

Abstract
Recent studies suggest that erythropoietin (EPO) has pleiotropic effects in several cell types in addition to hematopoietic cells; however, the role of EPO-mediated cell signaling in nonhematopoietic cells, including in cancer cells, remains controversial. Here, we report our findings of autocrine/paracrine production of EPO by breast cancer cells and its functional significance. We detected a significant level of autocrine/paracrine EPO in the conditioned medium from the culture of SKBR3 breast cancer cells, particularly when the cells were cultured in hypoxia. Through knockdown of EPO and EPO receptor expression and experimental elevation of EPO receptor expression in SKBR3 breast cancer cells, we demonstrated novel roles of autocrine/paracrine EPO-mediated cell signaling in regulating migration and invasion potential and stemness-like properties of breast cancer cells.
Keywords: erythropoietin, autocrine/paracrine loop, migration and invasion, stemness, breast cancer
Introduction
Erythropoietin (EPO), the main hemopoietic hormone, produced by a special group of cells in the kidney in adults, is critical for mammalian erythropoiesis in the bone marrow.1-3 EPO is a 34-kDa glycosylated protein belonging to the class I cytokine family. Production and secretion of endogenous EPO and the expression of its receptor (EpoR) are regulated by tissue oxygenation.4 Over the past decades, understanding of the physiologic functions of EPO has evolved significantly. EPO binds to EpoR and triggers formation of EpoR homodimers, thereby inducing a conformational change in EpoR so that receptor-associated Janus kinase-2 (JAK2) is activated. Activation of JAK2 leads to phosphorylation of tyrosine residues in EpoR and recruitment of Src homology 2 domain-containing proteins. Signaling proteins activated downstream of EpoR and JAK2 include signal transducer and activator of transcription-3 (STAT3), phosphatidylinositol 3-kinase (PI3K), Akt, extracellular signal-regulated kinase (Erk), and others.1,5
Since the isolation and purification of EPO from urine of patients with aplastic anemia in 1977,6 the essential role of EPO in regulating mature red blood cell production has been well established. EPO increases red blood cell mass by stimulating proliferation, inhibiting apoptosis, and inducing differentiation of erythroid progenitors in the bone marrow. The cloning of the EPO gene and production of recombinant human EPO (rHuEPO) led to the widespread use of rHuEPO in treating patients with anemia, including cancer- and chemotherapy-related anemia.7 However, the biological activity of EPO is not restricted to regulation of erythropoiesis.8,9 EpoR expression is also found in several nonhematopoietic normal tissues and in cancerous tissues, although at levels considerably lower than the level in erythroid progenitor cells.10,11
Therefore, although EPO was originally known only to be a critical component in the regulation of erythropoiesis, EPO has now been shown to act on multiple normal and cancerous nonhematopoietic tissues via binding to EpoR found in these tissues, suggesting that EPO has pleiotropic effects. Moreover, functional autocrine/paracrine EPO/EpoR systems have been identified on human cancer cells, including breast carcinoma, melanoma, cervical cancer, and prostate cancer cells, suggesting that the EPO/EpoR axis may contribute to tumor growth, progression, and metastasis.12-16 Randomized clinical trials in patients with cancer have produced controversial findings related to EPO and EpoR: some studies showed that rHuEPO may adversely impact disease progression and patient survival,17-20 whereas other studies did not show a significant detrimental effect of EPO on overall survival of cancer patients.21-23
In this report, we present evidence of autocrine/paracrine production of EPO in breast cancer cells in culture. We found that the EPO levels were higher in hypoxic culture than in normoxic culture. Silencing of EPO or EpoR by RNA interference led to marked inhibition of cell signaling and cell migration and invasion. Furthermore, we found that autocrine/paracrine production of EPO also played a role in stimulating tumorsphere growth of breast cancer cells. Our data are consistent with a few early reports of the existence of a functional autocrine/paracrine EPO/EpoR system in human cancer cells12-16 and expand on this previous knowledge by demonstrating a role of autocrine/paracrine EPO in regulating the stemness of breast cancer cells.
Results
EPO is present in the conditioned culture medium of SKBR3 breast cancer cells cultured in normoxia and hypoxia
By using a quantitative, commercially available EPO ELISA kit for in vitro diagnostic detection of EPO in human plasma, we measured the levels of EPO in the conditioned culture medium of four breast cancer cell lines, SKBR3, MDA468, MDA453, and MCF7, in both normoxia and hypoxia (Fig. 1). We found that SKBR3 cells secreted a significantly higher level of EPO in conditioned medium than other three cell lines. In normoxia, the total amount of EPO secreted into the culture medium of SKBR3 cells during a 40 h culture period was 4.48 mIU per 4 × 106 cells, compared with only 1.52, 1.15, and 0.85 per 4 × 106 cells for MDA468, MDA453 and MCF7 cells, respectively. The total amount of EPO secreted into the culture medium during same period in hypoxia increased to 6.83 mIU per 4 × 106 SKBR3 cells but only to 1.84, 1.29, and 1.03 mIU per 4 × 106 cells for MDA468, MDA453, and MCF7 cells, respectively.
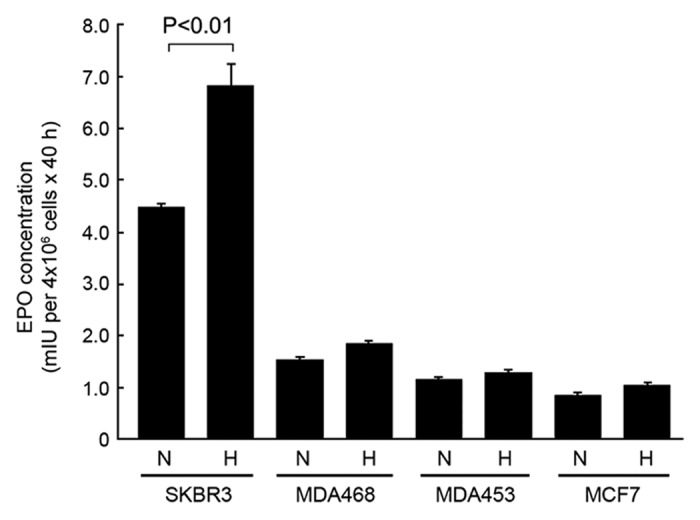
Figure 1. SKBR3 breast cancer cells express high levels of autocrine/paracrine EPO in normoxia and hypoxia compared with other breast cancer cell lines. The indicated breast cancer cells were cultured in medium with 0.5% FBS in normoxia (N) and hypoxia (H) (1% O2) for 40 h. Concentrated cell-free medium and conditioned medium were used for detection of EPO by ELISA as described in Materials and Methods.
