

Abstract
Congenital heart disease (CHD) is the most common birth defect affecting the structure and function of fetal hearts. Despite decades of extensive studies, the genetic mechanism of sporadic CHD remains obscure. Deleted in liver cancer 1 (DLC1) gene, encoding a GTPase-activating protein, is highly expressed in heart and essential for heart development according to the knowledge of Dlc1-deficient mice. To determine whether DLC1 is a susceptibility gene for sporadic CHD, we sequenced the coding region of DLC1 isoform 1 in 151 sporadic CHD patients and identified 13 non-synonymous rare variants (including 6 private variants) in the case cohort. Importantly, these rare variants (8/13) were enriched in the N-terminal region of the DLC1 isoform 1 protein. Seven of eight amino acids at the N-terminal variant positions were conserved among the primates. Among the 9 rare variants that were predicted as “damaging”, five were located at the N-terminal region. Ensuing in vitro functional assays showed that three private variants (Met360Lys, Glu418Lys and Asp554Val) impaired the ability of DLC1 to inhibit cell migration or altered the subcellular location of the protein compared to wild-type DLC1 isoform 1. These data suggest that DLC1 might act as a CHD-associated gene in addition to its role as a tumor suppressor in cancer.
Introduction
Congenital heart disease (CHD) presents a variety of structural malformations of the heart or great vessels at birth, constituting a major cause of birth defect-related deaths [1]. Although decades of research have revealed that both environmental and genetic factors contribute to the etiology of CHD, increasing evidence supports an important role of a genetic predisposition to the disease [1]–[4]. Indeed, many disease-causing genes, which follow Mendelian patterns of inheritance (e.g., TBX5, JAG1, NKX2-5, GATA4, NOTCH1), have been identified by pedigree analysis [5]–[10]; however, the genetic mechanism of most sporadic CHD cases remains elusive [11].
In our previous mutational screen in a Chinese sporadic CHD cohort, a low-coverage (100×) exome sequencing of 18 pooled samples identified a splice-site mutation (chr8:13072284, C>G, reference assembly: hg19) of the deleted in liver cancer 1 (DLC1) gene in a patient who has atrial septal defect (ASD). This variant is not recorded in The 1000 Genomes Project database and the dbSNP 137 database; after validation assays, it is absent in 800 control samples, suggesting that this splicing site mutation is unique in the CHD cohort (unpublished data).
DLC1, which encodes a GTPase-activating protein, is considered to be a tumor suppressor gene in several types of tumors (e.g., primary hepatocellular carcinoma, breast cancer, prostate cancer, non-small cell lung carcinoma and meningioma tumors) [12]–[18]. The migration and proliferation of some tumor cells are reported to be inhibited by DLC1 [19]–[22]. DLC1 can interact with tensin family proteins [23], [24] and is localized to focal adhesions [25], which together indicate that DLC1 is essential for the cytoskeletal organization and morphology of cells. Interestingly, Dlc1 −/− mice are embryonic lethal, and histologically, the heart is incompletely developed with a distorted architecture of the chambers [26]. Another study reported that Dlc1 homozygous gene-trapped mice demonstrated abnormalities in the embryonic heart and blood vasculature of the yolk sac [27]. These results, which were derived from observations of knockout mice, unequivocally prove that DLC1 is of paramount importance to the developmental events occurring in the embryonic heart.
The human DLC1 gene encodes four transcript variants: isoforms 1–4 encode protein products of 1528 aa, 1091 aa, 463 aa and 1017 aa, respectively. Although there have been numerous investigations focused on characterizing the multi-faceted function of DLC1 isoform 2, the properties of the other isoforms remain unclear. In particular, DLC1 isoform 1, the longest isoform of the DLC1 gene (NCBI Reference Sequence: NM_182643.2), is abundantly expressed in human heart tissues [28].
The evidence described above logically leads to the hypothesis that, in addition to its role as a tumor suppressor in cancer, DLC1 might play another role in the pathogenesis of CHD. Therefore, to verify the rare variant frequency of DLC1 isoform 1 in a CHD cohort, we sequenced the coding regions and intron boundaries of DLC1 isoform 1 in 151 CHD patients (not including the initial screening CHD cohort of our previous work). Functional experiments were then performed to determine the consequences of the identified mutations.
