

Abstract
During storage stability studies of a monoclonal antibody (mAb) it was determined that the primary route of degradation involved fragmentation into lower molecular weight species. The fragmentation was characterized with size-exclusion high performance liquid chromatography (SE-HPLC), SDS-PAGE and matrix assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectrometry. Fragmentation proceeded via hydrolysis, likely catalyzed by trace metal ions, of a peptide bond in the hinge region of the mAb’s heavy chain, which produced two prominent low molecular weight species during storage: a single, free Fab fragment and a Fab+Fc fragment. The fragmentation is observed in phosphate-buffered solutions at two ionic strengths but not in histidine-buffered solutions at identical ionic strengths. Chaotrope-induced and thermally-induced unfolding studies of the mAb indicated differences in the unfolding pathways between the two buffer solutions. This folding intermediate observed during chaotrope-induced unfolding was further characterized by intrinsic fluorescence quenching, which suggested that a small portion of the molecule is resistant to chaotrope-induced unfolding in histidine buffer systems. The thermally-induced unfolding indicates a reduction in cooperativity of the unfolding process in the presence of histidine relative to phosphate. A relationship between the histidine-induced effects on unfolding pathway and the relative resistance to fragmentation is suggested.
Introduction
Monoclonal antibodies (mAbs) represent a growing class of protein therapeutics, and the formulation of mAbs has become an important issue for many biopharmaceutical companies.1,2,3 Antibodies may exhibit instabilities including aggregation, fragmentation, oxidation, deamidation and other chemical degradations;3 thus, development of formulations in which antibodies are stable represents a challenge for the industry.
The current study focuses on fragmentation of a model mAb during storage in liquid formulations. Several fragmentation mechanisms for mAbs have been characterized, including disulfide bond disruption,4 hydrolysis in the hinge region at and near an aspartate-lysine bond5,6 and other cleavage routes favored by specific temperature, chemical and pH conditions.7,8,9,10,11 Here, we examine the effects of buffer species on fragmentation of a mAb. Choice of buffer is critical for successful development of stable protein formulations.11,12,13,14,15 We study the interdependence of buffer type, mAb fragmentation and mAb conformational stability. Conformational stability has been shown to significantly affect the rates of chemical degradation of proteins, as was shown in studies of oxidation of subtilisin.16 Fragmentation was monitored during isothermal incubation studies conducted in histidine or phosphate buffers at two ionic strengths, two buffers used for in antibody formulations.3 The mAb is shown to fragment into to primary species with molecular weights of approximately 100 and 50 kDa and the degradation process is demonstrated to occur more readily in phosphate buffer systems relative to histidine buffer systems. Fragments were characterized by SDS gel electrophoresis, size exclusion chromatography coupled with light scattering and matrix-assisted laser desorption ionization mass spectrometry. Conformational stability was assessed by differential scanning calorimetry, urea-induced unfolding curves and fluorescence quenching.
Materials and Methods
The fully humanized monoclonal antibody of the IgG1 subclass was manufactured by MedImmune Inc, Baltimore (USA) and has a molecular weight of 148 kDa. The sample purity (>99%) was analyzed and confirmed by size exclusion chromatography and gel electrophoresis. All other chemicals and reagents were of reagent grade. Protein concentrations were determined by UV absorption using an extinction coefficient of 1.61 cm2/mg at 280 nm (MedImmune, personal communication).
Incubation and chromatography
Protein samples (5 mg/mL) were prepared by dialysis into the desired buffer with four replacements of buffer over a minimum of 24 hours. The final dialysate was used for dilutions and blanks as necessary. Four buffered solutions at pH of 6 were used for the incubation experiments: 10 mM histidine, 10 mM histidine with 150 mM sodium chloride, 2.2 mM sodium phosphate or 2.2 mM sodium phosphate with 150 mM sodium chloride. The sodium phosphate concentration was chosen to match the ionic strength of 10 mM histidine. For the incubation studies with chelator, the same four buffers conditions were achieved by dialysis with 2.5 mM ethylenediaminetetraacetic acid (EDTA). All buffers contained 0.2 mg/mL sodium azide to prevent bacterial growth during storage.
A separate 0.5 mL Eppendorf centrifuge tube containing 50 μL of protein solution was used for each sample replicate. Samples were stored were stored at 40 ± 1°C for various lengths of time. At each time point, three tubes of each formulation were removed and centrifuged at 15,400 g (13,200 RPM) for 5 minutes to remove particulate. The supernatant was analyzed with size-exclusion high performance liquid chromatography (SE-HPLC) for soluble protein. A Tosoh Biosciences (Montgomeryville, PA) model G3000SWXL column was used for SE-HPLC. In addition to absorbance at 280 nm, a Wyatt Technology Corp. (Santa Barbara, CA) Optilab DSP interferometric refractometer and a miniDawn light scattering detector were employed to yield molecular weight estimates. The total monomer and fragment concentrations were calculated by integrating the respective peak areas; all fragment extinction coefficients were assumed to be identical to that of the full-length mAb. The calculated extinction coefficients from the primary sequences for the intact mAb and the two primary fragments identified are 1.38, 1.38 and 1.37 cm2/mg. For this reason no correction to measured extinction coefficient for the full-length mAb was made for the fragments.
In order to obtain enriched fragmentation products several fractions of the eluate were collected. The first fraction collected was from 8.1–8.7 mL elution volume, the second from 9.2–9.8 mL and the third from 10.0–11.2 mL. The fractions were subsequently concentrated to approximately 2 mg/mL protein using 10,000 MWCO Millipore (Billerica, MA) centrifugal filters. These fractions were then analyzed with SDS-PAGE and MALDI-TOF.
SDS-PAGE
Because antibodies may be susceptible to fragmentation at the high temperatures typically used during sample preparation for SDS-PAGE,4 prior to electrophoresis the mAb samples (approximately 0.6 mg/mL) were incubated for 16 hours at room temperature in 1% (w/v) non-reducing sodium dodecyl sulfate solutions (Pierce Biotechnologies, Inc., Rockford, IL). Pre-cast 4–20% Tris-Glycine gels were used for electrophoresis (Invitrogen Corp., Carlsbad, CA). The GelCode Blue stain reagent was used to stain for protein and GelCode Glycoprotein staining kits were used to stain glycosylated protein (Pierce Biotechnologies, Inc., Rockford, IL).
Matrix Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF)
Aliquots (1 μL, approximately 0.8 mg/mL total protein concentration) of solutions containing mAb incubation products or fractionated incubation products collected after SE-HPLC separation were allowed to dry on a stainless steel sample plate designed for MALDI-TOF. A 1 μL aliquot of the matrix, which consisted of 22.4 mg/mL sinapinic acid dissolved in 8.0% de-ionized water, 36.0% methanol and 56.0% acetonitrile by volume, was applied to the dried mAb spot and allowed to dry. Spectra were collected on a Voyager-DE STR Biospectrometry Workstation (Applied Biosystems, Foster City, CA) with an accelerating voltage of 25 kV, a grid voltage of 92%, a guide wire voltage of 0.3% and 1700 ns extraction delay time. Gaussian smoothing was applied to the raw spectra in order to remove noise and the resulting smoothed spectra peaks were fit to Gaussian distributions. The masses reported are the centers of the Gaussian fit and the error is the standard deviation of the fit. Three individual samples were purified via SE-HPLC and their mass spectra collected. Error for these determinations is reported as the error associated with the Gaussian fit to individual samples, which was larger than the sample-to-sample variation.
Preparation of fragment antigen binding (Fab) and fragment crystallizable (Fc)
Fab and Fc fragments were produced from the mAb using a Pierce kit, # 44885 (Pierce Biotechnologies, Inc., Rockford, IL). The digestion of the full-length mAb with papain was conducted according to the included protocol. Because the Protein A column provided inadequate resolution of fragments, a Hi-Trap DEAE ion exchange column (Amersham Biosciences, Sweden) was used to separate the digested Fab and Fc fragments.
