, randomly permute the order of genes and terms while still preserving the original connections from the GO bipartite graph. By preserving the original connections, we retain the transitive annotation properties of the GO DAG. We then take annotations connected to the top randomly shuffled genes until we've selected Mg annotations, and annotations connected to the top randomly shuffled terms until we've selected Mt annotations, and determine 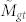 , the number of edges in the bipartite graph that extend between these top randomly shuffled genes and top randomly shuffled terms. In the (fairly common) case where selecting the top Mg/Mt annotations does not correspond to selecting a whole number of genes/terms, we take the top number of genes/terms whose total annotations is closest to Mg/Mt, respectively. We repeat the randomization process many times in order to determine a distribution of values for
, the number of edges in the bipartite graph that extend between these top randomly shuffled genes and top randomly shuffled terms. In the (fairly common) case where selecting the top Mg/Mt annotations does not correspond to selecting a whole number of genes/terms, we take the top number of genes/terms whose total annotations is closest to Mg/Mt, respectively. We repeat the randomization process many times in order to determine a distribution of values for 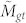 . We define a new p-value, pA(Mgt) which reflects the probability that
. We define a new p-value, pA(Mgt) which reflects the probability that 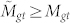 :
:
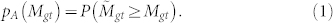 |
We determined the significance of all GO branches in our randomly generated gene sets with AEA (using 104 randomizations), and created a heat map of these values analogous to the one produced using standard set-overlap statistics (Figure 2(c)). The results of AEA are close to uniform across varying gene set degree (Figure 2(d)), demonstrating that AEA works well at eliminating annotation bias.
Experimental gene signatures are often highly-annotated
One of the most common applications of enrichment analysis is to ascertain the functional properties of a gene “signature” (an experimentally determined set of genes). Although we have demonstrated that AEA corrects for annotation bias with randomly generated gene sets, we also want to know how well this analysis can recapitulate biologically-relevant results. With this in mind, we downloaded signatures as recorded in the Gene Signatures Database (GeneSigDB)22. This database is a manual curation of previously published gene expression signatures, focusing primarily on cancer and stem cell signatures23. In the following analysis we will use all 309 human signatures from this database that contain at least 100 and less than 1000 genes that also are annotated to a term in the “Biological Process” ontology.
First, to assess whether annotation bias might play a role in evaluating the functional properties of these gene signatures, we determined the average number of annotations made by the genes occurring in each signature. Figure 4(a) shows the number of genes in a signature plotted against the average level of annotation for each signature. The expectation for a random selection of genes (the average number of annotations made by all genes – see above and Figure 1) is shown as a red line. The plot suggests that many genes belonging to these signatures are also more highly annotated in GO. Almost a third (99) of the signatures have an average level of annotation that is greater than any of our randomly generated gene sets and all but four signatures have an average level of annotation greater than expected by chance. Since we have shown that random gene sets with these annotation levels encounter a bias in traditional functional enrichment analysis, we believe these experimental signatures are an appropriate biological set with which to evaluate how AEA compares to FET when investigating and discovering the functions of genes derived from experimental biological data.
Figure 4. Annotation properties of experimental gene signatures.
(a) The number of genes versus the average number of annotations made to the genes in each signature. Genes from signatures generally contain many more GO annotations than one would expect if selecting genes randomly (red line). (b) The number of terms that are considered important (top 10% by rank) by one of the measures (either AEA or FET), but not important (bottom 80% by rank) by the other, plotted for each gene signature. The signatures are colored according to the average level of annotation (kavg = Mg/Ng).
It is common practice when evaluating the functional properties of a gene signature to focus on a set of “top” categories based on p-value rank. We investigate how different the results of AEA or FET might appear in this context. To this end, we selected the top 10% of terms based on their enrichment score in FET and AEA to designate as “important” according each to these measures. We compared this list of terms to the list of terms that are “not important” (in the bottom 80% of terms by rank) according to each measure. The number of terms considered important in AEA but not by FET versus the number of terms considered important by FET but not AEA for each signature is plotted in Figure 4(b). Complete agreement between FET and AEA on this plot is represented by a point at (0, 0), and complete disagreement is represented by a point at (1019, 1019). In order to see how annotation properties influenced any differences, we colored signatures based on the average level of annotation to their member genes.