Materials and Methods
Ethics statement
The written informed consent for the genetic analysis was obtained from all the subjects who participated in this study, and the research was approved by the ethics committee at Institute of Health Sciences, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences.
Sample preparation
A total of 151 patients with congenital heart disease were enrolled in the study at the First Hospital of Hebei Medical University. All the subjects were examined by experienced cardiologists, and the cardiac phenotypes were determined using standard transthoracic echocardiography and other tests according to the ICD-10 diagnostic criteria (Table S1 in File S1). The patients' basic medical situation and family history were recorded. The karyotypes of all patients were examined; with the exception of three individuals with trisomy 21, all others were normal. Most of the patients did not have extra-cardiac manifestations except the three individuals with Down syndrome. Most of the patients had undergone cardiac catheterization or surgery. After recruitment in Hebei and Shanghai of normal individuals without CHD, control blood samples (n = 500) were collected. Genomic DNA was extracted from peripheral blood using QIAamp DNA Blood Mini Kits.
Mutational analysis
The exons and portions of 5′UTR and 3′UTR regions of DLC1 isoform 1 were amplified using the primers shown in Table S2 in File S1. The PCR products were then purified using ExoSAP-IT reagent (USB) and sequenced with an ABI 3730 Genetic Analyzer. The results were analyzed using the ABI software suite and the identified variants were re-sequenced and validated.
Mutation simulation
The method of O'Roak et al. [29] was used to calculate the mutation weight of each base of the DLC1 isoform 1 coding sequence. Because the simulation only focused on the DLC1 gene, the locus-specific substitution rate was not considered. Thus the mutation weight for each base and each substitution can be calculated as follows:
where Wn is the weight measuring the nucleotide-specific substitution rates and has two values according to the base composition [30]:
For the weight Ws, which represents the relative transition or transversion substitution rates [31]:
We mutated each base to the other three bases and predicted the class of mutation (i.e., synonymous, missense or nonsense) that would be introduced. For the sake of convenience, only the missense and nonsense classes were considered. We then obtained the mutation weight of each base for missense and nonsense classes using:
To address whether the cluster of mutations we observed was identical to that expected by chance, after the common SNP sites were eliminated from the coding sequence, 13 non-synonymous rare mutations were randomly introduced into the gene based on the mutation weights in one simulation. We then recorded how often the number of mutations residing within the identical range of our cluster was larger than or equal to 8. The range of the cluster was defined as 639 bp (the length from substitution Ala220Val to Thr433Asn in the coding sequence). The significance was estimated as 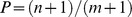 , where n is the number of instances where the randomized number was greater than the observed number and m was the number of randomizations (we employed
, where n is the number of instances where the randomized number was greater than the observed number and m was the number of randomizations (we employed 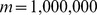 ). Thus, we could estimate the probability of the identical cluster occurring by chance.
). Thus, we could estimate the probability of the identical cluster occurring by chance.
Plasmids construction
The wild-type DLC1 isoform 1 expression plasmid was purchased from OpenBiosystems. Seven missense mutants of DLC1 isoform 1 (threonine substitution of alanine-350, lysine substitution of methionine-360, methionine substitution of leucine-413, lysine substitution of glutamic acid-418, valine substitution of aspartic acid-554, valine substitution of leucine-952 and leucine substitution of valine-1371) were generated by site-directed mutagenesis. The wild type DLC1 isoform 1 and these mutants were cloned into the pEGFP(N1) plasmid, and the DLC1-GFP fusion constructs were transferred into the retroviral plasmid pBabe-puro.
Cell culture
The human umbilical vein endothelial cell line (HUVEC, acquired from Lonza) was maintained in basal medium 199 (Invitrogen) with 20% fetal bovine serum (FBS), heparin (25 µg/mL, Sigma) and endothelial cell growth supplement (ECGS) (50 µg/mL, Sigma). The human bone marrow endothelial cell line (HBMEC-60) [32] was maintained in basal medium 200 (Invitrogen) with 20% FBS and a low-serum growth supplement (Invitrogen). The amphotropic Phenix packaging cell line, H29 was maintained in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) with 10% FBS (HyClone), 100 units/mL penicillin, 100 µg/mL streptomycin (Invitrogen) and 1 µg/mL tetracycline (Sigma) in 5% CO2 at 37°C.