Urea-induced equilibrium unfolding of the mAb
Urea stock solutions (10 M) were prepared in the desired buffer, and the urea concentrations were determined by measuring the refractive index of the solution.17 For unfolding experiments, samples were kept at a constant protein concentration of 0.1 g/L and the urea concentration was varied by mixing the proper amounts of urea from the stock solution with the corresponding buffer solution. A 10 mM Sodium Phosphate buffer was used as it provided a more consistent pH over the range of urea concentrations. For unfolding and refolding experiments the samples were incubated at 23°C for 12 h before measurements. This incubation time has been found to be sufficient to allow the protein to reach unfolding equilibrium, based on the reproducibility of the unfolding curves (data not shown). The refolding experiments were performed by successive dilution of an approximately 10 M urea sample and allowed to equilibrate at the new condition for 15 minutes prior to analysis. The resulting unfolding curves were fit using a global method as outlined by Bhuyan and Udgaonkar.18
Fluorescence spectroscopy
All spectroscopic measurements of the mAb were carried out at 23°C. Fluorescence measurements were carried out on a spectrofluorometer (SLM AMINCO, Urbana, IL, US) equipped with a temperature-controlled cell holder. Tryptophan fluorescence spectra were measured using excitation at 293 nm, and the emission spectra were collected between 310 to 400 nm. The spectral bandwidth was set at 4 nm for both excitation and emission.
Fluorescence quenching experiments
mAb solutions were placed in 0.2 cm cuvettes and the spectra were recorded between 310 – 400 nm after exciting at 293 nm. Aliquots of KI stock (6M) were added to the protein solution and mixed well. After incubation for a minimum of 3 minutes spectra were recorded. The solutions for quenching by KI contained 1 mM sodium thiosulfate to prevent formation of free iodine which can interfere with the tryptophan fluorescence signal.19 Tryptophan fluorescence quenching induced by KI was analyzed according to the Stern-Volmer and Lehrer equations. The quenching process can be described by the classical Stern-Volmer relationship.20
| (1) |
F0 and F are the fluorescence intensities in the absence and presence of quencher. [Q] is the concentration of the quencher, and KSV is the Stern-Volmer quenching constant. The fraction of total fluorophores accessible to the quencher can be calculated from the modified Stern-Volmer plot, also known as Lehrer’s plot.19
| (2) |
Where F0 is as defined earlier and ΔF is the change in the fluorescence intensity due to quenching, KQ is the Stern-Volmer quenching constant of the exposed tryptophan residues and fa is the fraction of the initial fluorescence which is accessible to the quencher. From this equation, a plot of F0/ΔF = f (1/[Q]) yields a straight line whose extrapolation at 1/[Q] = 0 gives the value of 1/fa on the axis F0/ΔF.
Differential scanning calorimetry measurements
Differential scanning calorimetry (DSC) experiments were performed on a VP-DSC Ultrasensitive Differential scanning calorimeter (Microcal, Northampton, MA). Protein solutions were dialyzed as described above and the respective dialysate was used in the reference cell. To ensure no bubble formation during the heating process, protein and buffer solutions were degassed with gentle stirring under vacuum for a minimum of 15 minutes before loading into the calorimeter. Samples were heated from 30–100°C at a rate of 1.5°C per minute. Normalized heat capacity (Cp) data were corrected for buffer baseline. Raw data from the DSC run were fit using Origin™ scientific plotting software which uses the Levenberg-Marquardt non-linear least square method.
Results
Thermally-accelerated degradation studies
Incubation of samples containing 5 mg/mL mAb in buffers containing 2.2 mM sodium phosphate or 2.2 mM sodium phosphate with 150 mM NaCl at 40°C resulted in mAb fragments (Figure 1). A trace amount of fragmentation was observed in samples buffered with 10 mM histidine. No insoluble aggregates were observed over the course of the incubation in any of the buffer systems tested. As can be seen from Figure 1B, for the 2.2 mM sodium phosphate formulation, only a minimal population of soluble species with a higher molecular weight than the monomer developed over the course of the study. Similar results were obtained with all the buffers tested (data not shown).
Figure 1.
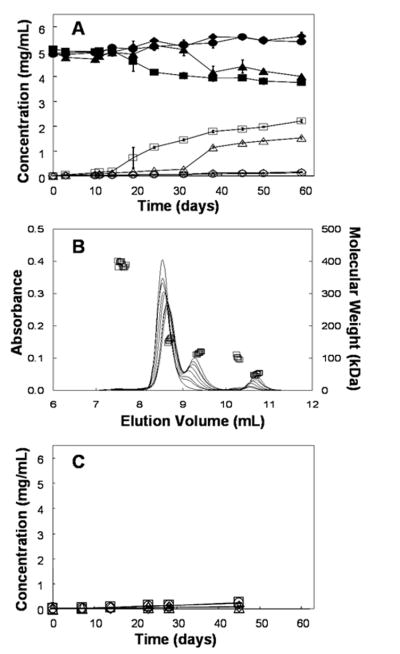
(A) Monomer concentration (closed symbols) and total fragment concentration (open symbols) during incubation at pH 6 and 40°C (●,○) 10 mM histidine, (◆,◇) 10 mM histidine, 150 mM NaCl, (■,□) 2.2 mM sodium phosphate (▲,△) 2.2 mM sodium phosphate, 150 mM NaCl (B) The curves are representative chromatograms measured by absorbance at 280 nm (left axis) from 0,10,14,24,31,38,50 and 59 days incubation at pH 6 and 40°C. The loss of full-length monomer and the growth of lower molecular weight fragment species are apparent over the course of the incubation. The data points (□) are molecular weight estimates from light scattering and correspond to the 50 day sample (right axis). (C) The total fragment concentration for the same incubation and buffer conditions as on panel A with the addition of 2.5 mM EDTA to all four buffer systems. The error bars represent the standard deviation from three independent samples; where they are not visible they are smaller than the symbols.
In Figure 1B, representative SE-HPLC chromatograms for samples incubated at 40°C in 2.2 mM sodium phosphate are shown with the corresponding molecular weights determined by online light scattering from the sample that had been incubated for 50 days. Several peaks are evident in the chromatograms: a small peak eluting at 7.5 mL, representing protein with a mass-averaged molecular weight of 392 ± 8 kDa (high molecular weight species HMWS), a large peak eluting at 8.6 ml for a species with a mass-averaged molecular weight of 157 ± 6 kDa (monomer), and peaks eluting at 9.3 ml, 10.2 ml, and 10.8 ml, representing species with mass-averaged molecular weights of 118 ± 3 kDa (fragment 1), 101 ± 3 kDa (fragment 2), and 51 ± 3 kDa (fragment 3), respectively.
2.2 mM sodium phosphate was chosen to match the ionic strength of 10 mM histidine because ionic strength can play an important role in many protein aggregation mechanisms.21,22 In this case, because fragmentation profiles were dramatically different in phosphate versus histidine-buffered solutions, even at equivalent ionic strengths, it appears that the buffer species itself affects fragmentation and not the ionic strength. From Figure 1C, it is clear that with the addition of 2.5 mM EDTA to phosphate buffer systems the fragmentation reaction is effectively eliminated over the time course of the experiment. To determine which metal ion contaminants are present inductively coupled plasma mass spectrometry (ICP-MS) was performed on the four buffer solutions. The primary metal ion contaminant identified was copper. In the 10 mM histidine solution the copper level was 7.3 ± 1.2 parts per billion (ppb), in the 10 mM histidine, 150 mM NaCl solution the copper level was 366 ± 26 ppb, in the 2.2 mM sodium phosphate solution the copper level was 15.2 ± 1.9 ppb and in the 2.2 mM sodium phosphate, 150 mM NaCl solution the copper level was 374 ± 21 ppb. Other metals tested such as nickel, zinc and cobalt were below detection limits with the exception of iron. The two sodium phosphate solutions contained and average of 1.6 ± 0.2 ppb iron while the two histidine samples were below the detection limit.
