

Abstract
Cystinosis is an autosomal recessive metabolic disease that belongs to the family of lysosomal storage disorders. The defective gene is CTNS, which encodes the lysosomal cystine transporter, cystinosin. Cystine accumulates in all tissues and leads to organ damage including end-stage renal disease. In this review, we outline the studies that support that genetic rescue of cystinosis could be an achievable goal, even though cystinosis is a multi-compartmental disease and cystinosin an intracellular transmembrane protein. Using the mouse model of cystinosis, the Ctns−/− mice, we showed that transplanted hematopoietic stem cells (HSCs) were able to act as vehicles for the delivery of a functional Ctns gene to the different organs and led to the significant decrease of the tissue cystine content and tissue preservation. Ex vivo gene-modified Ctns−/− HSC transplantation using a lentiviral vector containing CTNS complementary DNA (cDNA) was also successful in the Ctns−/− mice and built the foundations for a clinical trial for autologous HSC transplantation for cystinosis. The capacity of HSCs for rescuing non-hematopoietic disease is controversial, and new insights into regenerative medicine could be gained from unraveling the underlying mechanism of action.
Keywords: cystinosis, hematopoietic stem cells, kidney preservation, lentivirus vector, tissue cystine decrease
CYSTINOSIS: A MULTI-SYSTEMIC DISEASE DUE TO A DEFECTIVE LYSOSOMAL TRANSMEMBRANE PROTEIN
The clinical presentation and the protein involved in cystinosis do not make this disease an easy target for gene rescue. Cystinosis is an autosomal metabolic disease that belongs to the family of the lysosomal storage disorders (LSDs), characterized by a lysosomal accumulation of cystine in all the cells of the body [1]. Three allelic forms of the disease exist: infantile, juvenile and ocular. Clinically, they are distinguishable by the severity of symptoms and the age of onset. Patients with the infantile form typically present in their first year of life with symptoms of de Toni-debré-Fanconi syndrome characterized by severe fluid and electrolyte disturbances (vomiting, poor growth and rickets). Patients eventually progress to end-stage renal failure. Cystine accumulation also leads to multi-organ dysfunction and patients present with photophobia and blindness, hypothyroidism, hypogonadism, diabetes, myopathy and central nervous system defects. Patients with the juvenile form primarily develop progressive kidney dysfunction and photophobia around 12 years old, while the ocular form consists of photophobia and presents in adulthood. The gene underlying cystinosis, CTNS, is expressed in every cell and tissue and encodes a seven-transmembrane domain protein, cystinosin [2]. Cystinosin is a lysosomal H+-driven cystine transporter that allows cystine to exit the lysosomes [3, 4].
The only treatment for cystinosis is the drug cysteamine that reduces the intracellular concentration of cystine, and, if used early in the disease and in high doses, can delay the subsequent progression of renal glomerular damage and improve childhood growth [5]. However, the need for frequent dosing (every 6 h orally plus eye drops hourly) and severe side effects such as digestive intolerance and persistent odor render its chronic administration difficult, and compliance is a huge challenge in the pediatric population. Moreover, cysteamine does not prevent the proximal tubulopathy or the end-stage renal failure and only delays the onset of the complications [5, 6]. Patients with renal failure require dialysis or transplantation, both of which have significant negative health effects and due to the severe shortage of donor organs, patients may wait 3 to 6 years for transplantation. Thus, there is a pressing need for a new therapy with the potential to prevent the disease sequelae.
We tested gene rescue as a new therapeutic strategy in the mouse model for cystinosis, the Ctns−/− mice [7]. Ctns−/− mice accumulate cystine and cystine crystals in all organs tested, and developed similar anomalies as those observed in affected patients, such as the ocular defects [7, 8]. Ctns−/− mice backcrossed on a C57BL/6 background develop renal dysfunction from 6 months of age and incomplete proximal tubulopathy [9]. By 15 months of age, the mice develop end-stage renal failure. Histologically, cystine crystals accumulate in the same tissue cell types observed in human, such as kupffer cells in liver. In the kidney, crystals accumulate progressively within proximal tubular cells and capsular interstitial cells, and proximal tubular cells appeared to be de-differentiated; the cells are flat without brush borders and the tubules have a thick basal membrane [7, 9]. The proximal tubules also exhibit the ‘swan neck’ deformity, found in mice and humans with cystinosis, resulting in atubular glomeruli. Finally, heavy infiltration of inflammatory cells can be observed in the kidney of the Ctns−/− mice. This model is a unique instrument for testing emerging therapies for cystinosis as therapeutic impact can be objectively tested through the measurement of tissue cystine levels and kidney function.