Transwell migration assay
To test the effects of the DLC1 wild-type and mutant proteins on cell migration, pBabe-puro overexpression plasmids were transfected into the amphotropic Phenix packaging cell line, and the viruses were collected as previously described [32]. When the cells (HUVEC or HBMEC-60) grew to 30∼40% confluency, the culture medium was replaced with a 1∶1 mixture of fresh medium and the above virus-containing medium in the presence of 5 µg/mL polybrene for infection and this operation was repeated every 24 h until the infection rate of the target cells reached ∼80%, as judged by GFP-positive cells. After infection, 105 infected endothelial cells were resuspended in fresh media containing 0.5% serum, and the cells were seeded in inserts (Costar) containing 8 µm pores. These inserts were placed in Transwell cartridges that contained 300 µL of medium with 10% FBS in the bottom wells. At 24 h after seeding, the medium was aspirated, and 350 µL of trypsin was added into the wells to trypsinize the cells that had passed through the pores. After serum neutralization of the trypsin, the trypsinized cells were centrifuged for 4 min at 1000 rpm, resuspended in 100 µL phosphate-buffered saline (PBS) and counted using a hemocytometer.
Proliferation assay
When the virus infection rate reached ∼80%, 5×104 infected cells were seeded. After 2 days, the resulting cells were trypsinized and counted using a hemocytometer. Then, 5×104 of these cells were reseeded for another round of counting. The process was repeated for at least three cycles.
Active rho assay
Cells at 80% confluence were gently rinsed once with ice-cold Tris-buffered saline (TBS) and lysed. The lysate was centrifuged at 16,000×g at 4°C for 15 min, and the supernatant was subjected to active Rho purification and detection with the Active Rho Kit (Pierce, Cat No. 16116) according to the manufacturer's protocol.
Stress fiber staining and DLC1 subcellular localization
When the cells reached 40% confluence, they were transfected with pEGFP(N1) plasmids harboring DLC1 wild-type or mutant cDNA. After 24 h, the cells were fixed with 10% formalin for 15 min, permeated with 0.1% Triton X-100 for 10 min and stained with 5 units/mL rhodamine phalloidin (Invitrogen) for 20 min. The stained cells were imaged with using a laser confocal microscope. A total of 100 randomly selected transfected cells in each sample were assessed for subcellular localization of the DLC1-GFP fusion protein. The selected cells were also assessed for the percentage of cells with visible stress fibers as previously described [33].
Angiogenesis (tube-formation) assay
A total of 5×104 cells infected with DLC1-expressing viruses were suspended in 300 µL of DMEM supplemented with 10% FBS and 10 ng/mL FGF (Invitrogen). The cell suspension was seeded on 300 µL of pregelled Matrigel (10.8 mg/mL, Becton, Dickinson and Company). After 24 h, 10 microscopic fields were randomly selected for each well. Angiogenesis in each well was determined by counting the branch points of the formed tubes, as previously described [34].
Apoptosis assay
Cell apoptosis analysis was performed using an Apoptosis Assay Kit (Keygen Biotech) according to the manufacturer's instructions. Briefly, 1×106 cells infected with virus expressing wild-type or mutant DLC1 were trypsinized and resuspended in 500 µL of 1× binding buffer. Then, fluorochrome-conjugated Annexin V was added to the cell suspension and was incubated for 10 min at room temperature, followed by incubation with 5 µL of 7-AAD viability staining solution for 10 min at room temperature. The cells were then subjected to flow cytometry using a FACSAria (BD Biosciences).
Results
Identification of rare variants in the DLC1 gene of CHD patients
DLC1 isoform 1 contains 18 exons and spans 431,558 base pairs (bp). Each exon of DLC1 isoform 1 was amplified from the genomic DNA of 151 CHD patients and the PCR products were then sequenced by Sanger sequencing. After eliminating the common single-nucleotide polymorphisms (SNPs) (SNPs with minor allele frequency 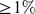 ) found in the dbSNP database, 13 rare non-synonymous variants were identified. One of these variants was found in 2 patients and each of the rest 12 variant was found in 1 patient. We then assessed the frequency of these rare variants in the control cohort by sequencing the corresponding sites in 500 normal samples using Sanger sequencing method. These data were combined with an additional exome sequencing dataset of 400 individuals (average depth 60×) (G.N., unpublished data) to widen the control cohort to 900 individuals. Consequently, only 3 rare variants identified in the CHD cohort were also found in the controls. In addition, 6 of the 13 variants were SNPs with very low frequency recorded in dbSNP build 137 (Table 1). Altogether, we identified 6 private variants that were absent in 900 controls and the dbSNP database (Table 1, Fig. 1A). The clinical information of 14 patients who carried these rare variants of DLC1 were reviewed, and ten of the fourteen patients had septal defects. We also reviewed the health status information of the parents of these patients, and all of them had no cardiac defects. However, it's a great pity that we could not obtained the blood samples of these parents because they came to the hospital years ago and we lost touch with these families.