A functional autocrine/paracrine EPO/EpoR loop exists in SKBR3 breast cancer cells
To understand the functional significance of EPO secreted by breast cancer cells into the culture medium, we first examined the changes in cell signaling at a basal level after knockdown of EPO expression in SKBR3 cells. Transient transfection of SKBR3 cells with each of three different EPO siRNAs significantly reduced the levels of EPO in the conditioned medium of SKBR3 cells compared with the levels of EPO in the conditioned medium of cells transfected with a control siRNA (Fig. 2A).
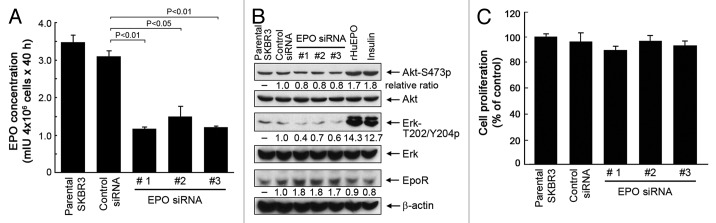
Figure 2. Knockdown of autocrine/paracrine EPO expression by siRNA inhibits cell signaling but not cell proliferation of SKBR3 cells. (A) SKBR3 cells were subjected to knockdown of EPO expression with three different siRNAs or control siRNA for 48 h. The cells were then cultured for an additional 40 h. The conditioned medium was harvested for detection of EPO as described in Figure 1. (B) SKBR3 cells were subjected to knockdown of EPO as described in (A). The cells were then harvested and lysed for western blotting analysis with the antibodies shown. Lysates from separate dishes of SKBR3 cells treated with rHuEPO (40 unit/mL) or insulin (10 nM) for 30 min prior to cell lysis were used as positive controls. (C) Parental SKBR3 cells and the SKBR3 cells with and without EPO knockdown were cultured for additional 5 d in normoxia, and then subjected to an MTT assay as described in Materials and Methods.
Stimulation of SKBR3 cells with rHuEPO or insulin, which served as a positive control, led to marked increases in the levels of Akt and Erk phosphorylation (Fig. 2B), indicating that SKBR3 cells express functional EpoR, which can mediate activation of the PI3K/Akt and MEK/Erk pathways, which are well-characterized EpoR downstream cell signaling pathways that are also activated by other growth factors, including insulin.
In contrast to stimulation of SKBR3 cells with rHuEPO or insulin, knockdown of EPO expression in SKBR3 cells led to decreases in the levels of Akt and Erk phosphorylation (Fig. 2B). Interestingly, knockdown of EPO expression in SKBR3 cells also led to a noticeable compensatory increase in the level of EpoR compared with the level of EpoR in untreated parental cells or control-siRNA-treated cells.
When SKBR3 cells were analyzed for cell survival and proliferation after a 5-d culture in normoxia following transfection with EPO siRNA or control siRNA as described in Figure 2A, we did not find significant differences between the two groups with respect to proliferation and survival (Fig. 2C), suggesting that knockdown of EPO expression alone has minimal effect on the proliferation and survival of the cells in the whole population. We were unable to continuously culture SKBR3 cells in a hypoxic chamber for 5 d with or without knockdown of EPO expression because the cells did not survive well after 5 d of culture in hypoxia.
Knockdown of autocrine/paracrine EPO with siRNA inhibits cell migration and invasion
We then examined the impact of autocrine/paracrine EPO on cell migration and invasion potential in SKBR3 cells. SKBR3 cells were transfected with EPO siRNA or control siRNA as described in Figure 2 and then cultured for 48 h in normoxia or hypoxia. Similar to the results in Figures 1 and 2A, the level of EPO detected was significantly higher in the conditioned medium of hypoxic cultures than in the conditioned medium of normoxic cultures, and knockdown of EPO expression led to marked decreases in the levels of EPO in both the conditioned medium of normoxic cultures and the conditioned medium of hypoxic cultures (Fig. 3A). Next, the conditioned media harvested from normoxic and hypoxic cultures of SKBR3 cells with and without knockdown of EPO expression were added into the lower wells of a transwell chamber, and fresh SKBR3 cells were placed in the upper wells. As shown in Figure 3B, the number of cells that passed through the matrix-coated membrane in the transwell was significantly higher with conditioned medium from hypoxic culture of control siRNA-treated SKBR3 cells than with conditioned medium from normoxic culture of control siRNA-treated SKBR3 cells. After SKBR3 cells were subjected to knockdown of EPO with either of two different EPO siRNAs, the number of cells that passed through the membrane continued to be higher for medium from hypoxic cultures than for medium from normoxic cultures, but compared with the number of cells migrated in control-siRNA-treated cells, the number of cells migrated in EPO siRNA-treated cells was markedly decreased (by 30% to 60%). To further confirm the effect of autocrine/paracrine EPO on cell migration in culture, we used scratch wound healing assay to compare the extent of cell migration in SKBR3 cell culture between treatment with control siRNA and with either of the two EPO siRNA used in Figure 3A and B. Figure 3C shows that the cell migration potential was markedly decreased in the cells treated with EPO siRNA. Because of the transient silencing effect of siRNA on knocking down gene expression, we performed the scratch assay only on day 3 after EPO knockdown by siRNA.