Figure 4(b) shows overall agreement between AEA and FET, as many points fall fairly close to the origin and, at most, reflect only a 10% difference in identified “important” terms. However, annotation bias is evident. In signatures containing the highest levels of annotation, i.e. those represented by reddish marks, the terms deemed most “important” by FET are more likely to be considered “unimportant” according to AEA, and vice versus. These results are consistent with the previous analysis in random gene sets that showed a bias by FET to place more significance between gene sets and terms with a higher number of annotations (see Figure 2). It also demonstrates that annotation bias is present when evaluating experimentally-derived gene signatures and is not an artifact of how we constructed our random gene sets.
In the supplement we also directly compare FET and AEA p-values and observe that, in these experimental signatures, a high annotation level is correlated with increased significance by FET compared to AEA and vice versus (Supplemental Figure S3(c)), consistent with the results shown in Figure 4(b).
Annotation enrichment analysis uncovers meaningful biological associations
Next, we investigated the specific biology that is highlighted using AEA and FET. For each measure, we chose approximately forty signatures having the most significant enrichment scores across all terms. Similarly, for each measure, we chose forty terms having the most significant enrichment scores across all signatures. For AEA a small number (981 out of 3149328 possible) of term-signature pairs have an estimated p-value of p < 10−6 after one million randomizations, therefore, when necessary, we broke ties by the number of signatures/terms enriched in the terms/signatures at this level. Using the selected sets of terms and signatures and the p-values associating all pairs in these sets, we then performed a standard hierarchical clustering analysis. The results are shown in Figure 5.
Figure 5. Clustergrams representing enriched term-signature pairs.

(a) A clustering of signatures and terms selected based on their enrichment-score according to FET. These signatures include those reported in25,27,29,32,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64. (b) A clustering of signatures and terms selected based on their enrichment-score according to AEA. The signatures and terms break into several, biologically distinct units. One is associated with immune-response, and includes signatures published in25,26,27,36,37,39,41,42,65,66. A second includes signatures related to cellular-differentiation published in29,44,46,48,67,68,69,70,71,72. Another cluster includes breast cancer signatures published in53,54,55,56,58,59,60,61,63. Finally three lymphoma32,64,73 and a viral signature33 associated with proliferation are also included. The colorscales for the p-values were chosen to give approximately the same red/green balance in each clustergram.
Clustering the FET results gives rise to a weak visual segregation of terms and signatures into groups (Figure 5(a)). These groups highlight the relationship between the gene signatures and several important biological processes. For example, the FET clustering shows an enrichment of cell-cycle related processes in breast cancer signatures24 and includes immune-related terms enriched in immune gene signatures. These two groups, however, account for only about half of the selected terms; the clustergram also includes a number of functional categories related to “proteins” and “phosphorylation” that are only enriched in a small number of signatures. From this analysis we suggest that the results of FET might be muddled by a signal driven by annotation bias, highlighting either highly-annotated signatures or more general biological processes.
In contrast, when using AEA distinct clusters of signatures and terms emerge (Figure 5(b)). The first includes signatures from immune-systems, lymphoma and leucocytes, and is logically also enriched in terms such as “immune system” and “response to stimulus” as well as terms related to “biological regulation”. Interestingly, one of the breast signatures associated with this cluster25 represents a list of genes defined based on immune response in breast cancer and the stem cell signature26 is from a study on patients with systemic sclerosis, a type of autoimmune disorder. In addition, the inclusion of a protein-kinase signature27 is interesting as MAP kinases have been shown to play an important role in immune response28.