) found in the dbSNP database, 13 rare non-synonymous variants were identified. One of these variants was found in 2 patients and each of the rest 12 variant was found in 1 patient. We then assessed the frequency of these rare variants in the control cohort by sequencing the corresponding sites in 500 normal samples using Sanger sequencing method. These data were combined with an additional exome sequencing dataset of 400 individuals (average depth 60×) (G.N., unpublished data) to widen the control cohort to 900 individuals. Consequently, only 3 rare variants identified in the CHD cohort were also found in the controls. In addition, 6 of the 13 variants were SNPs with very low frequency recorded in dbSNP build 137 (Table 1). Altogether, we identified 6 private variants that were absent in 900 controls and the dbSNP database (Table 1, Fig. 1A). The clinical information of 14 patients who carried these rare variants of DLC1 were reviewed, and ten of the fourteen patients had septal defects. We also reviewed the health status information of the parents of these patients, and all of them had no cardiac defects. However, it's a great pity that we could not obtained the blood samples of these parents because they came to the hospital years ago and we lost touch with these families.
Table 1. The rare variants identified in DLC1 isoform 1.
| Variant type | Patient ID | Gender | Age of diagnosis | Diagnosis | Exon | Nucleotide alterationa | Amino acid alteration | SIFT score | SIFT prediction (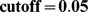 ) ) |
Number of mutations in patients | Number of mutations in controls | In dbSNPb | ALT allele frequency in dbSNPc |
| Private variants | 67 | M | 5 | VSD&PFO | 2 | c.797G>A | p.Gly266Glu | 0.406 | Tolerated | 1/151 | 0/900 | Na | Na |
| 153 | F | 8 | VSD | 3 | c.1048G>A* | p.Ala350Thr | 0.368 | Tolerated | 1/151 | 0/900 | Na | Na | |
| 168 | F | 5 | ASD | 3 | c.1079T>A* | p.Met360Lys | 0.001 | Damaging | 1/151 | 0/900 | Na | Na | |
| 169 | F | 22 | PS | 4 | c.1252G>A* | p.Glu418Lys | 0.027 | Damaging | 1/151 | 0/900 | Na | Na | |
| 89 | F | 2 | PDA | 4 | c.1298C>A | p.Thr433Asn | 0.02 | Damaging | 1/151 | 0/900 | Na | Na | |
| 131 | F | 8 | PDA | 9 | c.[1661A>T(+)1662T>C]* | p.Asp554Val | 0.014 | Damaging | 1/151 | 0/900 | Na | Na | |
| 190 | F | 7 | VSD | 9 | c.[1661A>T(+)1662T>C]* | p.Asp554Val | 0.014 | Damaging | 1/151 | 0/900 | Na | Na | |
| Other rare variants | 49 | F | 9 | TOF | 2 | c.659C>T | p.Ala220Val | 1 | Tolerated | 1/151 | 1/900 | Na | Na |
| 61 | F | 6 | TOF | 3 | c.1051C>T | p.Arg351Trp | 0 | Damaging | 1/151 | 2/900 | rs144283917 | 2.324/5869 | |
| 42 | F | 17 | VSD | 4 | c.1237T>A* | p.Leu413Met | 0.005 | Damaging | 1/151 | 0/900 | rs143447199 | 1/4545 | |
| 55 | F | 26 | PDA | 9 | c.1683C>A | p.Asp561Glu | 0.171 | Tolerated | 1/151 | 2/900 | rs201661577 | 5/2174 | |
| 124 | F | 4 | VSD | 9 | c.2854C>G* | p.Leu952Val | 0.003 | Damaging | 1/151 | 0/900 | rs184157214 | 1/2000 | |
| 28 | M | 1 | VSD | 16 | c.4111G>C* | p.Val1371Leu | 0.016 | Damaging | 1/151 | 0/900 | rs142865083 | 1/2000 | |
| 8 | M | 12 | VSD | 18 | c.4533C>G | p.Ile1511Met | 0.001 | Damaging | 1/151 | 0/900 | rs78322853 | Na |
Note. Na, no available data; M, male; F, female; VSD, ventricular septal defect; PFO, patent foramen ovale; ASD, atrial septal defect; PS, pulmonary stenosis; PDA, patent ductus arteriosus; TOF, tetralogy of Fallot. a, Nucleotide numbering is according to the RefSeq database NM_182643.2. b, The version of dbSNP used in the table is dbSNP build 137. c, The alternative allele frequency form the dbSNP database is calculated by the alternative allele count/two times the number of individuals assayed. *The mutant vectors were constructed according to these variants.