PROOF OF CONCEPT FOR USING HEMATOPOIETIC STEM CELLS AS A VEHICLE FOR GENE THERAPY IN CYSTINOSIS
Numbers of reports have established a proof of principle for allogeneic hematopoietic stem cell (HSC) transplantation in several LSDs [10]. However, a majority of them involve a secreted protein that can be recaptured by the adjacent cells and/or the main clinical phenotype is a neurodegenerative disease [11]. In contrast, the protein involved in cystinosis is a lysosomal transmembrane protein and the clinical entity is characterized by the progressive degeneration of multiple organs [2]. The genetic rescue of cystinosis would require the delivery of a functional CTNS gene to many cell types in most of the tissue compartments to address the serious global effects of this genetic disease. To date, no viral vector or gene-delivery system is capable of efficiently transducing cells in every tissue compartment. Thus, the use of a vehicle to bring the healthy gene to multiple organs is necessary. Adult bone marrow (BM) stem cells are an attractive vehicle as they are theoretically capable of integrating into every tissue compartment and are currently used in clinic [12]. Several BM cells can be used, the whole BM, the Hematopoietic Stem Cells (HSCs) and the Mesenchymal Stem Cells (MSCs). MSCs are multipotent stromal cells that can differentiate into a variety of cell types such as osteoblasts, adipocytes and chondrocytes [13, 14]. Clinical case reports for the use of MSCs in orthopedic applications have been published, though the effectiveness of these methods remains to be demonstrated [15]. HSCs are stem cells that give rise to blood cells. Their plasticity, i.e. their conversion to non-hematopoietic cell types, has been reported but is very controversial [16, 17]. The advantage of the HSCs is that they are routinely used in clinic, mainly to treat patients with cancers and other disorders of the blood and immune systems.
We tested these three types of adult BM stem cells in the mouse model of cystinosis [18]. We transplanted syngeneic BMCs, HSCs and MSCs isolated from GFP-transgenic mice into 2 months old Ctns−/− mice. As controls, Ctns−/− mice were transplanted with BMCs from Ctns−/− mice. All animals were lethally irradiated before transplantation. MSCs did not integrate efficiently and stably into any organ and only led to a short-term beneficial effect on the kidney function. This outcome is relevant with their paracrine mode of action rather than with cellular differentiation [19]. At 4 months post-transplantation in the mice treated with BMCs or HSCs, confocal microscopy and quantitative-PCR revealed a large quantity of transplanted BM-derived cells in all organs tested, from 5 to 19% of the total tissue cells (Figure 1; Table 1). Most of these cells were part of the intrinsic structure of the organ and were mainly resident-phagocytic cells such as kupffer cells in the liver and dendritic cells in the kidney. The organ-specific cystine content was reduced by 57–94% in all organs tested (Table 1). The natural progression of renal dysfunction was prevented, and deposition of corneal cystine crystals was significantly improved. We also transplanted BMCs and HSCs from luciferase-transgenic mice in Ctns−/− and wild-type mice to follow the fate of transplanted cells expressing a functional Ctns gene in live animals as a function of time. We observed abundant tissue colonization by BM-derived cells specifically in cystinosis mice, proving that tissue engraftment of BM-derived cells is a dynamic process and is linked to the underlying and progressive tissue injury.
FIGURE 1:

Confocal microscopy and histology analyses of kidney from Ctns−/− mouse control or transplanted with GFP-HSC. (A and B) Representative confocal microscopy pictures of kidney sections from Ctns−/− control (A) or transplanted with GFP-HSC (B) [18]. Nuclei stained by dapi are seen in blue, F-actin intermediate filament staining by Bodipy-Phalloidin is seen in red, and direct eGFP fluorescence is seen in green. Abundant GFP-positive BM-derived cells are observed in the Ctns−/− mice transplanted with GFP-HSC. (C and D) Representative kidney histology sections stained with hematoxylin/eosin from 15 to 17 months old Ctns−/− control (C) or transplanted with GFP-HSC (D) [20]. (C) Ctns−/− mice controls exhibit severe kidney anomalies characterized by atrophic proximal tubules, collapsed or sclerotic glomeruli, and large and multiple mononuclear infiltrates. (D) Treated Ctns−/− mice with high blood engraftment exhibited only focal renal anomalies and significantly less lymphoid infiltrates.
Table 1.