Another cluster is enriched in categories such as “system development” and “developmental process” and includes several signatures associated with stem cells or identified based on their role in cellular differentiation. It also includes a signature of oncogenes29, as well as a signature of homeodomain proteins, known to initiate cascades of genes that in turn will induce cellular differentiation into tissues and organs (e.g.30,31). The next cluster, associated primarily with breast cancer signatures, shows a strong enrichment for terms related to the cell cycle and cellular component organization, processes known to be differentially regulated in breast cancer24. Finally, two lymphoma and one viral signature that were identified based on cell proliferation (for example, by association with Myc targeting32,33) are enriched for terms such as “cellular metabolic process.” This is consistent with expectation since there is evidence that a connection exists between proliferation and metabolic pathways in cancer cells34,35.
Some predictions made by FET are likely a consequence of annotation bias in experimental gene signatures
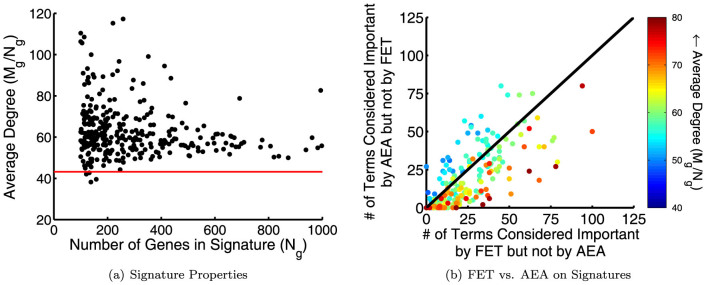
Next, we created random term sets, constructing each such that it has approximately the same number of unique genes annotated to its members as a real GO branch (for more details see Methods). We used these random term sets to study the application of functional enrichment analysis methods to experimental gene signatures and to systematically determine if annotation database properties might be a source of false positives. Specifically, using both the traditional FET and our proposed AEA, we investigated the enrichment of experimental gene signatures in randomly constructed term sets as well as real GO branches. We determined the number of term-signature comparisons considered significant at several different thresholds and present the results in Figure 6.
Figure 6. A plot of the number of term-signature comparisons deemed “significant” at various p-value thresholds.
A dotted line indicates only one comparison with a p-value less than or equal to the indicated threshold, and cases where no significant comparisons were found for the corresponding p-value are indicated by a bar not exceeding this line. Evaluations using gene annotations to GO branches are shown as solid colors, whereas evaluations using genes annotated to random term sets are striped. AEA-A refers to the results when using a quantitative approximation to AEA.
Surprisingly, using FET, there is almost no difference between the number of significant comparisons made using real GO branches and using the randomly generated term sets. This striking similarity can be understood as follows. When calculating the significance between two gene sets, FET assumes all genes in those sets have an equal probability of being chosen. This is a false assumption as some genes are actually more likely to be annotated to any given term in GO. Just as high degree genes are more likely to be annotated to a randomly chosen GO branch, so too are they more likely to be annotated to a random set of GO terms. As noted previously, experimental gene signatures include an abundance of genes with higher levels of annotations. Combined together, this bias means that these signatures are likely to be enriched in random sets of functional categories, just because their members have more annotations overall. We believe this illustrates a fundamental flaw of using FET for functional enrichment analysis, as it will predict significant associations, not because of biological signal, but as a result of a bias in signature annotation properties.
Compared to FET, AEA finds fewer enriched pairs at each threshold, but, unlike FET, finds no signatures enriched in the random term sets, demonstrating its ability to correct for annotation biases introduced from the hierarchical relationships between those terms in the ontology. These results give us confidence that AEA is highlighting the connections between gene sets and branches that are most likely to be truly biologically relevant and is robust against biases introduced by annotation properties. For more analysis comparing the effects of term set properties on FET and AEA see the supplementary material.
A quantitative approximation to annotation enrichment analysis partially corrects for annotation bias
One significant strength of AEA is that it makes no assumptions regarding the structure of gene-term annotations; however, because it uses a randomization scheme to estimate the null hypothesis, the precision of the estimated p-values is dependent upon the number of randomizations, and each run of the algorithm will give slightly different results. Therefore, we sought an analytic approximation of AEA in order to overcome these limitations.