Figure 1. Rare variants identified in DLC1 isoform 1.

(A) The locations of the rare variants are indicated by black lines on the DLC1 isoform 1 protein. FAT (focal adhesion targeting) region, SAM (sterile alpha motif), Rho-Gap (Rho-GTPase-activating protein) and START (steroidogenic acute regulatory protein related lipid transfer) domains are indicated by different colors. Stars denote the private variants identified in the CHD cohort. (B) DLC1 isoform 1 possesses an extended N-terminal region compared to isoform 2. The first 437 residues of isoform 1 are missing in isoform 2, and the sequence ‘TAIQGISEKEKAE’ is replaced by ‘MCRKKPDTMILTQ’ in isoform 2. The yellow box indicates the SAM domain in DLC1, and the green box shows the N-terminal region. (C) The conservation of residues in the N-terminal region was analyzed in different species. The primates and non-primates are separated by the blue lines in the boxes. Asterisks indicate the residues that are conserved among the primates. The residues that are conserved in the primates and non-primates locate in the red boxes. The UniProt accession ID is followed by a colon and the corresponding species name. (D) The private variants that altered the regulation of cell migration function of DLC1 are shown.
DLC1 rare variants cluster in the N-terminus of the protein
Compared to DLC1 isoform 2, which is the most studied isoform, the coding product of isoform 1 has an N-terminal end of 447 amino acids prior to the SAM domain (including an extended region of 437 amino acids and 10 amino acids which are different from the corresponding parts of DLC1 isoform 2) (Fig. 1B). Although several domains have been identified in the DLC1 protein, the function of the N-terminus is still undefined. Interestingly, 8 (61.5%) of the amino acid-altering variants identified in sporadic CHD were located in this region (Fig. 1A). To evaluate the rare variant frequency of this region in other populations, the rare variant information of DLC1 in the 1000 Genomes Project [35] and the Exome Sequencing Project [31] were collected and analyzed (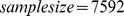 ). As described before, we defined amino acids 1-447 as the N-terminal region and found that 60 (29.6%) of the 203 rare protein-altering variants were localized in this region (Table S3 in File S1). Consequently, Fisher's exact test (two-tail) showed that, compared to variants found in the 1000 Genomes Project and the Exome Sequencing Project mentioned above, the rare variants identified in our CHD cohort significantly clustered at the N-terminus (
). As described before, we defined amino acids 1-447 as the N-terminal region and found that 60 (29.6%) of the 203 rare protein-altering variants were localized in this region (Table S3 in File S1). Consequently, Fisher's exact test (two-tail) showed that, compared to variants found in the 1000 Genomes Project and the Exome Sequencing Project mentioned above, the rare variants identified in our CHD cohort significantly clustered at the N-terminus (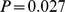 ), revealing that this might be a disease-associated mutation hot spot. We then used the methods from O'Roak et al. [29] to measure the mutation weight of each base of the DLC1 isoform 1 coding sequence. Subsequently 13 missense or nonsense mutations were randomly introduced into the gene in a simulation according to the mutation weights. After one million simulations, we found that the probability of mutation enrichment similar to the observed cases (at least 8 mutations in a range of 639 bp) was very low (
), revealing that this might be a disease-associated mutation hot spot. We then used the methods from O'Roak et al. [29] to measure the mutation weight of each base of the DLC1 isoform 1 coding sequence. Subsequently 13 missense or nonsense mutations were randomly introduced into the gene in a simulation according to the mutation weights. After one million simulations, we found that the probability of mutation enrichment similar to the observed cases (at least 8 mutations in a range of 639 bp) was very low (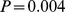 ), which illustrated that the existence of this mutation cluster in the case cohort was not a spontaneous phenomenon.