Ctns expression and cystine content decrease in tissues of Ctns−/− mice treated with wild-type BM-stem cells at 4 months post transplant
| Brain | Eye | Heart | Kidney | Liver | Muscle | Spleen | |
|---|---|---|---|---|---|---|---|
| Ctns expression, percentage of total cells | 5.9 | 13.4 | 11.0 | 12.9 | 9.6 | 5.4 | 19.3 |
| Percentage decreased of cystine content | 57.3 | 70.4 | 81.9 | 70.0 | 94.4 | 65.6 | 86.8 |
We investigated the long-term effects of HSC transplantation in Ctns−/− mice 7–15 months post-transplantation [20].The cystine content was significantly decreased in all tissues (from 54% in the kidney to 96.5% in the liver), proving that the treatment led to long-term and stably low levels of tissue cystine. We showed that syngeneic transplantation of HSC expressing a functional Ctns gene could provide long-term protection to the kidney and prevent the progression of the renal disease (Figure 1). However, we demonstrated that kidney preservation depends on achieving a relatively high level of donor-derived blood cell engraftment of Ctns-expressing cells, which is directly linked to the quantity of Ctns-expressing cells found within the kidney. These data suggest that the therapeutic benefits in the kidney after HSC transplantation are correlated with the quantity of transplanted stem cells expressing Ctns in the BM. These data also highlight the importance of testing the BM stem cell engraftment after transplantation to prevent the controversial data existing in the field of using adult stem cells for kidney repair [21]. In contrast, kidney preservation was not dependent on the age of the mice at the time of transplant, suggesting that if kidney injury is not too advanced the tissue could be rescued in older patients. Moreover, few to no cystine crystals were observed in all kidneys from treated mice, even the ones that were transplanted after 6 months of age. In contrast, abundant cystine crystals were consistently observed in the kidney from non-treated Ctns−/− mice. Thus, these results show that BM-derived cells specifically lead to the correction of cells containing cystine crystals.
This work represents the first proof of concept that transplantation of Ctns-expressing HSCs may lead to the successful treatment of cystinosis and could represent a new therapy for this disease. On 19 April 2012, Institutional Review Board approval was granted for an allogeneic transplantation protocol at the University of California, Los Angeles, to test the efficacy and toxicity of stem cell transplantation in patients affected with cystinosis. The clinical trial will include a maximum of six patients, aged ≥18 years, with significant signs of disease progression, or they can be children aged 13–17 years who do not tolerate cysteamine. Patients must also have a related BM donor who is HLA-matched on 10 of 10 alleles to minimize the risks of Graft-versus-host disease (GVHD). However, the strict HLA matching criterion considerably narrows down the pool of potential trial participants. To date, none of the candidates for the cystinosis allogeneic transplantation trial has had a full-match sibling. Moreover, in the case of cystinosis, where the introduction and regular use of the drug cysteamine have permitted patients to live to adulthood, albeit with significant medical problems [22], the risk–benefit ratio may not justify allogeneic HSC transplantation in young patients. Indeed, the risk of morbidity and mortality associated with allogeneic transplantation is significant, the major complication being GVHD [23, 24]. In recent studies, acute GVHD grade II-IV occurred in 20–32% of patients and chronic GVHD in 16–59%, both significantly impacting survival of the recipients [25–27]. Moreover, high risks of infection related to the myeloablative regimen and immunosuppressive medications account for 16–19% of deaths [28].
LENTIVIRAL-MODIFIED HEMATOPOIETIC STEM CELL TRANSPLANTATION FOR CYSTINOSIS: A SAFER STRATEGY
For the reasons described above, an autologous transplantation of HSCs ex vivo modified to express functional CTNS would be more appropriate for cystinosis. With regards to gene therapy, vectors derived from lentiviruses have replaced γ-retroviral vector due to their superior gene transfer efficiency and better biosafety profile [29–31]. Specifically, lentiviral vectors show greatly improved ability to transduce human HSCs compared to Murine Leukemia Virus (MLV)-based retroviral vectors, and deletions in their Long Terminal Repeats (LTRs) make the Self-Inactivated (SIN)-lentivirus vector a safer vector. Indeed, all cases of leukemogenic complications observed to date in clinical trials or animal models of gene therapy involved the use of retroviral vectors with LTR containing strong enhancer/promoters that can trigger distant enhancer activation [32–34]. In contrast, SIN-lentivirus vectors contain only one internal enhancer/promoter, which reduces the incidence of interactions with nearby cellular genes, and thus, decreases the risk of oncogenic integration [35, 36]. SIN-LTRs are also designed to prevent the possibility of developing replication competent lentivirus during production of the viral supernatants. Recently, a clinical trial using a SIN-LV to correct ex vivo HSCs in patients with X-adrenoleukodystrophy has achieved stable gene correction of ∼20% of hematopoietic cells [37]. Cerebral demyelination was arrested in the two patients without further progression over 3 years of follow-up, which represents a clinical outcome that is comparable with that observed after allogeneic transplantation [38]. No adverse event and no evidence of clonal dominance have been reported so far.