To further characterize the fragmentation products, parallel, non-reducing, SDS-PAGE analysis was performed on the incubation products before and after separation of fragments from monomer via SE-HPLC. One gel was stained for protein and the other for glycoprotein. Because the CH2 domains of the heavy chains (located in the Fc fragment) are the only portions of the mAb that are glycosylated, glycoprotein staining reveals the location of the Fc portion of the heavy chains and possibly Fc fragments. Figure 2A shows the results for the control (not incubated) mAb, and the gels pictured in Figures 2B and C correspond to the mAb sample in the 2.2 mM sodium phosphate buffer at pH 6 after 50 days of incubation at 40°C. Panels A and B are stained for protein while panel C is stained for glycoprotein. In lanes 2–4 of panel B the incubated samples show strong bands at molecular weights of approximately 150, 110, 50 and 25 kDa. The 25 kDa species seen in SDS-PAGE analysis is not evident in SE-HPLC chromatograms. The roughly 150, 110 and the 25 kDa species appear as double bands of nearly equivalent molecular weight, either suggesting that there are multiple fragmentation sites that generate species of similar molecular weights or that there are at least two primary isoforms of the intact, purified mAb. The latter explanation appears more likely, because the non-incubated mAb control shown in Figure 2A also exhibits a double band. In addition to the double bands, there are at least three additional faint bands that are most apparent in the highest loading (lane 3 figure 2B) between the 110 and 50 kDa species. These additional fragmentation products are not detected by SE-HPLC analysis.
Figure 2.
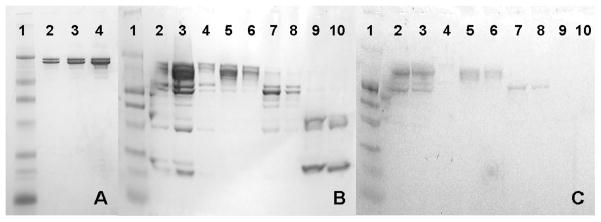
(A,B) Non-heated, non-reducing, SDS-PAGE gel stained for protein of the mAb in the 2.2 mM sodium phosphate, pH 6 formulation. (C) Same conditions as panels A and B except stained for glycoprotein (molecular weight marker is prestained for protein). The molecular weight marker in all three panels is in lane 1 and has bands at 210 kDa, 110 kDa, 80 kDa, 47 kDa, 32 kDa, 25 kDa and 16.5 kDa. Panel A consists of the control mAb (no incubation) in lanes 2–4 at loadings of 1, 2 and 5 μg protein. In panels B and C lanes 2–4 are the mAb incubation products after 50 days at 40°C as injected for SE-HPLC at three loadings. From left to right the loadings are approximately 5, 10 and 1 μg total protein respectively. Lanes 5 and 6 are the enriched monomer fraction collected during SE-HPLC (peak eluting at ~8.6 mL in Figure 1B) at loadings of 2 and 1 μg total protein respectively. Lanes 7 and 8 are the enriched large fragment fraction (fragment 1, the peak eluting at ~9.3 mL in Figure 1B) as collected from SE-HPLC at loadings of 2 and 1 μg total protein respectively. Lanes 9 and 10 are the enriched smaller fragment species that elute near 10.8 mL (fragment 3) as collected from SE-HPLC at loadings of 2 and 1 μg total protein respectively.
The fractions containing protein representing the major peaks were collected as they eluted from the SE-HPLC column, thus enriching each species, and were analyzed by SDS-PAGE (lanes 5–10, Fig. 2B). Again, the same primary species are identified at approximately 150, 110, 50 and 25 kDa, the primary bands appear as double bands and there are small amounts of other fragmentation products detected on the SDS-PAGE gels that are not observed during SE-HPLC analysis. Fragmentation during SDS-PAGE due to disulfide bond disruption, even in the absence of reducing agent, can result in low molecular weight artifacts and could be responsible for some fragmentation in addition to the fragmentation that occurs during incubation.4 From the sugar-stained gel in Figure 2C it is clear that the only species that are glycosylated are the monomer species, as expected, with an apparent molecular weight of approximately 150 kDa, and fragment 1, with an apparent molecular weight of approximately 110 kDa. Though less obvious in the sugar-stained gels both bands appear as double bands. The remaining smaller degradation products, including fragments 3 and the smaller fragment, with apparent molecular weights of approximately 50 and 25 kDa, are not glycosylated.
To determine the location of one of the primary fragmentation sites, purified fragment 3 from SE-HPLC was subjected to MALDI-TOF analysis which yielded a molecular weight of 47.48 ± 0.62 kDa (Figure 3A). This value compares well to the results from SDS page, where fragment 3 had an apparent molecular weight of approximately 50 kDa. Fragment 3 is the only major fragment which does not appear as a clear double band in the SDS-PAGE gel, but its MALDI-TOF spectrum exhibits a somewhat broad peak with a hint of an unresolved double feature. This suggests that the variability for a given fragment species is likely due to multiple species very similar in mass. The unfractionated incubation products and the other two fractions from SE-HPLC collected were also characterized with MALDI-TOF (Figure 3). The monomer fraction yielded a molecular weight of 148.2 ± 1.9 kDa in the control (not incubated), the unfractionated incubated sample and the enriched fraction. Although the MALDI-TOF spectrum for each sample shows somewhat broad peaks, the molecular weights of the isoforms resulting in double bands in the SDS-PAGE analysis could not be resolved by MALDI-TOF. The MALDI-TOF spectrum from the monomer fraction collected with SE-HPLC after 50 days incubation at 40°C (not shown) is nearly identical to that of the control (not incubated) mAb suggesting that much of the polydispersity in the fragment sizes is due to isoforms present in the initial purified mAb that result in small differences in the molecular weight. The largest fragment gives a molecular weight 101.0 ± 2.1 kDa in both the unfractionated and enriched fraction. The difference between the monomer and the large fragment is 47.2 kDa which corresponds well to the fragment 3 molecular weight of 47.48 ± 0.62 kDa.
Figure 3.
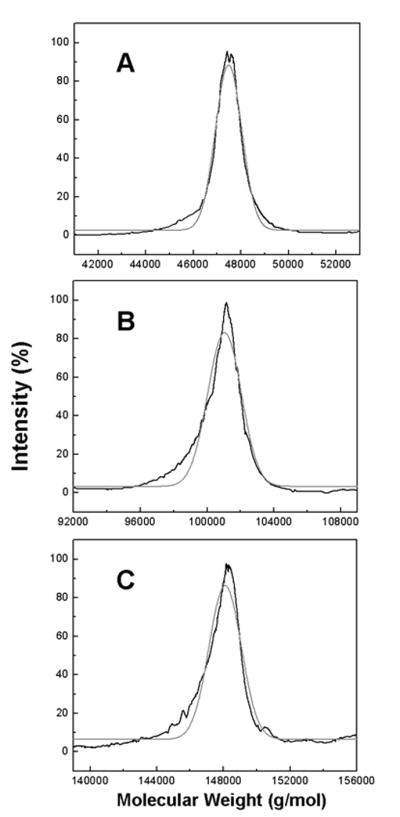
MALDI-TOF mass spectrum of the enriched species collected from SE-HPLC of the mAb after 50 days of incubation at 40°C in the 2.2 mM sodium phosphate, pH 6 formulation. Panel A displays enriched fragment 3 (the peak that eluted near 10.8 mL in Figure 1B). Panel B contains the enriched fragment 1 (the peak that eluted near 9.2 mL in Figure 1B). Panel C displays the enriched monomer fraction (the peak that eluted near 8.6 mL in Figure 1B). The black lines represent the smoothed raw intensity. The gray curves are the Gaussian fits of the intensity. The centroided masses are 47.48 ± 0.62 kDa for fragment 3, 101.0 ± 2.1 kDa for fragment 1 and 148.2 ± 1.9 kDa for the monomer fraction.