Figure 3. Knockdown of autocrine/paracrine EPO expression by siRNA inhibits migration and invasion potential of SKBR3 cells. (A) SKBR3 cells were subjected to knockdown of EPO expression as described in Figure 2A. The cells were then cultured in normoxia or hypoxia for an additional 40 h. The conditioned medium was used for detection of EPO as described in Figure 1. (B). SKBR3 cells were subjected to knockdown of EPO with siRNA #1 and #2 shown in (A) for 40 h. The conditioned medium was harvested, concentrated, and then mixed with fresh medium at a 1:1 ratio. A total volume of 700 μL 1:1 mixed medium (350 μL:350 μL) was added into the bottom wells in a Boyden transwell chamber, and 6 × 104 fresh SKBR3 cells (in 150 μL) were added into the upper well (insert). The transwell chamber was placed in a 37°C incubator filled with 5% CO2 and 95% air; 24 h later, the number of cells passed through the membrane in the transwell was analyzed. (C) SKBR3 cells were subjected to knockdown of EPO as in (B). After 2 d when cells reached 90% confluency, the cells were subjected to a scratch wood healing assay as described in Materials and Methods.
These findings strongly indicate that autocrine/paracrine EPO plays an important role in regulating breast cancer migration and invasion potential.
Knockdown of EpoR expression with shRNA inhibits cell migration and invasion
In addition to interrupting the autocrine/paracrine loop by knocking down EPO expression, we attempted to interrupt the autocrine/paracrine loop by knocking down EpoR expression in SKBR3 cells by transducing the cells with lentivirus-based shRNA constructs. We tested five different EpoR shRNA constructs for their efficiency in knocking down EpoR expression in SKBR3 cells (Fig. 4A). Of the five EpoR shRNA constructs, constructs 1, 2, and 4 strongly silenced EpoR expression in SKBR3 cells at 48 h after lentiviral transduction of the cells, compared with the results for the cells infected with lentiviral particles containing vector backbone control. The lentiviruses containing EpoR shRNA constructs 1 and 4 were used, along with the control lentivirus, for further studies testing the effect of knockdown of EpoR expression in SKBR3 cells.

Figure 4. Knockdown of EpoR expression inhibits cell migration and invasion potential of SKBR3 cells. (A) Five lentiviral constructs containing different EpoR-targeting sequences were used to transduce SKBR3 cells as described in Materials and Methods. After the transduction, the cells were harvested for detection of the efficiency of EpoR knockdown by western blotting. The level of β-actin was used for protein loading control. (B) Parental SKBR3 cells and SKBR3 cells transduced with lentivirus containing control vector or EpoR shRNA (#1 or #4) were added into the upper wells of transwell chambers at 6 × 104 cells in 150 μL. The bottom wells were filled with 700 μL regular cell culture medium. The transwell chambers were placed in a 37 °C incubator filled with 5% CO2 and 95% air; 24 h later, the number of cells passed through the membrane in the transwell was analyzed. (C) SKBR3 cells transduced with lentivirus containing control shRNA vector or EpoR shRNA (construct #1 or #4) were subjected to a scratch wood healing assay as described in Materials and Methods.
In experiments similar to those we conducted to examine the effect of EPO knockdown on cell migration and invasion potential (Fig. 3), we examined the effect of EpoR knockdown on the migration potential of SKBR3 cells. We found that SKBR3 cells transduced with the lentivirus containing EpoR shRNA constructs 1 and 4, particularly cells transduced with construct 1, had a significantly lower migration and invasion potential than SKBR3 cells transduced with the control lentivirus (Fig. 4B). We also confirmed the effect of EpoR knockdown on cell migration in culture by using the scratch wound healing assay. Compared with the cells transduced with the control virus, the cells transduced with lentivirus containing EpoR shRNA 1 or 4 migrated much slower toward the scratched acellular area on day 3 and day 5 (Fig. 4C). These observations provided further evidence that autocrine/paracrine EPO contributes to breast cancer cell migration and invasion potential.
Together with the findings from the experiments examining the effects of EPO expression knockdown on cell migration and invasion, these data strongly indicate that autocrine/paracrine EPO affects cell migration and invasion potential.
Overexpression of EpoR confers stemness-like properties on breast cancer cells, and knockdown of EpoR expression inhibits the stemness of breast cancer cells
Our observation that interruption of autocrine/paracrine EPO-mediated cell signaling had minimal effect on cell survival and proliferation of SKBR3 cells in the whole population does not exclude a possibility that autocrine/paracrine EPO may act on a small subpopulation of cells, such as stem-cell-like or tumor-initiating cells. To test our hypothesis that the autocrine/paracrine EPO/EpoR loop plays a role in regulating the stemness of breast cancer cells, we established stable pooled SKBR3 cells overexpressing wild-type EpoR and a constitutively active EpoR mutant (EpoR-R129C). Figure 5A shows that SKBR3 cells with lentiviral transduction of wild-type EpoR and EpoR-R129C had substantially increased levels of EpoR. Compared with the cells transduced with lentiviral control, SKBR3-EpoR-wt and SKBR3-EpoR-R129C cells, particularly SKBR3-EpoR-R129C cells, not only contained higher levels of activation-specific phosphorylated cell signaling molecules downstream of EpoR/Jak2 (i.e., S473-phosphorylated Akt, T202/Y204-phosphorylated Erk, and Y705-phosphorylated STAT3) but also displayed an increased ratio of CD44 to CD24, which is a marker of the stemness of cells.24,25 To confirm that overexpression of EpoR conferred a stemness-like phenotype, we performed tumorsphere assays using a standard protocol of tumorsphere culture. As shown in Figure 5B, compared with the size and number of tumorspheres in cells transduced with lentiviral control, both the size and the number of tumorspheres were markedly increased in SKBR3-EpoR-wt and SKBR3-EpoR-R129C cells, particularly in SKBR3-EpoR-R129C cells. Flow cytometry analysis showed that over 99% of SKBR3 cells were CD24-positive (Fig. 5C). Overexpression of wild-type EpoR or EpoR-R129C did not change the percentage of CD24+ population significantly, but increased the percentage of CD44+ population from the range of 10.6% to 10.9% in control virus-transduced cells to the range of 16.2% to 17.2% in wild-type EpoR virus-transduced cells and to the range of 28.0% to 28.7% in EpoR-R129C virus-transduced cells.