Using the Ctns−/− murine model for cystinosis, we tested the use of HSCs genetically modified ex vivo to express a functional CTNS transgene using a SIN-lentiviral vector (pCCL-CTNS) [39]. We are working with human CTNS complementary DNA (cDNA) in this animal study and using a vector backbone currently in application in clinical trials [37] to establish the preclinical proof of concept for a human trial. We showed that transduced cells were capable of decreasing the cystine content in all tissues, kidney function improved and cystine crystals resolved within the kidney (Figure 2). However, in most tissues, the impact of wild-type HSC transplantation was superior to that of pCCL-CTNS-transduced HSCs, demonstrating that the quantity of progenitors expressing CTNS is more critical than the over-expression of CTNS per cell. These data also confirm our previous statement, suggesting a correlation between the therapeutic benefits and the quantity of stem cell expressing Ctns in the BM [20]. Ex vivo modified HSCs retained their differentiative capabilities, populating all tissue compartments examined, and allowing long-term expression of the transgene. Thus, ex vivo cytokine activation of HSCs did not alter the lineage commitment pathways of progenitor cells. As seen for the wild-type HSCs, we observed that the BM-derived cells were mainly tissue-resident phagocytic cells such as dendritic cells in the kidney, Kupffer cells in the liver and microglial cells in the brain. Direct correlation between the levels of lentiviral DNA present in the peripheral blood and the levels present in tissues was demonstrated, suggesting that this could be a useful clinical marker for following future transplant in patients.
FIGURE 2:
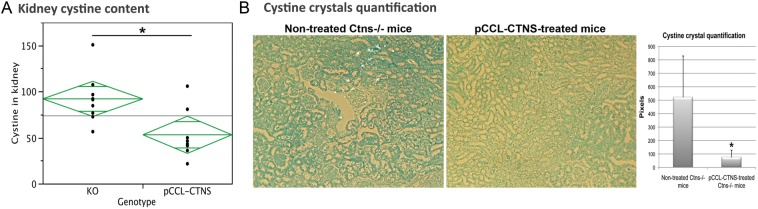
Kidney cystine and cystine crystal levels in Ctns−/− mouse controls or transplanted with Ctns−/− HSCs ex vivo modified with a pCCL-CTNS lentivirus vector [39]. (A) Cystine content levels (nmol half cystine/mg protein) in male Ctns−/− mice transplanted with pCCL-CTNS-transduced Ctns−/− HSCs (pCCL-CTNS) are significantly decreased compared with control non-treated Ctns−/− mice (KO) at 8 months post-transplant. (B) Kidney sections stained with methylene blue in ethanol were used to observe and quantify cystine crystals. Abundant cystine crystals were observed in kidney sections from non-treated Ctns−/− mice in contrast to pCCL-CTNS-treated mice. Error bars are defined as mean ± SD, *P < 0.05.
It is our long-term goal to develop a new therapy for cystinosis using autologous transplantation of HSCs ex vivo modified using a lentiviral vector to introduce a functional version of CTNS. Successful engraftment of the gene-modified stem cells into the BM compartment of cystinosis patients could serve as a source of healthy stem cells for any injured tissue for the life of the patient. Thus, autologous transplantation of gene-modified HSCs could potentially represent a life-long therapy that may prevent kidney transplantation and long-term complications associated with cystinosis. We recently concluded a pre-Investigational New Drug (IND) meeting for this product with the Food and Drug Administration (FDA) and in order to obtain an IND for a phase I clinical trial for cystinosis, we are now starting the pharmacology/toxicology studies on test product safety.
INSIGHT INTO THE MECHANISM FOR HSC-MEDIATED THERAPY FOR CYSTINOSIS
The observation that HSCs can rescue progressive tissue injury in multiple compartments in the context of non-hematopoietic genetic diseases is still controversial and the mechanism is unknown. Do the cells transdifferentiate within the tissues? Do the cells fuse with the host cells? Do they differentiate in hematopoietic lineage cells and have a paracrine effect? Could a combination of all these processes be involved?