Given that we want to estimate the significance of annotation overlap, one logical approach is to simply count the number of annotations made to a gene set, the number of annotations made to a branch in GO, and the number of annotations extending between that gene set and branch, and use the hypergeometric probability to determine the significance of this overlap. We point out that this approach makes the false assumption that annotations are independent, implying that a gene could be annotated to the same term multiple times. Another more limiting problem is that, unlike AEA, this approach erases the hierarchical organization of annotations encoded in the GO DAG. Because of these assumptions, predictions made under this framework will not have the reliability of the randomization protocol specified by AEA; however, they can be computed quickly and without the need for any randomization to generate a null hypothesis.
Acknowledging that we are making some false assumptions regarding the structure of gene-term annotations, we propose an analytic framework for evaluating functional enrichment which we call Annotation Enrichment Analysis Approximation (AEA-A). This approximation makes use of the hypergeometric probability to calculate the significance (or p-value approximating AEA, pa(Mgt)) of overlap between annotations made to a given gene set and branch in the GO hierarchy. Given Mg annotations to a gene set, Mt annotations to terms belonging to a GO branch, and Mtot annotations made in the GO ontology, the probability of finding Mgt or more annotations in common between these two sets can be written as:
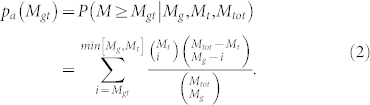 |
We point out that the equation for AEA-A is equivalent to performing an FET on annotation overlap instead of gene overlap (compare to equation 3). We tested the performance of this approximation by determining the functional enrichment of GO terms in our randomly generated gene sets. The results of AEA-A are uniform across varying gene set degree (see Supplemental Figure S5), demonstrating that AEA-A works well at eliminating annotation bias. However, the predicted p-values are often misleadingly low due to the independence assumption. This limitation is evident in analysis performed on the experimental signatures (Figure 6) – many more comparisons are deemed “significant” at each threshold using AEA-A than either AEA or the traditional FET looking at gene overlap. Furthermore, compared to AEA, the approximation is only partially able to discern between real GO branches and random term sets. However, we note that it does significantly outperform the traditional FET in this regard.
This analytic approximation may be appealing to many since it is conceptually cleaner than the randomization protocol specified by AEA. Furthermore, since it is mathematically equivalent to the more traditional FET analysis, it may also be simpler to implement in current functional enrichment tools. However, although AEA-A is conceptually appealing and has some advantages over traditional FET, it does not provide results that are as discerning as AEA. Therefore, we believe AEA is a better approach for analyzing functional enrichment in gene sets, but provide AEA-A as an alternative that combines many of the advantages of AEA with an analytical form that will be easier to implement in practice.
Discussion
We have demonstrated that evaluating the functional enrichment of gene sets using traditional set-overlap statistics, such as FET, is susceptible to producing false positives as a result of certain annotation database properties. We offer a solution, Annotation Enrichment Analysis, or AEA, that fully considers these properties, eliminating potential annotation bias in the predicted enrichment scores. The importance of using this approach is highlighted by the fact that many published gene-signatures include a large number of highly-annotated genes. This is likely in part due to a non-independence between identified signatures and functional annotations, since genes that are involved in a well-studied phenomena such as cancer are also more likely to be frequently annotated in these databases. Although it is possible that newly-derived gene signatures may not exhibit the same level of annotation-bias as these previously-published signatures, it is also very probable that highly annotated genes are important in a wide variety of well-studied systems and will continue to show up and influence the results of functional enrichment analysis on newly generated gene sets.