Calorimetric analysis of the mAb and fragments
Calorimetric detection of thermally-induced transitions was employed to gauge the conformational stability of the mAb in histidine or phosphate-buffered solutions in both the presence and absence of 150 mM NaCl. In these solutions none of the thermal unfolding events were fully reversible, and so all reported transitions must be regarded as apparent transition temperatures. The antibody aggregated during the thermal scan as was evidenced by the cloudy, particulate-containing solutions removed at the end of the DSC experiment. Therefore the aggregation reaction could have affected the apparent Tm measurements. Figure 4A–D displays the DSC profiles for the intact mAb and its isolated Fc and Fab fragments. Panels A–D are for solution conditions comprising 10 mM histidine, 10 mM sodium phosphate, 10 mM histidine plus 150 mM NaCl, and 10 mM sodium phosphate plus 150 mM NaCl, respectively.
Figure 4.
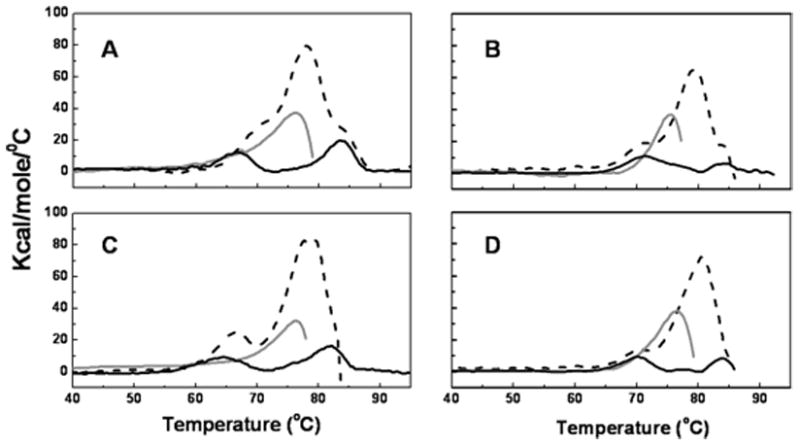
Differential Scanning Calorimetry (DSC) of the mAb at pH 6. Dashed black lines correspond to the full-length mAb, solid gray lines correspond to the Fab fragment of the mAb and the solid black lines correspond to the Fc fragment of the mAb. Panel A corresponds to the 10 mM histidine formulation. Panel B corresponds to the 10 mM sodium phosphate formulation. Panel C represents the 10 mM histidine, 150 mM NaCl formulation. Panel D represents the 10 mM sodium phosphate, 150 mM NaCl formulation.
In solutions of the intact mAb that contain 10 mM histidine (Figure 4A), three overlapping transitions centered at 69.6 ± 1.1, 77.5 ± 0.8 and 85.4 ± 1.4 °C are apparent in the thermogram. All errors represent standard deviations from three independent experiments. In the presence of 150 mM NaCl, (Figure 4C), only two transitions, one at 66.6 ± 0.6 and the other at 78.7 ± 0.5 °C, can be resolved. In contrast, the two resolved thermal transitions (occurring at 70.6 ± 0.1 and 79.7 ± 0.5 °C, respectively) that are observed in solutions of the intact mAb containing 10 mM sodium phosphate are unaffected by 150 mM NaCl and the averages reported here are the combined results for both formulations (Figures 4B and 4D).
For the Fab fragments, a single transition with a Tm near 76 °C was observed in all four buffer systems (Figure 4A–D). The average of the four is 75.6 ± 0.5 °C. The Fc fragments exhibit two thermal transitions, the second of which has a Tm of approximately 83 °C in all four buffer systems. The average is 83.1 ± 1.1 °C. The first transition temperature of the Fc fragments, however, is affected by the buffer system. In both the histidine and phosphate buffer systems the Tm of the first transition is reduced slightly in the presence of 150 mM NaCl. In the 10 mM histidine formulation the first transition occurs at 66.7 ± 0.8 °C and the 10 mM histidine, 150 mM NaCl formulation has an initial transition at 64.1 ± 1.0 °C. The first transition in the 10 mM sodium phosphate formulation occurs at 71.5 ± 0.3 °C, and the 10 mM sodium phosphate, 150 mM NaCl formulation produces the first transition at 69.8 ± 0.4 °C. However, the more significant difference is that the apparent melting temperature for the first transition is approximately 5 °C lower in histidine than in phosphate-buffered solutions at both ionic strengths.
The lower-temperature transition observed in the thermograms of the Fc fragments can be assigned to the CH2 domain, whereas the higher-temperature transition is that of CH3 domain.23 From the DSC studies of Fab and Fc fragments, the thermal transitions for the full-length mAb can be assigned. The first transition in Figure 4A with a Tm near 70 °C can be assigned to CH2 domain, the main transition (Tm of 78 °C) is from the domains of Fab region and the last transition with a Tm of around 83 °C is due to the unfolding of the CH3 domains.
Interestingly, in the thermograms for experiments conducted in NaCl-free histidine buffer, shown in Figure 4A, the melting temperature of the CH2 domain in the full-length mAb is approximately 4°C higher than the melting temperature for the isolated Fc fragment. However, in the presence 150 mM NaCl (Figure 4C), the stabilizing effect of the intact mAb on the melting temperature of the CH2 domain is less significant. In contrast, Fc melting temperatures in phosphate systems both with and without 150 mM NaCl (Figure 4B and 4D) are very similar for the full-length mAb and for isolated Fc fragments.
Melting transitions for isolated Fab fragments (Figures 4A–D) were insensitive to solution conditions. However, the Fab melting temperatures in the full length mAb were about 2–3°C higher than those for isolated Fab fragments in the same solutions.
Urea-induced equilibrium unfolding of mAb
Figure 5 shows the urea-induced unfolding of the mAb as studied by intrinsic tryptophan fluorescence measurements. The center of spectral mass (CSM) was determined as a function of denaturant concentration to monitor mAb unfolding. The mAb used in this study contains 22 tryptophan residues, which are nearly evenly distributed along the polypeptide chains. The native molecule has a maximum fluorescence emission at 330 nm. Upon unfolding the maximum is red-shifted to 352 nm.
Figure 5.
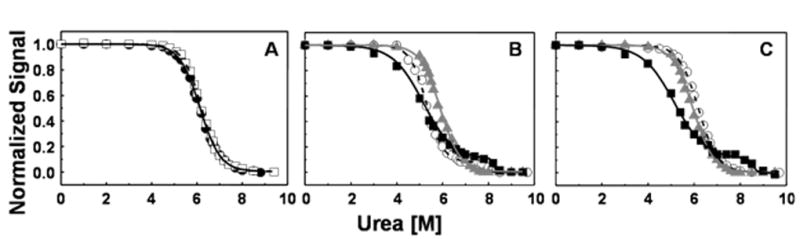
Denaturant unfolding curves utilizing intrinsic fluorescence, all samples at pH 6. Panel A is the full-length mAb in the 10 mM sodium phosphate (solid black circles and solid black line) and 10 mM sodium phosphate, 150 mM NaCl (open squares and dashed line). In panels B and C the solid black squares with the solid black lines correspond to the Fc fragment, the solid gray triangles with solid gray lines correspond to the Fab fragment and the open circles with dashed line correspond to the full-length mAb. Panel B the 10 mM Histidine, 150 mM NaCl formulation and panel C contains the 10 mM histidine formulation. The Fc fragments and the full-length mAb in the 10 mM histidine, 150 mM NaCl are fit to three-state models; all remaining unfolding curves are fit with a two-state model.