In the context of cystinosis, we observed differentiation of the transplanted HSCs into tissue-resident phagocytic cells within tissues [18, 20, 39]. Tissue-resident phagocytic cells, macrophages and dendritic cells, are known to contribute to tissue homeostasis by, for instance, clearing apoptotic material [40]. We suspect that these cells have a major role in the pathogenesis of cystinosis. It is plausible that the demonstrated high level of cystine clearance after HSC transplantation is attributable to the phagocytosis of cystine-overloaded cells by the BM-derived cells expressing CTNS (Figure 3). Indeed, it has been shown that Kupffer cells contain 75% of the total cystine content in the liver, whereas they only represent <10% of the total cells in the liver [41].
FIGURE 3:
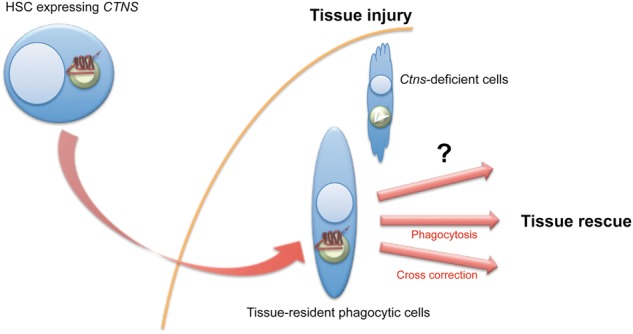
Fate of the HSCs after transplantation in the Ctns−/− mice. HSCs migrate to the injured tissues and differentiate into tissue-resident phagocytic cells. The question that remains unsolved is how these cells lead to tissue rescue? Is it by phagocytizing the cystine-overloaded cells, by transferring the functional cystinosin to adjacent cells or by another pathway?
Alternatively, these cells might transfer the functional cystinosin from the CTNS-expressing cells to the adjacent deficient cells. Cross correction has been well demonstrated in the case of secreted enzymes in several lysosomal storage disorders such as Hurler syndrome [42]. In this case and in the case of other lysosomal enzyme-deficient LSDs, the uptake of the enzymes from the extracellular milieu by neighboring cells occurs via the mannose-6-phosphate receptor that subsequently mediates endocytosis and lysosomal targeting [43, 44]. However, the protein involved in cystinosis is a seven-transmembrane lysosomal protein. To test the existence of cross corrections in this context, we generated a new cystinosis mouse model that ubiquitously expressed the DsRed reporter gene. DsRed Ctns−/− HSCs were transduced with a lentiviral vector containing the CTNS-eGFP fusion cDNA and transplanted in our regular Ctns−/− mice [39]. Cystinosin-eGFP was observed in the lysosomes of DsRed-expressing BM-derived cells within the tissues. Remarkably, we also observed host cells containing eGFP-positive intracellular vesicles. In vitro, we also showed that cystinosin could be transferred to adjacent Ctns-deficient cells leading to their significant cystine content decrease. These data showed that, although cystinosin is a lysosomal transmembrane protein, a transfer to adjacent cells is possible and functionally active (Figure 3).
However, direct secretion of cystinosin is not possible. Microvesicles or exosomes containing CTNS genetic material, both released from the endosomal compartment, could be involved [45, 46]. Recently, Dr Iglesias et al. showed that human MSCs could release microvesicles containing cystinosin protein and mRNA that led to cystine decrease in human CTNS-deficient cells [47]. Additional studies are still necessary to demonstrate the route for cystinosin transfer in vitro and in vivo and the mechanisms by which HSCs can lead to tissue preservation in cystinosis.
CONCLUSION: WHAT DOES THIS WORK BRING TO THE FIELD?
This work is important given the conflicting data and non-reproducible results that have led to negative pronouncements such as ‘unmanipulated HSCs are unlikely to provide organ repair beyond hematopoiesis’ [48]. Indeed, HSC plasticity is still not commonly accepted and whether HSCs can rescue progressive tissue injury in multiple compartments, including the kidney, is under debate [49]. Thus, we started with some reasonable skepticism that simply transplanting Ctns-expressing HSCs in the Ctns−/− mice would not be sufficient to address the serious global effects of this genetic disease. This was particularly a concern, given that cystinosin is an ubiquitous, intracellular, transmembrane lysosomal protein [2–4]. However, we were able to demonstrate that transplantation of HSCs expressing a functional Ctns gene could be therapeutic. The challenge is now to test this treatment in patients with cystinosis. HSC gene therapy will be the safest approach, compared with allogeneic HSC transplantation; however, it will require several years to conduct the safety studies required by the FDA.