), which illustrated that the existence of this mutation cluster in the case cohort was not a spontaneous phenomenon.
Most rare variants are predicted to be deleterious
We then BLAST-searched the N-terminal sequence in the UniProt database and aligned the homologous sequences [36]. The alignment showed that, seven of eight amino acids at the N-terminal variant positions were conserved among the primates, and it's worth noting that Arg351, Met360 and Leu413 were conserved in the primates and non-primates (Fig. 1C). The SIFT scores were also calculated to predict the effects of the rare variants on protein function [37] (Table 1, Table S3 in File S1). Among the 9 rare variants that were predicted as “damaging” in the case cohort (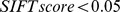 ), 5 were located at the N-terminal region. As for other five rare variants beyond the N-terminal end, there were three amino acid substitutions in the region between the sterile alpha motif (SAM) and Rho-GTPase-activating protein (GAP) domains, but none in the focal adhesion targeting region [38], [39]. The other two amino acid substitutions (Val1371Leu and Ile1511Met) were located in the steroidogenic acute regulatory protein related lipid transfer (START) domain. All of these substitutions were predicted to be deleterious except the c.1683C>A transition (Table 1). We also evaluated the effects of these 13 rare variants found in the case cohort by multiple prediction methods (PolyPhen-2, LRT, Mutation Taster, etc.), and the prediction results from PolyPhen-2 were similar to the SIFT results (Table S4 in File S1).
), 5 were located at the N-terminal region. As for other five rare variants beyond the N-terminal end, there were three amino acid substitutions in the region between the sterile alpha motif (SAM) and Rho-GTPase-activating protein (GAP) domains, but none in the focal adhesion targeting region [38], [39]. The other two amino acid substitutions (Val1371Leu and Ile1511Met) were located in the steroidogenic acute regulatory protein related lipid transfer (START) domain. All of these substitutions were predicted to be deleterious except the c.1683C>A transition (Table 1). We also evaluated the effects of these 13 rare variants found in the case cohort by multiple prediction methods (PolyPhen-2, LRT, Mutation Taster, etc.), and the prediction results from PolyPhen-2 were similar to the SIFT results (Table S4 in File S1).
Three mutations affect the role of DLC1 in cell migration
To study whether the rare variants identified in the CHD cohort affect the protein function of DLC1, we cloned 7 of the variants, including 4 private variants and 3 other rare variants, by introducing the point mutations into the wild-type DLC1 isoform 1. These variants are as the following: Mutant 1, Ala350Thr; Mutant 2, Met360Lys; Mutant 3, Leu413Met; Mutant 4, Glu418Lys; Mutant 5, Asp554Val; Mutant 6, Leu952Val; and Mutant 7, Val1371Leu. These seven variants were selected because they were absent in 900 control samples (altogether 10 rare variants were absent in 900 control samples, but mutant vectors of Gly266Glu, Thr433Asn and Ile1511Met were failed to construct for technical reasons). Cell migration inhibition is one of the most studied functions of DLC1. However, most studies focused on the isoform 2 of DLC1 (1091 aa) and the effect of isoform 1 and its mutants on cell migration has not been reported. Therefore, we assessed the functions of DLC1 isoform 1 and its mutants on migration in human umbilical vein endothelial cells (HUVEC) and human bone marrow endothelial cells 60 (HBMEC-60), the two cell lines widely used in cardiovascular disease studies. The wild-type isoform 1, mutants 1–7, and the control vector were transfected into HUVEC and HBMEC-60 cells (Fig. 2A), following by transwell migration assays to analyze the migratory abilities of the cells. As shown in Figure 2, DLC1 isoform 1 suppressed the migration abilities of HUVEC and HBMEC-60 in vitro. Mutants 2, 4 and 5 (Fig. 1D), which either changed the polarity (Met360 and Asp554) or altered the electric charge (Glu418) of the amino acids, rescued the migration suppression by the wild-type DLC1 protein, as the migration of the cells transfected by these mutants was similar to the control cells. The other mutants appeared to have no significant differences from the wild type to suppress cell migration (Fig. 2B, 2C). In addition, the migration rescue effect of Mutants 2, 4 and 5 could not be accounted for by their effect on cell proliferation, because the mutants and the wild-type protein similarly suppressed the growth of endothelial cells (Fig. S1 in File S1).
Figure 2. DLC1 isoform 1 mutants had different effects on cell migration compared with the wild type protein.