In 10 mM phosphate buffer, both in absence and presence of 150 mM NaCl (Figure 5A) the unfolding transition of the mAb can be described by a single sigmoidal curve, consistent with a simple 2-state N ↔ U transition with a ΔG value 7.35 ± 0.12 kcal/mole and an m value of 1.19 ± 0.02 kcal/mole/M. The errors reported here represent the standard deviation from fits to the data from three separate experiments. Usually, for small, single domain proteins, the m value correlates very strongly with the amount of protein surface exposed to solvent upon unfolding.24 In the 10 mM histidine, 150 mM NaCl formulation (Figure 5B) the unfolding transition of the full-length mAb can be described by 2 successive sigmoidal curves showing a clear 2-step transition from native (N) to unfolded (U) state with the formation of an intermediate (I) state around 7.2 M urea. The Cm values are 5.8 and 8.2M for first and second transitions, respectively. For the N to I transition, a ΔG value of 8.1 ± 0.3 kcal/mole and an m value of 1.40 ± 0.06 kcal/mole/M were obtained. For the I to U transition, a ΔG value of 18.4 ± 6.0 kcal/mole and an m value of 2.3 ± 0.8 kcal/mole/M were obtained from the curve fit. In 10 mM histidine at pH 6 without added NaCl, the mAb exhibits an apparent single unfolding transition (Figure 5C) that can be fit to a simple 2-state model to yield a free energy of unfolding (ΔG) value of 8.01 ± 0.20 kcal/mole, a half transition (Cm) value of 6.2 M urea and an m value of 1.30 ± 0.03 kcal/mole/M, where the m value is the dependence of the free energy of unfolding on denaturant concentration. Under all the conditions tested, the mAb unfolding by urea was completely reversible, as determined by refolding curves generated by a step-wise dilution of a solution containing an initial urea concentration of approximately 10M (data not shown).
Urea-induced unfolding conducted on isolated Fab fragments of the mAb resulted in a nearly identical unfolding curve to that of the full-length mAb in 10 mM histidine and are not affected by NaCl concentration (Figures 5B and C). However, urea-induced unfolding of isolated Fc fragments exhibited a clear three-state unfolding process, with the first transition occurring at lower urea concentration than the single step observed in unfolding experiments using isolated Fab fragments. The second transition occurs at a higher urea concentration than that required to unfold isolated Fab fragments. As with the unfolding curves for the Fab fragments, those for the Fc were not significantly affected by NaCl concentration.
Solvent exposure of tryptophan during unfolding
In order to gain further understanding of the intermediate state observed during unfolding of the full-length mAb, KI fluorescence quenching studies were employed. These studies can provide information on the relative tryptophan exposure to aqueous environment in the folded, intermediate and unfolded states. Stern-Volmer plots for the quenching of tryptophan fluorescence in 10 mM sodium phosphate or 10 mM histidine, both with 150 mM NaCl are shown in Figure 6 for the native protein (at 0 M Urea) and the protein in 7.5 M urea. The insets are the corresponding Lehrer’s plot, which were used to calculate fa, the fraction of initial tryptophan fluorescence accessible to the quencher. The reported values for the Stern-Volmer constant and fa value for free tryptophan as obtained by KI quenching are 11.3 and 1.0 respectively.25
Figure 6.
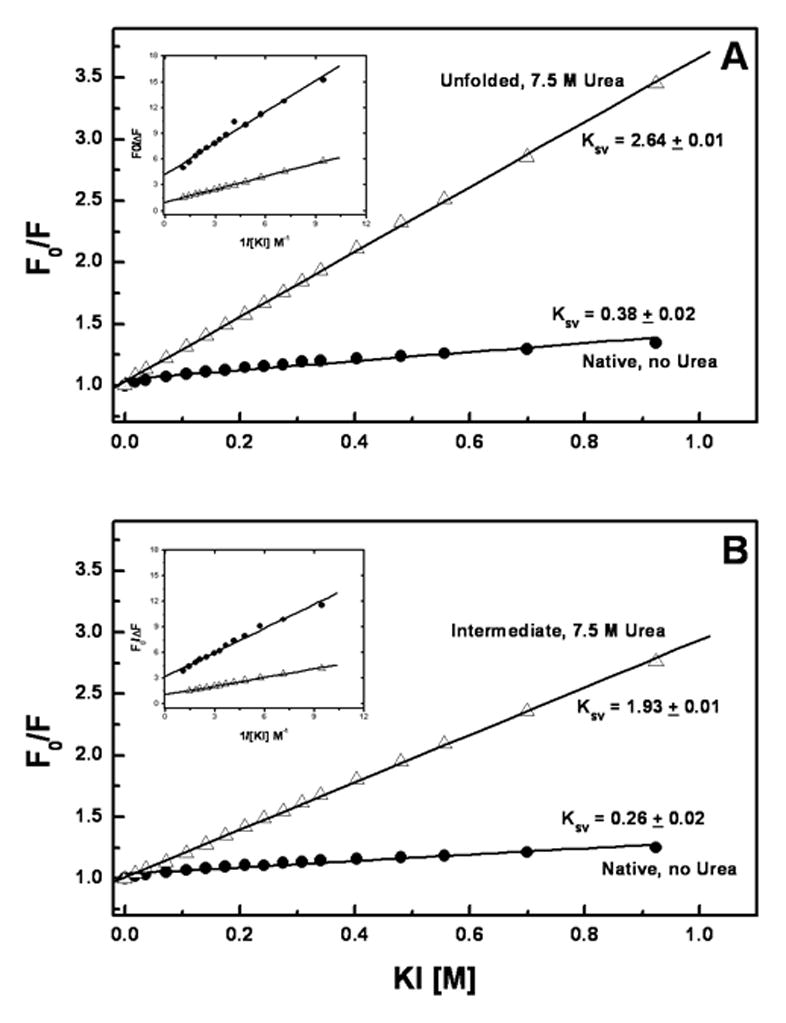
Panel A contains the Stern-Volmer plot of the mAb in the absence (●) and presence of 7.5 M urea (△) for the 10 mM phosphate, 150 mM NaCl, pH 6 formulation. Panel B contains the Stern-Volmer plot of the mAb in the absence (●) and presence of 7.5 M urea (△) for the 10 mM histidine, 150 mM NaCl, pH 6 formulation. Here F0 is the fluorescence intensity in absence of KI and F is the intensity in presence of KI. In both the panels, the solid lines correspond to the linear regression of the data set and the corresponding Stern-Volmer constants (Ksv) are displayed on the graphs. The inset shows the Lehrer’s plots for iodide quenching data, which were used to calculate the fraction of initial tryptophan fluorescence accessible to the quencher (fa).
Figure 6A shows the Stern-Volmer plots of the mAb in 10 mM phosphate and 150 mM NaCl, both in the absence and presence of 7.5 M urea. Under these conditions the Ksv values for native (no urea) and unfolded (7.5 M urea) protein were 0.36 and 2.94 M−1, respectively. The fa value obtained from Lehrer’s plot at 7.5M urea in 10 mM phosphate, 150 mM NaCl is 1.07 ± 0.02, which indicates that all tryptophan residues are accessible to quenching by iodide and hence exposed to solvent, which is indicative of essentially complete unfolding of the protein molecule. The fa value for the native (no urea) mAb was 0.23 ± 0.03 suggesting that approximately 77% of the tryptophan fluorescence residues are not accessible to the quencher.
The Stern-Volmer constant (Ksv) for the native mAb in 10 mM histidine, 150 mM NaCl is 0.26 ± 0.02 (Figure 6B). Upon addition of 7.5 M urea, a Ksv value of 1.93 ± 0.02 M−1 is observed. This intermediate Ksv value is consistent with the presence of a partially folded intermediate seen in urea induced unfolding experiments at 7.5 M urea (Figure 5). Lehrer’s plot analysis for the mAb in 10 mM histidine, 150 mM NaCl gave an fa value of 0.29 ± 0.02, indicating that under native conditions only 29% of the tryptophan fluorescence is accessible to physical contact with iodide. In the presence of 7.5 M urea, where the intermediate state is observed, a fa value of 0.82 ± 0.01 was obtained. This suggests that a small part of the mAb, perhaps one domain, is conformationally more stable in the 10 mM histidine, 150 mM NaCl buffer system than in the phosphate-buffered solutions. The two formulations without NaCl yielded similar results to those seen for the 10 mM phosphate, 150 mM NaCl formulation as there were no unfolding intermediates observed (data not shown).