The annotation-bias associated with FET results and the bias for higher annotation-levels among experimentally-derived gene signatures is largely unrecognized. Although significant p-values for functional enrichment in experimental signatures may initially seem compelling for the bioinformatician, we suggest that these results do not always reflect biological properties but instead have a high potential to be a result of statistical bias. In light of our analysis we suggest using the AEA approach either alongside or in place of other traditional measures, especially for gene signatures that are known to contain significantly more or less annotations than one would expect by chance. Furthermore, we urge the bioinformatics community to consider annotation properties of gene signatures and annotation databases before utilizing results from the wide variety of available gene set enrichment tools. We believe that considering annotation enrichment will allow biologists to better interpret the functional roles of genes identified as important in their experimental system.
Methods
Calculating functional enrichment using set-overlap statistic
In this analysis we used Fisher's Exact Test (FET) to perform a “traditional” functional enrichment analysis. FET is related to the hypergeometric probability and can be used to calculate the significance, or p-value estimated using FET (pF(Ngt)), of the overlap between two independent sets. For example, given a gene set containing Ng genes, a GO term with annotations to kt different genes, and Ntot total genes annotated in GO, the probability that Ngt or more genes belong both to this gene set and are annotated to the GO term can be calculated as:
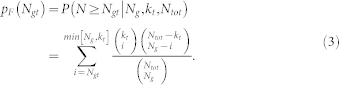 |
Note that in this equation Ng and kt are mathematically interchangeable.
Together with the FET, we also sometimes determine the False Discovery Rate (FDR) of the tests in order to account for Type I errors (20,21). In these cases, we calculate the FDR using the matlab function “mafdr” and report the associated q-values.
Constructing biased random gene sets and random term sets
In order to investigate potential bias due to the annotation properties, we constructed random gene sets with the same number of members, but with varying amounts of annotations made by those members. Each set with a desired total number of annotations, Mg, was created by first randomly selecting Ng genes. We then randomly selected one gene in this gene set (gene i) and one gene not in the gene set (gene j). If replacing gene i with gene j caused the total number of annotations made by genes in the gene set to approach Mg, we replaced gene i with gene j with a high probability (p = 0.95), but if the replacement caused the average degree of the gene set to move farther away from Mg we replaced gene i with gene j with a low probability (p = 0.05). This swapping continued until the total number of annotations made by the gene set was within 0.1% of Mg. In this way we created 200 gene sets with Ng = 200 genes each, but whose average degree (kavg = Mg/Ng) varies from approximately 21 to 65, or from around half to 1.5 times the expected average degree of 43 (see Figure 1).
We also constructed sets of random GO terms. Specifically, to build a random term set for comparison with a branch in the GO DAG, we determined the number of annotations made to the parent term of the GO branch (kt), we then randomly ordered all the terms in GO and selected the top Nt terms until the number of unique genes annotated to those Nt random terms 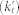 was within a small percentage of
was within a small percentage of 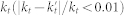 . In the case where selecting both Nt and Nt+1 terms were within this limit we chose Nt to minimize the absolute difference between kt and
. In the case where selecting both Nt and Nt+1 terms were within this limit we chose Nt to minimize the absolute difference between kt and 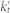 . If selecting the top Nt terms did not lead to a situation within this limit, we reshuffled the terms and selected the top Nt terms in this new list, repeating until a suitable random collection of terms could be chosen. In this way we created 10192 random term sets with approximately the same number of unique genes annotated to each as to real GO branches.
. If selecting the top Nt terms did not lead to a situation within this limit, we reshuffled the terms and selected the top Nt terms in this new list, repeating until a suitable random collection of terms could be chosen. In this way we created 10192 random term sets with approximately the same number of unique genes annotated to each as to real GO branches.
Clustering AEA and FET results
Hierarchical clustering of the AEA and FET results was performed using the “clustergram” function in Matlab with default settings.
Author Contributions
K.G. and M.G. contributed to the model conception and design; K.G. performed the analysis; K.G. and M.G. wrote the manuscript.
Supplementary Material
Supplemental Information
Acknowledgments
We would like to thank Emanuele Mazzola for helpful discussions regarding this work.