If this therapy is successful in humans, this work might change the course of treatment for other genetic diseases for which curative therapy requires gene addition to many cells in multiple tissue compartments, where the protein involved is an intracellular transmembrane protein, and for which HSC transplantation has never been considered as an option. This work might also bring new insights into the field of tissue repair by HSC transplantation as applied to non-hematopoietic disorders. Our data suggest that no transdifferentiation occurs with HSCs, but rather differentiation in tissue-resident phagocytic cells. Unraveling the mechanisms by which HSCs could lead to tissue repair in the context of cystinosis could lead to new concepts such as the existence of cross correction in the case of a lysosomal transmembrane protein. Therefore, not only do we believe that genetic rescue is indeed an achievable goal for cystinosis, this work might take regenerative medicine in a different direction that has not yet been explored.
CONFLICT OF INTEREST STATEMENT
None declared.
ACKNOWLEDGEMENTS
The author is funded by the Cystinosis Research Foundation and NIH RO1-DK090058 and R21-DK090548.
REFERENCES
- 1.Gahl WA, Thoene JG, Schneider JA. Cystinosis. N Engl J Med. 2002;347:111–121. doi: 10.1056/NEJMra020552. [DOI] [PubMed] [Google Scholar]
- 2.Town M, Jean G, Cherqui S, et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat Genet. 1998;18:319–324. doi: 10.1038/ng0498-319. [DOI] [PubMed] [Google Scholar]
- 3.Cherqui S, Kalatzis V, Trugnan G, et al. The targeting of cystinosin to the lysosomal membrane requires a tyrosine-based signal and a novel sorting motif. J Biol Chem. 2001;276:13314–13321. doi: 10.1074/jbc.M010562200. [DOI] [PubMed] [Google Scholar]
- 4.Kalatzis V, Cherqui S, Antignac C, et al. Cystinosin, the protein defective in cystinosis, is a H(+)-driven lysosomal cystine transporter. EMBO J. 2001;20:5940–5949. doi: 10.1093/emboj/20.21.5940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gahl WA, Balog JZ, Kleta R. Nephropathic cystinosis in adults: natural history and effects of oral cysteamine therapy. Ann Intern Med. 2007;147:242–250. doi: 10.7326/0003-4819-147-4-200708210-00006. [DOI] [PubMed] [Google Scholar]
- 6.Brodin-Sartorius A, Tete MJ, Niaudet P, et al. Cysteamine therapy delays the progression of nephropathic cystinosis in late adolescents and adults. Kidney Int. 2011;81:179–189. doi: 10.1038/ki.2011.277. [DOI] [PubMed] [Google Scholar]
- 7.Cherqui S, Sevin C, Hamard G, et al. Intralysosomal cystine accumulation in mice lacking cystinosin, the protein defective in cystinosis. Mol Cell Biol. 2002;22:7622–7632. doi: 10.1128/MCB.22.21.7622-7632.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalatzis V, Serratrice N, Hippert C, et al. The ocular anomalies in a cystinosis animal model mimic disease pathogenesis. Pediatr Res. 2007;62:156–162. doi: 10.1203/PDR.0b013e31809fda89. [DOI] [PubMed] [Google Scholar]
- 9.Nevo N, Chol M, Bailleux A, et al. Renal phenotype of the cystinosis mouse model is dependent upon genetic background. Nephrol Dial Transplant. 2010;25:1059–1066. doi: 10.1093/ndt/gfp553. [DOI] [PubMed] [Google Scholar]
- 10.Krivit W, Aubourg P, Shapiro E, et al. Bone marrow transplantation for globoid cell leukodystrophy, adrenoleukodystrophy, metachromatic leukodystrophy and Hurler syndrome. Curr Opin Hematol. 1999;6:377–382. doi: 10.1097/00062752-199911000-00004. [DOI] [PubMed] [Google Scholar]
- 11.Platt FM, Boland B, van der Spoel AC. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol. 2012;199:723–734. doi: 10.1083/jcb.201208152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quesenberry PJ, Abedi M, Aliotta J, et al. Stem cell plasticity: an overview. Blood Cells Mol Dis. 2004;32:1–4. doi: 10.1016/j.bcmd.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 13.Brighton CT, Hunt RM. Early histological and ultrastructural changes in medullary fracture callus. J Bone Joint Surg Am. 1991;73:832–847. [PubMed] [Google Scholar]