Discussion
Conformational stability is known to play a significant role in protein aggregation13,22,26,27,28 and increased conformational stability of a protein has also been shown to correlate in some cases with increase resistance to chemical modification/degradation.29 Reaction site solvent accessibility is known to play a role in several pathways for chemical degradation of proteins including deamidation30 and oxidation.16,31 Additionally, Liu et al found that deamidation and fragmentation of antibodies typically occurs in the flexible regions, particularly the hinge region.10 Although it might be expected that increased solvent exposure results in increased fragmentation rates, little direct evidence is available to address this issue.5,11 In the present case, thermally-accelerated degradation studies revealed that fragmentation was the primary pathway by which the mAb degrades at 40°C, and that fragmentation levels were affected by buffer type. To explore the potential relationship of the observed buffer effects on fragmentation to the mAb conformational stability, the dominant site on the mAb where fragmentation occurred was determined and buffer effects on the mAb conformational stability were explored.
Two fragmentation mechanisms are responsible for the most prevalent forms of non-enzymatic mAb fragmentation: hydrolysis, primarily of the solvent accessible hinge region; and disruption of interchain disulfide bonds.3,4,5,6,10,32 There are two labile peptide bonds in the heavy chain of the mAb which are particularly susceptible to hydrolysis, namely the aspartate-proline30 and the bonds near the aspartate-lysine bond.5 The aspartate-proline bond is located just on the Fc side of the two interchain disulfide bonds of the hinge region whereas the aspartate-lysine bond is just on the Fab side, near the papain cleavage site. It has been recently noted that the location of hinge region fragmentation is dependent on the pH and tends to move towards the CH2 domain with decreasing pH.6 Furthermore, Gaza-Bulesco and Liu identified three bonds in the hinge region where hydrolysis occurs at the pH used in this study (pH 6), primarily the aspartate-lysine and the histidine-lysine bonds as well as the aspartate-threonine bond.6,10
The catalysis of protein hydrolysis by transition metals has long been known and is well described.33,34,35 This is due to the ability of transition metals to stabilize electrically excited transition states during hydrolysis reactions. To determine if any trace metal ions were responsible for the fragmentation observed in our studies, the incubation experiment was repeated for all buffers with the addition of a strong chelator, EDTA. The presence of the chelator significantly reduced the rate of fragmentation suggesting that trace metal ions are likely catalyzing the hydrolysis in the hinge region. The primary contaminating metal ion is copper which is known to catalyze hydrolysis of proteins.34 The sodium chloride is the primary source of the copper contamination. Both the phosphate solutions generate hydrolysis in the absence of chelator suggesting that 15 ppb copper contamination is sufficient for hydrolysis. However, the 10 mM histidine, 150 mM NaCl solution contains 366 ppb copper and yet no fragmentation was observed, even in the absence of chelator. Thus, the differences in trace metal ion levels do not explain why fragmentation is seen in one buffer system but is absent in the other.
There are two primary fragment species identified previously from Figure 1B, as fragment 1 and fragment 3, with apparent molecular weights of 118 and 51 kDa respectively. Both the hydrolysis and disulfide bond disruption mechanisms could potentially generate fragments of approximately these sizes, so the SE-HPLC results alone are not sufficient for inference of the identification of the cleavage sites. For this reason SDS-PAGE analysis of the incubation products (Figure 2) was performed. The gels were stained for either protein or carbohydrate. In the SDS-PAGE gels it is clear that there is another degradation product present with an approximate molecular weight of 25 kDa that is not observed during SE-HPLC. However, it is not clear whether this fragment is generated during incubation at 40°C or whether it is generated via the SDS-PAGE procedure, which is known to cause disulfide bond disruption in antibodies and generate a variety of low molecular weight artifacts.4 There are multiple faint bands in the gel that could represent small amounts of additional degradation products from the incubation that are not resolved with chromatography. Another possibility is that they are generated by the SDS-PAGE process. From Figure 2C it is clear that the only fragments that are glycosylated are the largest fragments with apparent molecular weights over 100 kDa. From this observation, certain deductions about which degradation mechanism is most likely responsible can be made. It is clearly not the hydrolysis of the aspartate-proline bond just below the hinge region, as this would result in a glycosylated fragment with an approximate molecular weight of 30 kDa. Furthermore, for disulfide bond disruption to produce a 50 kDa fragment the light chains and the heavy chains would all have to separate from one another, as is the case in the presence of a strong reducing agent, generating a glycosylated 50 kDa fragment. This is not observed. In fact, when the mAb is exposed to strong reducing agents such as dithiothreitol or tris(2-carboxyethyl)phosphine, it rapidly aggregates and forms visible particulates (data not shown). No significant soluble or insoluble aggregates were observed during the 40°C incubation. The SE-HPLC and the SDS-PAGE results are consistent with a fragmentation near the aspartate-lysine bond near the papain cleavage region. This would result in two fragments, a Fab and a Fab+Fc with molecular weights of approximately 47 kDa and 101 kDa respectively of which only the 101 kDa fragment would be glycosylated. This mechanism has been elucidated by Cordoba et al. and the chromatograms presented in their work as well as by Gaza-Bulesco and Liu are consistent with those presented here.5,6
MALDI-TOF was employed to confirm the hinge region fragmentation (Figure 3). Fragmentation of the aspartate-lysine bond in the hinge region of the mAb would generate a Fab fragment with a molecular weight of 47.4 kDa. This is nearly exactly what is measured via MALDI-TOF (47.48 ± 0.62 kDa) confirming that the fragmentation reaction is a hydrolysis occurring in the hinge region. There is clearly some polydispersity in the observed molecular weights but there is no clear evidence of two distinct populations as was observed with the appearance of double bands in SDS-PAGE. The hydrolysis is not necessarily occurring at only one site but all of the hydrolysis is occurring at or near the aspartate-lysine bond. Some of the polydispersity in the fragment size comes from the starting mAb material as there are clearly many isoforms of slightly different molecular weights that generate the broad peak seen for the monomer.
A critical question is why the mAb is more susceptible to the hydrolysis of the hinge region in phosphate as opposed to histidine buffer systems. To address this question we examined the conformational stability of the mAb in both buffer systems. Thermally-induced unfolding monitored by differential scanning calorimetry (DSC) and denaturant-induced unfolding can be used to elucidate information about individual domain stability and/or domain-domain interactions. In large, multi-domain proteins such as an IgG, domain-domain interactions make a significant contribution to the overall stability of the molecule.36,37,38,39,40
The initial expectation was that the phosphate buffer formulations destabilize the native conformation of the mAb, making it more susceptible to hydrolysis of the hinge region. This is not likely the case as the phosphate buffer system does not significantly alter the resistance to thermal unfolding (Figure 4B and D) of the Fc, Fab or the full-length mAb. The histidine buffer formulations, on the other hand, appear to differentially affect the stability of portions of the mAb as the melting temperatures of the Fc fragment, and particularly the CH2 domain, are altered relative to those observed in the phosphate-containing formulations. This suggests an interaction between histidine and the mAb that disrupts the normal domain-domain interactions, differentially affecting the CH2 domain. In fact, histidine has been shown to interact with antibodies during freeze-drying15 and to bind to interferon-tau (IFN-tau) in solution.13 The histidine interactions with the isolated Fc fragment are not significantly affected by NaCl concentration; however, the full-length mAb thermogram is affected by sodium chloride concentration (Figure 4A and C). In the histidine system without sodium chloride the Fab and Fc exhibit cooperative domain-domain interactions that mask the reduced stability of the CH2 domain. This type of compensation is common in multi-domain proteins.37,41,42 However, when the full-length mAb is in the presence of histidine and sodium chloride the same stabilizing effect is not observed and the thermogram appears essentially as an addition of the separate Fab and Fc thermograms (Figure 4C). This suggests that histidine interferes with the domain-domain interactions between the Fab and Fc. The histidine interaction is presumed to be a binding event which is manifested indirectly in the thermal and unfolding transitions; however, attempts to measure a binding constant with isothermal titration calorimetry (ITC) were unsuccessful (data not shown). This suggests that histidine binding to the mAb in its native conformation is weak and/or is not enthalpic in nature.