References
- Huang D. W. a. W. et al. DAVID bioinformatics resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucl. Acids Res. 35, W169–W175 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Nat. Acad. Sci. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- King O. D., Foulger R. E., Dwight S. S., White J. V. & Roth F. P. Predicting gene function from patterns of annotation. Genome Res. 13, 896–904 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafavi S. & Morris Q. Fast integration of heterogeneous data sources for predicting gene function with limited annotation. Bioinformatics 26, 1759–1765 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov R. L. et al. The COG database: an updated version includes eukaryotes. BMC bioinformatics 4, 41. Epub. (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M. & Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serres M. H., Goswami S. & Riley M. Genprotec: an updated and improved analysis of functions of escherichia coli k-12 proteins. Nucl. Acids Res. 32, D300–2 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R. et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucl. Acids Res. 38, D473–D479 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serres M. H. & Riley M. MultiFun, a multifunctional classification scheme for escherichia coli k-12 gene products. Microb. Comp. Genomics 5, 205–222 (2000). [DOI] [PubMed] [Google Scholar]
- Ashburner M. et al. Gene ontology: tool for the unification of biology. the gene ontology consortium. Nat. Genetics 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium T. G. O. The Gene Ontology in 2010: extensions and refinements. Nucl. Acids Res. 38, D331–D335 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beissbarth T. & Speed T. P. GOstat: find statistically overrepresented gene ontologies within a group of genes. Bioinformatics 20, 1464–1465 (2004). [DOI] [PubMed] [Google Scholar]
- Martin D. et al. GOToolBox: functional analysis of gene datasets based on gene ontology. Genome Biol. 5, (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexa A., Rahnenführer J. & Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607 (2006). [DOI] [PubMed] [Google Scholar]
- Glass K., Ott E., Losert W. & Girvan M. Implications of functional similarity for gene regulatory interactions. Jour. of the Royal Soc., Interface 9, 1625–1636 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- The_gene_ontology_consortium. Creating the gene ontology resource: design and implementation. Genome Res. 11, 1425–1433 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivals I., Personnaz L., Taing L. & Potier M.-C. Enrichment or depletion of a GO category within a class of genes: which test? Bioinformatics 23, 401–407 (2007). [DOI] [PubMed] [Google Scholar]
- Khatri P. & Drăghici S. Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics 21, 3587–3595 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn O. J. Multiple comparisons among means. Jour. of the Amer. Stat. Assoc. 56, 52–64 (1961). [Google Scholar]
- Storey J. D. A direct approach to false discovery rates. Jour. of the Royal Stat. Soc.: Series B 64, 479–498 (2002). [Google Scholar]
- Storey J. D. & Tibshirani R. Statistical significance for genomewide studies. Proc. Nat. Acad. Sci. 100, 9440–9445 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culhane A. C. et al. GeneSigDB: a manually curated database and resource for analysis of gene expression signatures. Nucl. Acids Res. 40, D1060–D1066 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culhane A. C. et al. GeneSigDB–a curated database of gene expression signatures. Nucl. Acids Res. 38, D716–D725 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loddo M. et al. Cell-cycle-phase progression analysis identifies unique phenotypes of major prognostic and predictive significance in breast cancer. British Jour. of Cancer 100, 959–970 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyal F. et al. A comprehensive analysis of prognostic signatures reveals the high predictive capacity of the proliferation, immune response and RNA splicing modules in breast cancer. Breast Cancer Res. 10, R93. Epub. (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohgaki T. et al. Up regulated expression of tumour necrosis factor alpha converting enzyme in peripheral monocytes of patients with early systemic sclerosis. Annals of the Rheumatic Diseases 64, 1165–1173 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G., Whyte D. B., Martinez R., Hunter T. & Sudarsanam S. The protein kinase complement of the human genome. Science 298, 1912–1934 (2002). [DOI] [PubMed] [Google Scholar]