Figure 5. Overexpression of wild-type EpoR and constitutively active mutant EpoR R129C confers stemness-like properties on SKBR3 breast cancer cells. (A) Parental SKBR3 cells and the SKBR3 cells transduced with the lentivirus containing control vector, wild-type EpoR cDNA or EpoR-R129C cDNA were harvested for western blotting analysis with the antibodies shown. (B) The indicated sublines of SKBR3 cells were cultured with MammoCult medium for 7 d. Cells grown into spheres were counted under a microscope and the number of tumorsphere formed was determined as described in Materials and Methods. Representative areas of tumorspheres were photographed. (C) The indicated sublines of SKBR3 cells were subjected to flow cytometry analysis after double staining of the cells with CD44-PE and CD24-FITC antibodies.
To confirm the role of EpoR in regulation of breast cancer stemness by the autocrine/paracrine EPO/EpoR loop, we performed tumorsphere assays with SKBR3 cells with knockdown of EpoR expression as described in Figure 4. For the tumorsphere assay, the culture time was longer than the culture time for the experiments with cells overexpressing EpoR (Fig. 5) to allow the tumorspheres to grow large enough to demonstrate the effect of EpoR knockdown. Figure 6A shows that knockdown of EpoR led to a decrease in total level of CD44 and a concomitant increase in total level of CD24 in SKBR3 cells transduced with lentiviruses containing EpoR shRNA construct 1 or construct 4. Figure 6B shows that compared with control-virus-transduced cells, SKBR3 cells with knockdown of EpoR with either the lentiviral construct 1 or construct 4 showed significant decreases in both the size and the number of tumorspheres. Figure 6C further shows the results of CD44/CD24 flow cytometry analysis of the cells with and without EpoR knockdown. Knockdown of EpoR decreased the percentage of CD44+/CD24− cells from the range of 0.106% to 0.203% in control virus-transduced cells to the range of 0.023% to 0.032% in the cells transduced with EpoR shRNA construct #1 and to the range of 0.013% to 0.046% in the cells transduced with EpoR shRNA construct #4.

Figure 6. Knockdown of EpoR expression inhibits stemness of SKBR3 breast cancer cells. (A) Parental SKBR3 cells and the SKBR3 cells transduced with the lentivirus containing control vector, EpoR shRNA construct #1 or #4 were harvested for western blotting analysis with the antibodies shown. (B) The indicated sublines of SKBR3 cells were cultured with MammoCult medium for 12 d. Cells grown into spheres were counted under a microscope and the number of tumorsphere formed was determined as described in Materials and Methods. Representative areas of tumorspheres were photographed. (C) The indicated sublines of SKBR3 cells were subjected to flow cytometry analysis after double staining of the cells with CD44-PE and CD24-FITC antibodies.
Taken together, these data indicate that autocrine/paracrine EPO signaling plays a novel role in regulating breast cancer cell stemness and the stemness regulated by the autocrine/paracrine EPO/EpoR loop may be a mechanism contributing to breast tumorigenesis.
Discussion
Autocrine signaling is a form of signaling in which a cell secretes a hormone or a chemical substance that binds to the receptors on the same cell, leading to functional changes in the cell, whereas paracrine signaling is a form of cell signaling in which the target cell is near the signal-releasing cell.26 In this study, we characterized the functions of autocrine/paracrine EPO produced by breast cancer cells in normoxic and hypoxic cultures. We found that the level of EPO produced by breast cancer cells was higher in hypoxia than in normoxia. This observation is consistent with the knowledge that EPO is a product of hypoxia-inducible gene expression.27 The experiments we performed in this study did not allow us to distinguish EPO produced as a result of autocrine signaling from EPO produced as a result of paracrine signaling; however, it is likely that both autocrine and paracrine mechanisms exist in the phenotypes we examined in the current study.
Tumor hypoxia is very common in solid tumors, including breast cancer.28-30 Hypoxia-inducible factor-1 (HIF-1) is a master gene transcription factor regulating a variety of cell responses to hypoxia.27,31,32 HIF-1 has been shown to play important roles in regulating epithelial-to-mesenchymal transition of cancer cells and enhanced migration and invasion potential of cancer cells.33-35 Our data presented in this paper showing that EPO/EpoR autocrine/paracrine signaling can mediate migration and invasion potential of breast cancer cells suggest that autocrine/paracrine EPO signaling could be one of the mechanisms by which HIF-1 regulates the invasive phenotype of cancer cells.33,36,37 Targeting of HIF-1, which is being actively investigated as a potential strategy for cancer therapy, could inhibit the effects of autocrine/paracrine EPO signaling on cell migration and invasiveness. Inhibition of EPO autocrine/paracrine signaling pathways in cancer cells could be one of the mechanisms explaining the anticancer effects of several anti-HIF-1 agents reported in the literature.38,39
An earlier study showed that stimulation of breast cancer cells with rHuEPO increased the proportion of tumor-initiating cells.40 Our current study further shows that autocrine/paracrine EPO can also enhance the proportion of tumor-initiating cells, an indicator of stemness of breast cancer cells. Stemness is considered to play a critical role in tumorigenesis through promoting cancer cell self-renewal and conferring resistance of cancer cells to anticancer treatment. Our findings suggest that the autocrine/paracrine EPO/EpoR loop is one of the mechanisms by which breast cancer cells maintain stemness. HIF-1, which regulates EPO expression, has recently been shown to play a role in cancer stemness.41,42
Our finding that autocrine/paracrine EPO seemed to have a greater effect on cell migration and invasion than on cell survival and proliferation is noteworthy. This finding is consistent with a role of EPO in regulating a small subpopulation of tumor-initiating cells. Conventional cell proliferation and survival assays are whole-cell population-based, but tumor-initiating cells represent only a very small proportion of cells in the total cell population. This may explain why the effect of EPO on cell proliferation measured by whole-cell population-based assays was not very obvious and may also explain why the published data on the role of EPO in regulating cancer cell proliferation are somewhat inconsistent.43-47 In contrast, the cell migration and invasion assay counts the absolute number of cells that penetrate through the transwell membrane and is thus technically different from the whole-cell population-based cell proliferation and survival assays. Indeed, in the tumorsphere culture experiment, which counts only the absolute number of tumorspheres formed, the effect of EPO/EpoR on stimulating the survival of tumor-initiating cells was clearly documented.