(A) Western blot analyses of DLC1 isoform 1 mutant overexpression in two endothelial cell lines, HUVEC and HBMEC-60. In HUVECs, the effect of DLC1 isoform 1 mutation on the GAP activity of the protein was detected by western blotting. (B) Representative images of the Transwell migration assay using HUVECs cells are shown. (C) The quantification of HUVEC and HBMEC-60 migration showed significant differences between wild-type DLC1 and Mutants 2, 4 and 5, whereas the other mutants showed no significant difference from wild-type DLC1. Wild-type DLC1 also showed an inhibitory effect on cell migration compared to the control vector. *Student's t-test 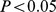 ; **
; ** 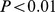 . Scale bars, 100 µm. Ns, not significant.
. Scale bars, 100 µm. Ns, not significant.
The Glu418Lys mutant changes subcellular localization of DLC1
DLC1 is an inhibitor protein of small GTPases including RhoA/B/C and CDC42. Such an inhibitory effect was thought to be mainly mediated by the GAP domain of DLC1. Interestingly, none of the variants identified in CHD lay within the GAP domain. Since a recent study reported that the protein sequences outside of GAP domain may also affect the Rho-inhibiting activity of DLC1 [40], we studied whether the CHD variants affect the GAP activity of DLC1. It was found all the mutants and the wild-type protein efficiently suppressed the activation of RhoA (Fig. 2A). Then we considered whether the small GTPases in the endothelial cells were regulated by DLC1 in situ by analyzing the formation of stress fibers in the cells, a process that is regulated by Rho activities. The DLC1 constructs were tagged with GFP, and the stress fiber formation was analyzed by the high-affinity F-actin probe Rhodamine phalloidin. The data showed that when the wild-type and mutant DLC1 were expressed in the endothelial cells, the formation of stress fibers were prevented to similar levels (Fig. 3A, Fig. S3 in File S1).
Figure 3. The subcellular localization of wild-type DLC1 and mutants in HUVECs.

(A) Images of wild-type and mutant DLC1 distribution in HUVECs using laser scanning confocal microscopy. (B) The percentage of cells with wild-type and mutant DLC1 proteins with exclusive cytoplasmic-localization. Mutant 4 showed a significant difference from the wild type, as opposed to the control vector. *Student's t-test 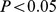 ; **
; ** 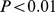 . Scale bar, 25 µm. Ns, not significant.
. Scale bar, 25 µm. Ns, not significant.
Although the variants in DLC1 did not lead to any difference in the regulation of endothelial cytoskeleton, we observed Mutant 4 (Glu418Lys) markedly altered the localization of the protein in the cells. Fluorescent confocal microscopy revealed that DLC1 isoform 1 was primarily located in the cytoplasm, as were Mutants 1–3 and 5–7. Mutant 4 was found in both the cytoplasm and nucleus. Compared to the wild type and the other 6 mutant proteins which were excluded from the nucleus of 73% – 84% endothelial cells, the Mutant 4 protein was not seen in only 11% of the nucleus, suggesting the protein nuclear translocation (PNT) caused by the Glu418Lys substitution (Fig. 3). It was previously reported that PNT occurred in 10% of tumor cells after transfection with DLC1 isoform 2 and was accompanied by morphological changes, and then these cells progressed to apoptosis stage [41]. Although no difference was observed between the cells transfected by Mutant 4 and those by other DLC1 constructs in our apoptosis analysis, all the wild type and mutant DLC1 led to markedly enhanced percentages of apoptotic cells (Fig. S2 in File S1).
Discussion
Congenital heart disease is complex. Although key mutations have been identified by pedigree research, the great heterogeneity of CHD makes it very difficult to identify the responsible genes, particularly among sporadic CHD cohorts. However, disease or deleterious alleles could be rare [42], and rare variants that have obvious functional consequences will show the largest effect size for the disease [43]. Therefore, we focused on the identification of rare variants in a case cohort. We successfully identified 13 rare variants in a sporadic CHD cohort and provide clear evidence that 8 rare variants are clustered in the N-terminal region of the protein. However, we should note that, the reference variant data from the 1000 Genomes Project and the Exome Sequencing Project were produced by different platforms, most of which were next generation sequencing platforms. The sequencing depth, coverage and data analysis pipelines might affect the variant detection rate. It is the consideration that the variant number from different platforms might not be compared directly. So we focused on the locations of the rare variants on the protein, and the analysis strategy is feasible in our study. More importantly, in our in vitro assays, three private variants (corresponding to Mutants 2, 4 and 5) were shown to alter the ability of DLC1 to inhibit cell migration or the subcellular localization of the protein, which supported the notion that private variants might also play major roles in the pathological process of complex diseases [43]. In addition, the extended N-terminal region of DLC1 isoform 1 harbors 83% (5/6) of the private variants identified in the CHD cohort in a non-random manner. The relatively high transcriptional level of DLC1 isoform 1 in human heart tissues [28] implies that the unique N-terminal region may possess a tissue-specific function in the cardiovascular system. However, future studies are necessary to elucidate the details.