- Dong C., Davis R. J. & Flavell R. A. MAP kinases in the immune response. Ann. Rev. of Immunology 20, 55–72 (2002). [DOI] [PubMed] [Google Scholar]
- Futreal P. A. et al. A census of human cancer genes. Nat. Rev. Cancer 4, 177–183 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepveu A. Role of the multifunctional CDP/Cut/Cux homeodomain transcription factor in regulating differentiation, cell growth and development. Gene 270, 1–15 (2001). [DOI] [PubMed] [Google Scholar]
- Magli M. C. The role of homeobox genes in hematopoiesis. Biotherapy 10, 279–294 (1998). [DOI] [PubMed] [Google Scholar]
- Gentles A. J. et al. A pluripotency signature predicts histologic transformation and influences survival in follicular lymphoma patients. Blood 114, 3158–3166 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairo S. et al. Hepatic stem-like phenotype and interplay of wnt/beta-catenin and myc signaling in aggressive childhood liver cancer. Cancer Cell 14, 471–484 (2008). [DOI] [PubMed] [Google Scholar]
- DeBerardinis R. J., Lum J. J., Hatzivassiliou G. & Thompson C. B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metabolism 7, 11–20 (2008). [DOI] [PubMed] [Google Scholar]
- Vander Heiden M. G., Cantley L. C. & Thompson C. B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speers C. et al. Identification of novel kinase targets for the treatment of estrogen receptor?negative breast cancer. Clin. Cancer Res. 15, 6327–6340 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balko J. M. et al. Gene expression patterns that predict sensitivity to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer cell lines and human lung tumors. BMC Genomics 7, 289 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillion J. et al. The high-mobility group a1a/signal transducer and activator of transcription-3 axis: an achilles heel for hematopoietic malignancies? Cancer Res. 68, 10121–10127 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Y. M. et al. Toward an ontology-based framework for clinical research databases. Jour. of Biomed. Infor. 44, 48–58 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiriou C. et al. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc. Nat. Acad. Sci. 100, 10393–10398 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Delgado B. et al. Differential expression of NF-kappaB pathway genes among peripheral t-cell lymphomas. Leukemia 19, 2254–2263 (2005). [DOI] [PubMed] [Google Scholar]
- Zola H. et al. CD molecules 2006–human cell differentiation molecules. Jour. of Immunological Methods 319, 1–5 (2007). [DOI] [PubMed] [Google Scholar]
- Bergamaschi A. et al. Extracellular matrix signature identifies breast cancer subgroups with different clinical outcome. The Jour. of Pathology 214, 357–367 (2008). [DOI] [PubMed] [Google Scholar]
- Moreland R. T., Ryan J. F., Pan C. & Baxevanis A. D. The homeodomain resource: a comprehensive collection of sequence, structure, interaction, genomic and functional information on the homeodomain protein family. Database: The Jour. of Biol. Databases and Curation 2009, Epub. (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S. X. et al. Gene expression profile and angiogenic marker correlates with response to neoadjuvant bevacizumab followed by bevacizumab plus chemotherapy in breast cancer. Clin. Cancer Res. 14, 5893–5899 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Eynden G. G. et al. Differential expression of hypoxia and (lymph)angiogenesis-related genes at different metastatic sites in breast cancer. Clin. & Exper. Metastasis 24, 13–23 (2007). [DOI] [PubMed] [Google Scholar]
- Rae M. T. et al. Steroid signalling in human ovarian surface epithelial cells: the response to interleukin-1alpha determined by microarray analysis. The Jour. of Endocrinology 183, 19–28 (2004). [DOI] [PubMed] [Google Scholar]
- Wood C. E., Kaplan J. R., Fontenot M. B., Williams J. K. & Cline J. M. Endometrial profile of tamoxifen and low-dose estradiol combination therapy. Clin. Cancer Res. 16, 946–956 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan K. A., Chen G., Kalemkerian G. P., Wicha M. S. & Beer D. G. An embryonic stem cell-like signature identifies poorly differentiated lung adenocarcinoma but not squamous cell carcinoma. Clin. Cancer Res. 15, 6386–6390 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason D. X., Jackson T. J. & Lin A. W. Molecular signature of oncogenic ras-induced senescence. Oncogene 23, 9238–9246 (2004). [DOI] [PubMed] [Google Scholar]