Recombinant EPO has been widely used in patients for prevention and alleviation of anemia, including cancer- and cancer therapy-induced anemia. Adverse effects of rHuEPO on cancer progression have been reported in several randomized clinical studies and experimental models.17-20,48 A subpopulation of stem-like cancer cells has been implicated in driving tumor formation, relapse, and metastasis.24 These stem-like cancer cells are generally more resistant to cancer treatments than non-stem-like cancer cells. Our studies investigating the roles of autocrine/paracrine EPO in cancer cell migration and invasiveness and in the stemness of breast cancer cells suggest that rHuEPO may promote tumor progression and metastasis through a similar mechanism in breast cancer patients.
In summary, we report two major findings concerning a functional autocrine/paracrine EPO/EpoR loop in breast cancer cells from the study of SKBR3 cells: (1) autocrine/paracrine EPO/EpoR activity was higher in breast cancer cells cultured in hypoxia than in breast cancer cells cultured in normoxia, and (2) autocrine/paracrine EPO/EpoR activity was linked to breast cancer cell migration/invasion potential and stemness. Our work suggests a novel paradigm wherein autocrine/paracrine EPO regulates tumorigenesis of breast cancer via stimulating stem-like cancer cells or tumor-initiating cells in at least a subset of breast cancers.
Materials and Methods
Cell lines and culture
SKBR3, MDA468, MDA453, and MCF7 breast cancer cells were obtained from American Type Culture Collection and maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) in a 37 °C incubator in a 5% CO2/95% air atmosphere.
Reagents, antibodies, and plasmids
rHuEPO was purchased from R&D Systems. The antibodies used in western blot analysis for detecting human EpoR (D5) were purchased from Santa Cruz Biotechnology, antibodies for detecting CD44 and CD24 by western blotting and flow cytometry were purchased from BD Biosciences, and antibodies for detecting total Akt, Erk, STAT3, and their activation-specific phosphorylated forms (Akt-S473, Erk-T202/Y204, and STAT3-Y705) were purchased from Cell Signaling Technology, Inc.
Western blot analysis
Cultured cells were lysed in a lysis buffer containing 50 mM TrisHCl (pH 7.4), 150 mM NaCl, 0.5% NP-40, 50 mM NaF, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 25 μg/mL aprotinin, and 25 μg/mL leupeptin and kept on ice for 15 min. The lysates were cleared by centrifugation, and the supernatants were collected. Equal amounts of protein lysate, as determined by the Pierce Coomassie Plus colorimetric protein assay (Thermo Fisher Scientific), were separated by SDS-PAGE, blotted onto nitrocellulose, and probed with the intended primary antibodies. The signals were visualized using the enhanced chemiluminescence detection kit (Amersham Biosciences). Quantification of western blotting data was performed by using ImageJ software (National Institutes of Health).
Detection of EPO in medium by ELISA
EPO secreted by breast cancer cells in the conditioned medium was measured by using the human EPO Quantikine IVD ELISA kit (R&D Systems). For normoxic culture, breast cancer cells were cultured in 6-well culture plates for 40 h in culture medium with 0.5% FBS in a 37 °C incubator filled with 5% CO2 and 95% air. For hypoxic stimulation, the cells were placed in an airtight chamber that was flushed with a gas mixture of 5% CO2 and 95% N2. The oxygen concentration inside the chamber was maintained at 1% using the Pro-Ox O2 regulator (Model 110; BioSpherix). The hypoxic chamber was placed in the same 37 °C incubator where parallel groups of cells were incubated in normoxic culture conditions. Concentrated conditioned medium was used for detection of EPO with the ELISA kit according to the protocol provided by the manufacturer. The optical density (OD) at 450 nm (the primary wavelength) and 600 nm (the reference wavelength) of each sample was recorded. Concentrated cell-free medium was used as background control for subtraction when the OD value was calculated. The concentration of EPO, expressed as mIU per 4 × 106 cells per 40 h in culture, was determined by comparing the difference of OD value at 450 nm and 600 nm with a standard curve generated with the rHuEPO provided in the kit.
RNA interference
EPO siRNA oligonucleotides and EpoR shRNA constructs were purchased from Sigma-Aldrich. For EPO siRNA transfection, 50 pmol siRNA, and 5 μL of FuGENE-6 (Roche) were mixed in 300 μL of serum-free medium for 20 min, and the resulting siRNA/FuGENE-6 mixture was added into 1 mL of cell culture overnight. After siRNA transfection, the cells were cultured for an additional 48 h to allow knockdown of expression of the targeted genes. The sequences of EPO siRNAs were as follows: #1 (SASI_HS01-00203044), sense strand: CAUUCAGAGA GCAGCUUUAd TdT, antisense strand: UAAAGCUGCU CUCUGAAUGd TdT; #2 (SASI_Hs01_00203045), sense strand: CUGACACUUU CCGCAAACUd TdT, antisense strand: AGUUUGCGGA AAGUGUCAGd TdT; #3 (SASI_Hs01_00203046), sense strand: GAGCAACUCU GAGAUCUAAd TdT, antisense strand: UUAGAUCUCA GAGUUGCUCd TdT.
For shRNA-mediated knockdown of EpoR, we used a lentiviral transduction approach to infect the cells with viral particles, which were produced in HEK293T packaging cells by co-transfection of sequence-verified MISSION shRNA clones with compatible packaging plasmids (pSPAX2 and pMD2.G). The sequences for EpoR shRNA clones (constructed in pLKO.1-puro vector) were as follows: #1 (TRCN0000058313), CGTGTCATCC ACATCAATG; #2 (TRCN0000058314), CCCTTATGAG AACAGCCTT; #3 (TRCN0000372140), AGCTGTGGCT GTACCAGAAT G; #4 (TRCN0000372195), GATGATCAGG GATCCAATAT G. #5 (TRCN0000058316), TGCCAGCTTT GAGTACACTA T. SKBR3 cells were transduced with the lentivirus particles containing various EpoR shRNA clones 2 times every other week.