Similar conclusions can be drawn from the denaturant-induced mAb unfolding experiments. In phosphate buffered solutions (either in the presence or absence of 150 mM NaCl), the mAb unfolds in a highly cooperative, apparent two-state fashion (Figure 5A). In contrast, in histidine-buffered solutions containing 150 mM NaCl, the unfolding pattern of the full-length mAb is a superposition of the separate Fab and Fc curves, indicating that in this buffer system there is minimal domain-domain cooperativity in the unfolding transition (Figure 5B). In histidine buffered solutions without added salt the full-length mAb unfolds in a cooperative, apparent two-state fashion, in spite of the fact that the isolated Fc fragment shows three state unfolding behavior (Figure 5C).
The unfolding intermediate seen at urea concentrations of approximately 7.5 M in solutions containing 10 mM histidine and 150 mM NaCl was further characterized with KI quenching (Figure 6). This analysis revealed that the intermediate state does not resemble a random coil but has some residual structure that leaves approximately 18% of the initial fluorescence inaccessible to quencher. At the same urea concentration in the phosphate buffered systems essentially all of the initial fluorescence is accessible to the quencher. Based on the unfolding curves of the fragments this is likely due to one of the domains of the Fc fragment having increased resistance to denaturant unfolding in the histidine system relative to the phosphate buffer system; yielding the second higher energy transition similar to the CH3 unfolding step in the Fc fragment. Once again this suggests a binding event between histidine and the mAb. It is possible that this event is only apparent once the native conformation is compromised.
With information on the nature of the fragmentation as well as the differential effects of histidine on the conformational stability of the mAb, deductions as to the mechanism of the buffer effects on fragmentation can be made. The possibility that the free histidine in the buffer acts as a chelator was not considered as the association constant for histidine Cu(II) binding is 1 M−1 while that of phosphate in the −2 charge state is 700 M−1.43,44 One plausible explanation is that the same interaction that results in the reduced cooperativity of unfolding in histidine buffer systems, due to weakened domain-domain interactions, directly competes with the fragmentation reaction. As the unfolding pathways are only affected in histidine buffer systems, there must be some degree of preferential interaction (binding) of the mAb with histidine relative to its interaction with phosphate. This binding could interfere with the fragmentation reaction imparting the resistance to fragmentation seen for the mAb in histidine buffer systems relative to phosphate. The domain that is most directly affected by the histidine interaction is the CH2 domain. In its crystal structure, this domain is known to cooperatively interact with the domains of the constant region of the Fab45 and that interaction is also suggested here by the dependency of CH2 unfolding behavior on the presence or absence of the Fab domains. However, this domain-domain interaction requires significant flexibility of the hinge region. It is that flexibility which has been implicated in IgGs susceptibility to hydrolysis in the hinge region.5 The histidine interaction reduces the flexibility of this mAb; which as a side-effect reduces domain-domain interactions and cooperative unfolding. The reduction in flexibility of the molecule decreases the solvent accessibility of the hydrolysis site and slows the rate of fragmentation.
Buffer species have been known to alter protein behavior, including fragmentation, during formulation yet these issues have only been dealt with empirically.11 This work not only describes how buffer species can differentially alter the conformational stability of a large multi-domain protein but how these effects are manifested from a formulation perspective. The current results further document the importance of determining the effects of different buffer species when developing stable formulations of therapeutic proteins.
Acknowledgments
We would like to thank the following people from MedImmune for helpful discussions and support of this work: Steven Bishop, Stephen Chang, Tom Leach, and Cindy Oliver.
References
- 1.Harris RJ, Shire SJ, Winter C. Commercial manufacturing scale formulation and analytical characterization of therapeutic recombinant antibodies. Drug Dev Res. 2004;61:137–154. [Google Scholar]
- 2.Daugherty AL, Mrsny RJ. Formulation and delivery issues for monoclonal antibody therapeutics. Adv Drug Deliv Rev. 2006;58:686–706. doi: 10.1016/j.addr.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 3.Wang W, Singh S, Zeng DL, King K, Nema S. Antibody structure, instability and formulation. J Pharm Sci. 2007;96:1–26. doi: 10.1002/jps.20727. [DOI] [PubMed] [Google Scholar]
- 4.Liu H, Gaza-Bulesco G, Chumase C, Newby-Kew A. Characterization of lower molecular weight artifact bands of recombinant monoclonal IgG1 antibodies on non-reducing SDS-PAGE. Biotechnol Lett. 2007;29:1611–1622. doi: 10.1007/s10529-007-9449-8. [DOI] [PubMed] [Google Scholar]
- 5.Cordoba AJ, Shyong B, Breen D, Harris AJ. Non-enzymatic hinge region fragmentation of antibodies in solution. J Chromatogr B. 2005;818:115–121. doi: 10.1016/j.jchromb.2004.12.033. [DOI] [PubMed] [Google Scholar]
- 6.Gaza-Bulesco G, Liu H. Fragmentation of a recombinant monoclonal antibody at various pH. Pharm Res. 2008;25:1881–1890. doi: 10.1007/s11095-008-9606-3. [DOI] [PubMed] [Google Scholar]
- 7.Kroon DJ, Baldwin-Ferro A, Lalan P. Identification of sites of degradation in a therapeutic monoclonal antibody by peptide mapping. Pharm Res. 1992;9:1386–1393. doi: 10.1023/a:1015894409623. [DOI] [PubMed] [Google Scholar]
- 8.Paborji M, Pochopin NL, Coppola WP, Bogardus JB. Chemical and physical stability of chimeric L6, a mouse-human monoclonal antibody. Pharm Res. 1994;11:764–771. doi: 10.1023/a:1018948901599. [DOI] [PubMed] [Google Scholar]
- 9.Alexander AJ, Hughes DE. Monitoring of IgG antibody thermal stability by micellar electrokinetic capillary chromatography and matrix-assisted laser desorption/ionization mass spectrometry. Anal Chem. 1995;67:3626–3632. doi: 10.1021/ac00116a002. [DOI] [PubMed] [Google Scholar]
- 10.Liu H, Gaza-Bulesco G, Sun J. Characterization of the stability of a fully human monoclonal IgG after prolonged incubation at elevated temperature. J Chrom B. 2006;837:35–43. doi: 10.1016/j.jchromb.2006.03.053. [DOI] [PubMed] [Google Scholar]
- 11.Zheng JY, Janis LJ. Influence of pH, buffer species, and storage temperature on physicochemical stability of a humanized monoclonal antibody LA298. Int J Pharm. 2006;308:46–51. doi: 10.1016/j.ijpharm.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 12.Kaushik JK, Bhat R. A mechanistic analysis of the increase in the thermal stability of proteins in aqueous carboxylic acid salt solutions. Protein Sci. 1999;8:222–233. doi: 10.1110/ps.8.1.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katayama DS, Nayar R, Chou DK, Valente JJ, Cooper J, Henry CS, Vander Velde DG, Villarete L, Liu CP, Manning MC. Effect of buffer species on the thermally induced aggregation of interferon-tau. J Pharm Sci. 2006;95:1212–1226. doi: 10.1002/jps.20471. [DOI] [PubMed] [Google Scholar]