- 14.Carroll SH, Ravid K. Differentiation of mesenchymal stem cells to osteoblasts and chondrocytes: a focus on adenosine receptors. Expert Rev Mol Med. 2013;15 doi: 10.1017/erm.2013.2. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 15.Wakitani S, Nawata M, Tensho K, et al. Repair of articular cartilage defects in the patello-femoral joint with autologous bone marrow mesenchymal cell transplantation: three case reports involving nine defects in five knees. J Tissue Eng Regen Med. 2007;1:74–79. doi: 10.1002/term.8. [DOI] [PubMed] [Google Scholar]
- 16.Ogawa M, Larue AC, Mehrotra M. Hematopoietic stem cells are pluripotent and not just ‘hematopoietic. Blood Cells Mol Dis. 2013;51:3–8. doi: 10.1016/j.bcmd.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagers AJ, Sherwood RI, Christensen JL, et al. Little evidence for developmental plasticity of adult hematopoietic stem cells. Science. 2002;297:2256–2259. doi: 10.1126/science.1074807. [DOI] [PubMed] [Google Scholar]
- 18.Syres K, Harrison F, Tadlock M, et al. Successful treatment of the murine model of cystinosis using bone marrow cell transplantation. Blood. 2009;114:2542–2552. doi: 10.1182/blood-2009-03-213934. [DOI] [PubMed] [Google Scholar]
- 19.Baer PC, Geiger H. Mesenchymal stem cell interactions with growth factors on kidney repair. Curr Opin Nephrol Hypertens. 2010;19:1–6. doi: 10.1097/MNH.0b013e328333062c. [DOI] [PubMed] [Google Scholar]
- 20.Yeagy BA, Harrison F, Gubler MC, et al. Kidney preservation by bone marrow cell transplantation in hereditary nephropathy. Kidney Int. 2011;79:1198–1206. doi: 10.1038/ki.2010.537. [DOI] [PubMed] [Google Scholar]
- 21.Yeagy BA, Cherqui S. Kidney repair and stem cells: a complex and controversial process. Pediatr Nephrol. 2011;26:1427–1434. doi: 10.1007/s00467-011-1789-x. [DOI] [PubMed] [Google Scholar]
- 22.Cherqui S. Cysteamine therapy: a treatment for cystinosis, not a cure. Kidney Int. 2012;81:127–129. doi: 10.1038/ki.2011.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnston L. Acute graft-versus-host disease: differing risk with differing graft sources and conditioning intensity. Best Pract Res Clin Haematol. 2008;21:177–192. doi: 10.1016/j.beha.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 24.Pallera AM, Schwartzberg LS. Managing the toxicity of hematopoietic stem cell transplant. J Support Oncol. 2004;2:223–237. discussion 237–8, 241, 246–247. [PubMed] [Google Scholar]
- 25.Cutler C, Li S, Ho VT, et al. Extended follow-up of methotrexate-free immunosuppression using sirolimus and tacrolimus in related and unrelated donor peripheral blood stem cell transplantation. Blood. 2007;109:3108–3114. doi: 10.1182/blood-2006-09-046219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geyer MB, Jacobson JS, Freedman J, et al. A comparison of immune reconstitution and graft-versus-host disease following myeloablative conditioning versus reduced toxicity conditioning and umbilical cord blood transplantation in paediatric recipients. Br J Haematol. 2011;155:218–234. doi: 10.1111/j.1365-2141.2011.08822.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schleuning M, Judith D, Jedlickova Z, et al. Calcineurin inhibitor-free GVHD prophylaxis with sirolimus, mycophenolate mofetil and ATG in Allo-SCT for leukemia patients with high relapse risk: an observational cohort study. Bone Marrow Transplant. 2009;43:717–723. doi: 10.1038/bmt.2008.377. [DOI] [PubMed] [Google Scholar]
- 28.Wingard JR, Hsu J, Hiemenz JW. Hematopoietic stem cell transplantation: an overview of infection risks and epidemiology. Infect Dis Clin North Am. 2010;24:257–272. doi: 10.1016/j.idc.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 29.Case SS, Price MA, Jordan CT, et al. Stable transduction of quiescent CD34(+)CD38(−) human hematopoietic cells by HIV-1-based lentiviral vectors. Proc Natl Acad Sci USA. 1999;96:2988–2993. doi: 10.1073/pnas.96.6.2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyoshi H, Smith KA, Mosier DE, et al. Transduction of human CD34+ cells that mediate long-term engraftment of NOD/SCID mice by HIV vectors. Science. 1999;283:682–686. doi: 10.1126/science.283.5402.682. [DOI] [PubMed] [Google Scholar]