Construction of wild-type EpoR and EpoR R129C mutant
Wild-type EpoR was subcloned by RT-PCR using the total RNA extracted from MCF7 breast cancer cells as the template. The sequences of PCR primers were as follows: forward primer, ATTAGGATCC GCCACCATGG ACCACCTCGG GGCGTC; reverse primer, TCGCTCTAGA CTAACTTCCG TGATGGTGAT GGTGATGAGA GCAAGCCACA TAG. The PCR product was inserted into LentiORF pLEX-MCS vector (Thermo Scientific Open Biosystems) through the BamHI and XbaI sites and appended with His tag at the 3′ end. EpoR R129C mutant was generated by using a site-directed mutagenesis kit (Agilent Technologies). The constructs were subcloned into pLEX-MCS lentiviral vector and transfected into in HEK293T packaging cells as described above. SKBR3 cells were transduced with the lentivirus particles containing pLEX construct with or without cDNA of wild-type EpoR or EpoR R129C mutant for 2 times every other week.
Cell proliferation assay
Cells were cultured in 24-well plates at 37 °C. Following the indicated treatments, 10 mg/mL methylthiazolyldiphenyl-tetrazolium bromide (MTT) was added (50 μL/well), and the cells were incubated for an additional 2 h. The cells were then lysed with a lysis buffer (500 μL/well) containing 20% sodium dodecyl sulfate (SDS) in dimethyl formamide/H2O (1:1, v/v; pH 4.7) at 37 °C for at least 6 h. The relative number of surviving cells in each group was determined by measuring the OD of the cell lysates at an absorbance wavelength of 570 nm. The OD value of each treatment group was expressed as a percentage of the OD value of the untreated control cells.
Transwell migration and invasion assay
In vitro transwell migration and invasion assays were performed using a modified Boyden chamber in which the surfaces of the filter were coated with Matrigel (Becton Dickinson). After rehydration of the filter membrane with serum-free culture medium, the lower chamber of the transwell was filled with 700 μL of 5% FBS medium supplemented with various additions for treatment, and to the upper chamber were added 6 × 104 cells suspended in 150 μL of 0.5% FBS medium. The chambers were placed in a 37 °C incubator for the desired time periods. After incubation, the upper-chamber medium was removed, and any residual cells in the chamber were gently wiped out with a cotton-tipped swab. The filter membranes in the chambers were rinsed with PBS and stained with HEMA3 solution according to the manufacturer’s instructions (Thermo Fisher Scientific). The cells penetrated into the membrane were counted under a microscope. Cells in several different microscope fields of each filter were counted. Each treatment group was set in triplicate inserts/wells. Representative areas for each treatment were photographed.
Scratch wound healing assay
The migration potential of cells in culture was performed by measuring the distance that cells traveled into an acellular area over time. After cells reached 90% confluency in monolayer culture, a scratch wound was generated using 100 µl pipette tip. The distance traveled by the cells between the two boundaries of the wound was observed under an inverted, phase-contrast microscope. Photographs showing the distance under same magnification were taken at various time points after the wounding.
Tumorsphere culture of human breast cancer cells
Tumorsphere culture was performed by plating 2 × 103 cells per well in triplicate onto ultralow-attachment 6-well plates (Corning Life Sciences) and culturing the cells in MammoCult medium (StemCell Technologies, Inc.) containing 0.9% methylcellulose (v/v). After 7 to 12 d, tumorspheres with diameters greater than 75 μm (for the cells with or without EpoR overexpression) or greater than 25 μm (for the cells with EpoR knockdown) were counted under a microscope, and the number of tumorspheres formed in each well was determined. Representative spheres were photographed.49,50
Flow cytometry analysis of CD44 and CD24 expression patterns
The expression pattern of CD44 and CD24 markers was analyzed by flow cytometry after double staining of the cells with fluorescently labeled CD44 and CD24 antibodies. In brief, 106 cells were pelleted by centrifugation at 1500 rpm for 5 min at 4 °C, resuspended in 100 μL of a buffer containing mouse monoclonal anti-human CD24–fluorescein isothiocyanate (FITC) antibody (1:50 dilution, BD PharMingen) and anti-human CD44–phytoerythrin (PE) antibody (1:50 dilution, BD PharMingen), and incubated for 30 min at 4 °C. The cells were then washed twice with cold PBS and subjected to analysis with LSRFortessa Cell Analyzer (BD Biosciences).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported in part by a National Institutes of Health R01 award (CA129036 to Z Fan) and a Breast Cancer Research Foundation award (to Z Fan). The work was also supported in part by the National Institutes of Health through MD Anderson’s Cancer Center Support Grant, CA016672. We thank Stephanie Deming of the Department of Scientific Publications at The University of Texas MD Anderson Cancer Center for editorial assistance.