- Lauss M. et al. Consensus genes of the literature to predict breast cancer recurrence. Breast Cancer Res. and Treatment 110, 235–244 (2008). [DOI] [PubMed] [Google Scholar]
- Mutarelli M. et al. Time-course analysis of genome-wide gene expression data from hormone-responsive human breast cancer cells. BMC Bioinformatics 9 Suppl 2, S12 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C. M. et al. AP-2gamma promotes proliferation in breast tumour cells by direct repression of the CDKN1A gene. The EMBO Jour. 28, 3591–3601 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiriou C. et al. Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. Jour. of the Nat. Cancer Inst. 98, 262–272 (2006). [DOI] [PubMed] [Google Scholar]
- Thorner A. R. et al. In vitro and in vivo analysis of B-Myb in basal-like breast cancer. Oncogene 28, 742–751 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loi S. et al. Predicting prognosis using molecular profiling in estrogen receptor-positive breast cancer treated with tamoxifen. BMC Genomics 9, 239 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller L. D. et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc. Nat. Acad. Sci. 102, 13550–13555 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirapati P. et al. Meta-analysis of gene expression profiles in breast cancer: toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res. 10, R65. Epub. (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford N. P. et al. Bromodomain 4 activation predicts breast cancer survival. Proc. Nat. Acad. Sci. 105, 6380–6385 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troester M. A. et al. Gene expression patterns associated with p53 status in breast cancer. BMC Cancer 6, 276 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dairkee S. H. et al. A molecular ‘signature' of primary breast cancer cultures; patterns resembling tumor tissue. BMC Genomics 5, 47 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H. Y. et al. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biology 2, E7. Epub. (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kopylow K. et al. Screening for biomarkers of spermatogonia within the human testis: a whole genome approach. Human reproduction 25, 1104–1112 (2010). [DOI] [PubMed] [Google Scholar]
- Tome M. E. et al. A redox signature score identifies diffuse large b-cell lymphoma patients with a poor prognosis. Blood 106, 3594–3601 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccaluga P. P. P. et al. Gene expression analysis of peripheral t cell lymphoma, unspecified, reveals distinct profiles and new potential therapeutic targets. The Jour. of Clin. Investigation 117, 823–834 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin M., Herscovitch M., Zhang N., Waxman D. J. & Gilmore T. D. Overexpression of an activated REL mutant enhances the transformed state of the human b-lymphoma BJAB cell line and alters its gene expression profile. Oncogene 28, 2100–2111 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leval L. et al. The gene expression profile of nodal peripheral t-cell lymphoma demonstrates a molecular link between angioimmunoblastic t-cell lymphoma (AITL) and follicular helper t (TFH) cells. Blood 109, 4952–4963 (2007). [DOI] [PubMed] [Google Scholar]
- Bertucci F. et al. Prognosis of breast cancer and gene expression profiling using DNA arrays. Annals of the New York Acad. of Sci. 975, 217–231 (2002). [DOI] [PubMed] [Google Scholar]
- Scott L. A. et al. Invasion of normal human fibroblasts induced by v-Fos is independent of proliferation, immortalization, and the tumor suppressors p16INK4a and p53. Mol. and Cell. Biology 24, 1540–1559 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godard S. et al. Classification of human astrocytic gliomas on the basis of gene expression: a correlated group of genes with angiogenic activity emerges as a strong predictor of subtypes. Cancer Res. 63, 6613–6625 (2003). [PubMed] [Google Scholar]
- Koçer S. S., Djurić P. M., Bugallo M. F., Simon S. R. & Matic M. Transcriptional profiling of putative human epithelial stem cells. BMC Genomics 9, 359 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran U. R. et al. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer 7, 64 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner R. G. et al. Kaposi's sarcoma-associated herpesvirus-infected primary effusion lymphoma has a plasma cell gene expression profile. Proc. Nat. Acad. Sci. 100, 10399–10404 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Information