- 31.Naldini L, Blomer U, Gallay P, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 32.Hacein-Bey-Abina S, Garrigue A, Wang GP, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Z, Dullmann J, Schiedlmeier B, et al. Murine leukemia induced by retroviral gene marking. Science. 2002;296:497. doi: 10.1126/science.1068893. [DOI] [PubMed] [Google Scholar]
- 34.Modlich U, Kustikova OS, Schmidt M, et al. Leukemias following retroviral transfer of multidrug resistance 1 (MDR1) are driven by combinatorial insertional mutagenesis. Blood. 2005;105:4235–4246. doi: 10.1182/blood-2004-11-4535. [DOI] [PubMed] [Google Scholar]
- 35.Modlich U, Bohne J, Schmidt M, et al. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood. 2006;108:2545–2553. doi: 10.1182/blood-2005-08-024976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Montini E, Cesana D, Schmidt M, et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J Clin Invest. 2009;119:964–975. doi: 10.1172/JCI37630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cartier N, Hacein-Bey-Abina S, Bartholomae CC, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 38.Cartier N, Hacein-Bey-Abina S, Bartholomae CC, et al. Lentiviral hematopoietic cell gene therapy for X-linked adrenoleukodystrophy. Methods Enzymol. 2012;507:187–198. doi: 10.1016/B978-0-12-386509-0.00010-7. [DOI] [PubMed] [Google Scholar]
- 39.Harrison F, Yeagy BA, Rocca CJ, et al. Hematopoietic stem cell gene therapy for the multisystemic lysosomal storage disorder cystinosis. Mol Ther. 2013;21:433–44. doi: 10.1038/mt.2012.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lech M, Grobmayr R, Weidenbusch M, et al. Tissues use resident dendritic cells and macrophages to maintain homeostasis and to regain homeostasis upon tissue injury: the immunoregulatory role of changing tissue environments. Mediators Inflamm. 2012;2012:951390. doi: 10.1155/2012/951390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hippert C, Dubois G, Morin C, et al. Gene transfer may be preventive but not curative for a lysosomal transport disorder. Mol Ther. 2008;16:1372–1381. doi: 10.1038/mt.2008.126. [DOI] [PubMed] [Google Scholar]
- 42.Di Domenico C, Villani GR, Di Napoli D, et al. Gene therapy for a mucopolysaccharidosis type I murine model with lentiviral-IDUA vector. Hum Gene Ther. 2005;16:81–90. doi: 10.1089/hum.2005.16.81. [DOI] [PubMed] [Google Scholar]
- 43.Kosuga M, Takahashi S, Sasaki K, et al. Adenovirus-mediated gene therapy for mucopolysaccharidosis VII: involvement of cross-correction in wide-spread distribution of the gene products and long-term effects of CTLA-4Ig coexpression. Mol Ther. 2000;1:406–413. doi: 10.1006/mthe.2000.0067. [DOI] [PubMed] [Google Scholar]
- 44.Tsuji D, Kuroki A, Ishibashi Y, et al. Metabolic correction in microglia derived from Sandhoff disease model mice. J Neurochem. 2005;94:1631–1638. doi: 10.1111/j.1471-4159.2005.03317.x. [DOI] [PubMed] [Google Scholar]
- 45.Camussi G, Deregibus MC, Bruno S, et al. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 2010;78:838–848. doi: 10.1038/ki.2010.278. [DOI] [PubMed] [Google Scholar]
- 46.Zomer A, Vendrig T, Hopmans ES, et al. Exosomes: fit to deliver small RNA. Commun Integr Biol. 2010;3:447–450. doi: 10.4161/cib.3.5.12339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iglesias DM, El-Kares R, Taranta A, et al. Stem cell microvesicles transfer cystinosin to human cystinotic cells and reduce cystine accumulation in vitro. PLoS One. 2012;7:e42840. doi: 10.1371/journal.pone.0042840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rovo A, Gratwohl A. Plasticity after allogeneic hematopoietic stem cell transplantation. Biol Chem. 2008;389:825–836. doi: 10.1515/BC.2008.103. [DOI] [PubMed] [Google Scholar]
- 49.Theise ND. Stem cell plasticity: recapping the decade, mapping the future. Exp Hematol. 2010;38:529–539. doi: 10.1016/j.exphem.2010.04.013. [DOI] [PubMed] [Google Scholar]