Glossary
Abbreviations:
- EPO
erythropoietin
- EpoR
EPO receptor
- Erk
extracellular signal-regulated kinase
- FITC
fluorescein isothiocyanate
- HIF-1
hypoxia-inducible factor-1
- JAK2
Janus kinase-2
- OD
optical density
- PE
phytoerythrin
- PI3K
phosphatidylinositol 3-kinase
- rHuEPO
recombinant human EPO
- STAT3
signal transducer and activator of transcription 3
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/26717
References
- 1.Lacombe C, Mayeux P. The molecular biology of erythropoietin. Nephrol Dial Transplant. 1999;14(Suppl 2):22–8. doi: 10.1093/ndt/14.suppl_2.22. [DOI] [PubMed] [Google Scholar]
- 2.Eckardt KU, Kurtz A. Regulation of erythropoietin production. Eur J Clin Invest. 2005;35(Suppl 3):13–9. doi: 10.1111/j.1365-2362.2005.01525.x. [DOI] [PubMed] [Google Scholar]
- 3.Jelkmann W. Regulation of erythropoietin production. J Physiol. 2011;589:1251–8. doi: 10.1113/jphysiol.2010.195057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang GL, Semenza GL. Molecular basis of hypoxia-induced erythropoietin expression. Curr Opin Hematol. 1996;3:156–62. doi: 10.1097/00062752-199603020-00009. [DOI] [PubMed] [Google Scholar]
- 5.Jelkmann W. Molecular biology of erythropoietin. Intern Med. 2004;43:649–59. doi: 10.2169/internalmedicine.43.649. [DOI] [PubMed] [Google Scholar]
- 6.Miyake T, Kung CK, Goldwasser E. Purification of human erythropoietin. J Biol Chem. 1977;252:5558–64. [PubMed] [Google Scholar]
- 7.Browne JK, Cohen AM, Egrie JC, Lai PH, Lin FK, Strickland T, Watson E, Stebbing N. Erythropoietin: gene cloning, protein structure, and biological properties. Cold Spring Harb Symp Quant Biol. 1986;51:693–702. doi: 10.1101/SQB.1986.051.01.082. [DOI] [PubMed] [Google Scholar]
- 8.Noguchi CT, Wang L, Rogers HM, Teng R, Jia Y. Survival and proliferative roles of erythropoietin beyond the erythroid lineage. Expert Rev Mol Med. 2008;10:e36. doi: 10.1017/S1462399408000860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jelkmann W, Bohlius J, Hallek M, Sytkowski AJ. The erythropoietin receptor in normal and cancer tissues. Crit Rev Oncol Hematol. 2008;67:39–61. doi: 10.1016/j.critrevonc.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 10.Hardee ME, Arcasoy MO, Blackwell KL, Kirkpatrick JP, Dewhirst MW. Erythropoietin biology in cancer. Clin Cancer Res. 2006;12:332–9. doi: 10.1158/1078-0432.CCR-05-1771. [DOI] [PubMed] [Google Scholar]
- 11.Hardee ME, Rabbani ZN, Arcasoy MO, Kirkpatrick JP, Vujaskovic Z, Dewhirst MW, Blackwell KL. Erythropoietin inhibits apoptosis in breast cancer cells via an Akt-dependent pathway without modulating in vivo chemosensitivity. Mol Cancer Ther. 2006;5:356–61. doi: 10.1158/1535-7163.MCT-05-0196. [DOI] [PubMed] [Google Scholar]
- 12.Acs G, Chen M, Xu X, Acs P, Verma A, Koch CJ. Autocrine erythropoietin signaling inhibits hypoxia-induced apoptosis in human breast carcinoma cells. Cancer Lett. 2004;214:243–51. doi: 10.1016/j.canlet.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 13.Kumar SM, Acs G, Fang D, Herlyn M, Elder DE, Xu X. Functional erythropoietin autocrine loop in melanoma. Am J Pathol. 2005;166:823–30. doi: 10.1016/S0002-9440(10)62303-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lopez TV, Lappin TR, Maxwell P, Shi Z, Lopez-Marure R, Aguilar C, Rocha-Zavaleta L. Autocrine/paracrine erythropoietin signalling promotes JAK/STAT-dependent proliferation of human cervical cancer cells. Int J Cancer. 2011;129:2566–76. doi: 10.1002/ijc.25935. [DOI] [PubMed] [Google Scholar]
- 15.Jeong JY, Hoxhaj G, Socha AL, Sytkowski AJ, Feldman L. An erythropoietin autocrine/paracrine axis modulates the growth and survival of human prostate cancer cells. Mol Cancer Res. 2009;7:1150–7. doi: 10.1158/1541-7786.MCR-08-0243. [DOI] [PubMed] [Google Scholar]
- 16.Pelekanou V, Kampa M, Kafousi M, Dambaki K, Darivianaki K, Vrekoussis T, Sanidas E, Tsiftsis DD, Stathopoulos EN, Castanas E. Erythropoietin and its receptor in breast cancer: correlation with steroid receptors and outcome. Cancer Epidemiol Biomarkers Prev. 2007;16:2016–23. doi: 10.1158/1055-9965.EPI-06-1023. [DOI] [PubMed] [Google Scholar]
- 17.Henke M, Laszig R, Rübe C, Schäfer U, Haase KD, Schilcher B, Mose S, Beer KT, Burger U, Dougherty C, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double-blind, placebo-controlled trial. Lancet. 2003;362:1255–60. doi: 10.1016/S0140-6736(03)14567-9. [DOI] [PubMed] [Google Scholar]
- 18.Bohlius J, Langensiepen S, Schwarzer G, Seidenfeld J, Piper M, Bennett C, Engert A. Recombinant human erythropoietin and overall survival in cancer patients: results of a comprehensive meta-analysis. J Natl Cancer Inst. 2005;97:489–98. doi: 10.1093/jnci/dji087. [DOI] [PubMed] [Google Scholar]
- 19.Tonelli M, Hemmelgarn B, Reiman T, Manns B, Reaume MN, Lloyd A, Wiebe N, Klarenbach S. Benefits and harms of erythropoiesis-stimulating agents for anemia related to cancer: a meta-analysis. CMAJ. 2009;180:E62–71. doi: 10.1503/cmaj.090470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bohlius J, Schmidlin K, Brillant C, Schwarzer G, Trelle S, Seidenfeld J, Zwahlen M, Clarke M, Weingart O, Kluge S, et al. Recombinant human erythropoiesis-stimulating agents and mortality in patients with cancer: a meta-analysis of randomised trials. Lancet. 2009;373:1532–42. doi: 10.1016/S0140-6736(09)60502-X. [DOI] [PubMed] [Google Scholar]
- 21.Littlewood TJ, Bajetta E, Nortier JW, Vercammen E, Rapoport B, Epoetin Alfa Study Group Effects of epoetin alfa on hematologic parameters and quality of life in cancer patients receiving nonplatinum chemotherapy: results of a randomized, double-blind, placebo-controlled trial. J Clin Oncol. 2001;19:2865–74. doi: 10.1200/JCO.2001.19.11.2865. [DOI] [PubMed] [Google Scholar]