- 14.Chen B, Bautista R, Yu K, Zapata GA, Mulkerrin MG, Chamow SM. Influence of histidine on the stability and physical properties of a fully human antibody in aqueous and solid forms. Pharm Res. 2003;20:1952–1960. doi: 10.1023/b:pham.0000008042.15988.c0. [DOI] [PubMed] [Google Scholar]
- 15.Tian F, Sane S, Rytting JH. Calorimetric investigation of protein/amino acid interactions in the solid state. Int J Pharm. 2006;310:175–186. doi: 10.1016/j.ijpharm.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 16.DePaz RA, Barnett CC, Dale DA, Carpenter JF, Gaertner AL, Randolph TW. The excluding effects of sucrose on a protein chemical degradation pathway: methionine oxidation in subtilisin. Arch Biochem Biophys. 2000;384:123–132. doi: 10.1006/abbi.2000.2106. [DOI] [PubMed] [Google Scholar]
- 17.Pace CN. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 18.Bhuyan AK, Udgaonkar JB. Observation of multistate kinetics during the slow folding and unfolding of barstar. Biochemistry. 1999;38:9158–9168. doi: 10.1021/bi990285w. [DOI] [PubMed] [Google Scholar]
- 19.Lehrer SS. Solute Perturbation of Protein Fluorescence - Quenching of Tryptophyl Fluorescence of Model Compounds and of Lysozyme by Iodide Ion. Biochemistry. 1971;10:3254–63. doi: 10.1021/bi00793a015. [DOI] [PubMed] [Google Scholar]
- 20.Stern OaMV. On the quenching time of fluorescence. Z Phys. 1919;20:183–199. [Google Scholar]
- 21.Chi EY, Krishnan S, Randolph TW, Carpenter JF. Physical stability of proteins in aqueous solution: Mechanism and driving forces in nonnative protein aggregation. Pharm Res. 2003;20:1325–1336. doi: 10.1023/a:1025771421906. [DOI] [PubMed] [Google Scholar]
- 22.Chi EY, Krishnan S, Kendrick BS, Chang BS, Carpenter JF, Randolph TW. Roles of conformational and colloidal stability in the aggregation of recombinant human granulocyte colony-stimulating factor. Protein Sci. 2003;12:1903–1913. doi: 10.1110/ps.0235703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vermeer AWP, Norde W. The thermal stability of immunoglobulin: Unfolding and aggregation of a multi-domain protein. Biophys J. 2000;78:394–404. doi: 10.1016/S0006-3495(00)76602-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pace CN, Shaw KL. Linear extrapolation method of analyzing solvent denaturation curves. Proteins. 2000;4:1–7. doi: 10.1002/1097-0134(2000)41:4+<1::aid-prot10>3.3.co;2-u. [DOI] [PubMed] [Google Scholar]
- 25.Narayanaswami V, Yamauchi Y, Weers PM, Maekawa H, Tsuchida K, Oikawa K, Kay CM, Sato R, Ryan RO. Spectroscopic charcterization of the conformational adaptability of Bombyx mori apolipophorin III. Eur J Biochem. 2000;267:728–736. doi: 10.1046/j.1432-1327.2000.01050.x. [DOI] [PubMed] [Google Scholar]
- 26.Carpenter JF, Kendrick BS, Chang BS, Manning MC, Randolph TW. Inhibition of stress-induced aggregation of protein therapeutics. In: Abelson JN, Simon MI, Wetzel R, editors. Amyloid, Prions, and Other Protein Aggregates. San Diego: Academic Press; 1999. pp. 236–255. [DOI] [PubMed] [Google Scholar]
- 27.Kim YS, Wall JS, Meyer J, Murphy C, Randolph TW, Manning MC, Solomon A, Carpenter JF. Thermodynamic modulation of light chain amyloid fibril formation. J Biol Chem. 2000;275:1570–1574. doi: 10.1074/jbc.275.3.1570. [DOI] [PubMed] [Google Scholar]
- 28.Kim YS, Cape SP, Chi E, Raffen R, Wilkins-Stevens P, Stevens FJ, Manning MC, Randolph TW, Solomon A, Carpenter JF. Counteracting effects of renal solutes on amyloid fibril formation by immunoglobulin light chains. J Biol Chem. 2001;276:1626–1633. doi: 10.1074/jbc.M007766200. [DOI] [PubMed] [Google Scholar]
- 29.Meyer JD, Ho B, Manning MC. Effects of conformation on the chemical stability of pharmaceutically relevant polypeptides. Pharm Biotechnol. 2002;13:85–107. doi: 10.1007/978-1-4615-0557-0_4. [DOI] [PubMed] [Google Scholar]
- 30.Manning MC, Patel K, Borchardt RT. Stability of proteins. Pharm Res. 1989;6:903–918. doi: 10.1023/a:1015929109894. [DOI] [PubMed] [Google Scholar]
- 31.Pan B, Abel J, Ricci MS, Brems DN, Wang DIC, Trout BL. Comparative oxidation studies of methionine residues reflect a structural effect on chemical kinetics in rhG-CSF. Biochemistry. 2006;45:15430–15443. doi: 10.1021/bi061855c. [DOI] [PubMed] [Google Scholar]
- 32.Usami A, Ohtsu A, Takahama S, Fujii T. Effect of pH, hydrogen peroxide and temperature on the stability of human monoclonal antibody. J Pharm Biomed Anal. 1996;14:1133–1140. doi: 10.1016/s0731-7085(96)01721-9. [DOI] [PubMed] [Google Scholar]
- 33.Lieben F. The hydrolysis of proteins and peptones at high temperatures and the catalytic effect of metal ions on the rate of hydrolysis. J Biol Chem. 1943;151:117–21. [Google Scholar]
- 34.Sigel A, Sigel H. Metal Ions in Biological Systems. New York: Marcel Dekker, Inc; 2001. [Google Scholar]
- 35.Grant KB, Kassai M. Major advances in the hydrolysis of peptides and proteins by metal ions and complexes. Curr Org Chem. 2006;10:1035–49. [Google Scholar]
- 36.Wörn A, Plückthun A. Mutual stabilization of VL and VH in single-chain antibody fragments, investigated with mutants engineered for stability. Biochemistry. 1998;37:13120–13127. doi: 10.1021/bi980712q. [DOI] [PubMed] [Google Scholar]
- 37.Jaenicke R. Stability and folding of domain proteins. Prog Biophys Mol Biol. 1999;71:155–241. doi: 10.1016/s0079-6107(98)00032-7. [DOI] [PubMed] [Google Scholar]
- 38.Jäger M, Plückthun A. Domain interactions in antibody Fv and scFv fragments: effects on kinetics and equilibria. FEBS Lett. 1999;462:307–312. doi: 10.1016/s0014-5793(99)01532-x. [DOI] [PubMed] [Google Scholar]
- 39.Pedroso I, Irún MP, Machicado C, Sancho J. Four-state equilibrium unfolding of scFv antibody fragment. Biochemistry. 2002;41:9873–9884. doi: 10.1021/bi025742e. [DOI] [PubMed] [Google Scholar]
- 40.Garber E, Demarest SJ. A broad range of Fab stabilities within a host of therapeutic IgGs. Biochem Biophys Res Commun. 2007;355:751–757. doi: 10.1016/j.bbrc.2007.02.042. [DOI] [PubMed] [Google Scholar]
- 41.Brandts JF, Hu CQ, Lin L. A simple model for proteins with interacting domains. Applications to scanning calorimetry data. Biochemistry. 1989;28:8588–8596. doi: 10.1021/bi00447a048. [DOI] [PubMed] [Google Scholar]
- 42.Zhang L, Guo P, Zhang H, Jing G. Cooperative unfolding of Escherichia coli ribosome recycling factor originating from its domain-domain interaction and its implication for function. Arch Biochem Biophys. 2006;450:191–202. doi: 10.1016/j.abb.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 43.Velez S, Nair NG, Reddy VP. Transition metal ion binding studies of carnosine and histidine: Biologically relevant antioxidants. Coll and Surf B. 2008;66:291–4. doi: 10.1016/j.colsurfb.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 44.Sigel H. Metal-DNA Chemistry. Washington DC: American Chemical Society; 1989. [Google Scholar]
- 45.Saphire EO, Parren PWHI, Pantophlet R, Zwick MB, Morris GM, Rudd PM, Dwek RA, Stanfield RL, Burton DR, Wilson IA. Crystal structure of a neutralizing human IgG against HIV-1: a template for vaccine design. Science. 2001;293:1155–1159. doi: 10.1126/science.1061692. [DOI] [PubMed] [Google Scholar]