- 22.Savonije JH, van Groeningen CJ, van Bochove A, Honkoop AH, van Felius CL, Wormhoudt LW, Giaccone G. Effects of early intervention with epoetin alfa on transfusion requirement, hemoglobin level and survival during platinum-based chemotherapy: Results of a multicenter randomised controlled trial. Eur J Cancer. 2005;41:1560–9. doi: 10.1016/j.ejca.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 23.Chang J, Couture F, Young S, McWatters KL, Lau CY. Weekly epoetin alfa maintains hemoglobin, improves quality of life, and reduces transfusion in breast cancer patients receiving chemotherapy. J Clin Oncol. 2005;23:2597–605. doi: 10.1200/JCO.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 24.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Beça FF, Caetano P, Gerhard R, Alvarenga CA, Gomes M, Paredes J, Schmitt F. Cancer stem cells markers CD44, CD24 and ALDH1 in breast cancer special histological types. J Clin Pathol. 2013;66:187–91. doi: 10.1136/jclinpath-2012-201169. [DOI] [PubMed] [Google Scholar]
- 26.Sporn MB, Roberts AB. Autocrine, paracrine and endocrine mechanisms of growth control. Cancer Surv. 1985;4:627–32. [PubMed] [Google Scholar]
- 27.Maxwell PH, Pugh CW, Ratcliffe PJ. Inducible operation of the erythropoietin 3′ enhancer in multiple cell lines: evidence for a widespread oxygen-sensing mechanism. Proc Natl Acad Sci U S A. 1993;90:2423–7. doi: 10.1073/pnas.90.6.2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knowles HJ, Harris AL. Hypoxia and oxidative stress in breast cancer. Hypoxia and tumourigenesis. Breast Cancer Res. 2001;3:318–22. doi: 10.1186/bcr314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown NS, Bicknell R. Hypoxia and oxidative stress in breast cancer. Oxidative stress: its effects on the growth, metastatic potential and response to therapy of breast cancer. Breast Cancer Res. 2001;3:323–7. doi: 10.1186/bcr315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goonewardene TI, Sowter HM, Harris AL. Hypoxia-induced pathways in breast cancer. Microsc Res Tech. 2002;59:41–8. doi: 10.1002/jemt.10175. [DOI] [PubMed] [Google Scholar]
- 31.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–4. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem. 1994;269:23757–63. [PubMed] [Google Scholar]
- 33.Semenza GL. Hypoxia, clonal selection, and the role of HIF-1 in tumor progression. Crit Rev Biochem Mol Biol. 2000;35:71–103. doi: 10.1080/10409230091169186. [DOI] [PubMed] [Google Scholar]
- 34.Semenza GL. Involvement of hypoxia-inducible factor 1 in human cancer. Intern Med. 2002;41:79–83. doi: 10.2169/internalmedicine.41.79. [DOI] [PubMed] [Google Scholar]
- 35.Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29:625–34. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–90. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- 37.Peinado H, Cano A. A hypoxic twist in metastasis. Nat Cell Biol. 2008;10:253–4. doi: 10.1038/ncb0308-253. [DOI] [PubMed] [Google Scholar]
- 38.Garber K. New drugs target hypoxia response in tumors. J Natl Cancer Inst. 2005;97:1112–4. doi: 10.1093/jnci/dji261. [DOI] [PubMed] [Google Scholar]
- 39.Onnis B, Rapisarda A, Melillo G. Development of HIF-1 inhibitors for cancer therapy. J Cell Mol Med. 2009;13(9A):2780–6. doi: 10.1111/j.1582-4934.2009.00876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Phillips TM, Kim K, Vlashi E, McBride WH, Pajonk F. Effects of recombinant erythropoietin on breast cancer-initiating cells. Neoplasia. 2007;9:1122–9. doi: 10.1593/neo.07694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, Liu Y, Malek SN, Zheng P, Liu Y. Targeting HIF1α eliminates cancer stem cells in hematological malignancies. Cell Stem Cell. 2011;8:399–411. doi: 10.1016/j.stem.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harrison H, Rogerson L, Gregson HJ, Brennan KR, Clarke RB, Landberg G. Contrasting hypoxic effects on breast cancer stem cell hierarchy is dependent on ER-α status. Cancer Res. 2013;73:1420–33. doi: 10.1158/0008-5472.CAN-12-2505. [DOI] [PubMed] [Google Scholar]
- 43.Westenfelder C, Baranowski RL. Erythropoietin stimulates proliferation of human renal carcinoma cells. Kidney Int. 2000;58:647–57. doi: 10.1046/j.1523-1755.2000.00211.x. [DOI] [PubMed] [Google Scholar]
- 44.Arcasoy MO, Amin K, Karayal AF, Chou SC, Raleigh JA, Varia MA, Haroon ZA. Functional significance of erythropoietin receptor expression in breast cancer. Lab Invest. 2002;82:911–8. doi: 10.1097/01.LAB.0000020415.72863.40. [DOI] [PubMed] [Google Scholar]
- 45.Lai SY, Grandis JR. Understanding the presence and function of erythropoietin receptors on cancer cells. J Clin Oncol. 2006;24:4675–6. doi: 10.1200/JCO.2006.08.1190. [DOI] [PubMed] [Google Scholar]
- 46.LaMontagne KR, Butler J, Marshall DJ, Tullai J, Gechtman Z, Hall C, Meshaw A, Farrell FX. Recombinant epoetins do not stimulate tumor growth in erythropoietin receptor-positive breast carcinoma models. Mol Cancer Ther. 2006;5:347–55. doi: 10.1158/1535-7163.MCT-05-0203. [DOI] [PubMed] [Google Scholar]
- 47.Della Ragione F, Cucciolla V, Borriello A, Oliva A, Perrotta S. Erythropoietin receptors on cancer cells: a still open question. J Clin Oncol. 2007;25:1812–3, author reply 1815. doi: 10.1200/JCO.2006.09.7212. [DOI] [PubMed] [Google Scholar]
- 48.Liang K, Esteva FJ, Albarracin C, Stemke-Hale K, Lu Y, Bianchini G, Yang CY, Li Y, Li X, Chen CT, et al. Recombinant human erythropoietin antagonizes trastuzumab treatment of breast cancer cells via Jak2-mediated Src activation and PTEN inactivation. Cancer Cell. 2010;18:423–35. doi: 10.1016/j.ccr.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar A, Xu J, Sung B, Kumar S, Yu D, Aggarwal BB, Mehta K. Evidence that GTP-binding domain but not catalytic domain of transglutaminase 2 is essential for epithelial-to-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 2012;14:R4. doi: 10.1186/bcr3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shaw FL, Harrison H, Spence K, Ablett MP, Simões BM, Farnie G, Clarke RB. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. J Mammary Gland Biol Neoplasia. 2012;17:111–7. doi: 10.1007/s10911-012-9255-3. [DOI] [PubMed] [Google Scholar]