Cell migration is an evolutionarily conserved mechanism that includes four steps: polarization, protrusion, adhesion and retraction [44]. Actin is primarily involved in the last three steps. Studies have confirmed that DLC1 can function in the regulation of actin cytoskeletal organization and cell migration [45], suggesting that DLC1 acts as an important regulator of migration. It is essential for endothelial cells in the outflow tract (OT) and atrioventricular (AV) regions to migrate into the cardiac jelly during embryonic heart development [46]. Similarly, the migration of cardiac neural crest cells is also a crucial event during heart development, and the inappropriate timing or path of cardiac neural crest cell migration will cause cardiac congenital anomalies [47]. Thus, if the migration regulatory ability of DLC1 is impaired in the early stage of fetal cardiac development, it is reasonable to speculate that inaccurate developmental consequences, such as defects or malformations, will occur. Although DLC1 is generally considered to affect cell motility and focal adhesion via the Rho-Gap domain and focal adhesion targeting region, respectively [38], [39], [45], the SAM domain has also been reported to regulate cell migration [48]. We demonstrated that three private variants near the SAM domain could reduce the inhibitory effect of wild-type DLC1, suggesting that these mutations might be implicated in regulating the function of the SAM domain.
Although DLC1 isoform 2 has been well studied during the past ten years, the functions of DLC1 isoform 1 still need to be characterized. A series of assays were performed to verify whether DLC1 isoform 1 had a function similar to isoform 2. As shown above, all the mutant and wild-type protein had suppression effects on Rho (Fig. 2A), and similarly regulated the cytoskeleton rearrangement and prevented the formation of stress fiber in the endothelial cells (Fig. 3A, Fig. S3 in File S1). Considering that endocardium formation in the primitive heart tube is affected by vasculogenesis [49], we conducted an angiogenesis assay in vitro, and DLC1 isoform 1 and the mutants had similar prohibitive effects on angiogenesis (Fig. S4 in File S1). Although the mutants showed no difference from the wild-type protein, these negative results only indicate that the variations did not affect these specific features in certain cells. Indeed, the variants might impair the function of DLC1 in other ways or in other cardiac cells. Furthermore, to the best of our knowledge, this is the first report using in vitro assays to demonstrate that DLC1 isoform 1 manifests a function analogous to isoform 2. In conclusion, our mutational analysis of DLC1 isoform 1 presents a spectrum of rare variants in a CHD cohort and shows a mutation cluster in the N-terminus of the DLC1 protein. Our functional assays prove that the ability to inhibit cell migration or the subcellular localization of the protein are altered by three private variants. These findings provide novel insight that DLC1 may be a high-priority candidate gene associated with CHD.
Supporting Information
Tables S1–S4 and Figures S1–S4. Table S1. The statistics of phenotype information of 148 non-trisomy CHD patients; Table S2. The primers for PCR to amplify the exons and portions of 5′UTR and 3′UTR regions of DLC1 isoform 1; Table S3. Rare variants of DLC1 isoform 1 identified in The 1000 Genomes project and Exome sequencing project; Table S4. The effects of 13 rare variants identified in the CHD cohort were predicted using multiple prediction algorithms; Figure S1. Effect of wild-type DLC1 isoform 1 and mutants on HUVEC proliferation; Figure S2. The apoptosis analysis of wild-type DLC1 isoform 1 and mutants in HUVECs; Figure S3. Percentage of cells overexpressing wild-type DLC1 isoform 1 and mutants that exhibited stress fibers; Figure S4. Wild-type DLC1 isoform 1 and mutants had similar effects on angiogenesis.
(DOC)
Acknowledgments
We are grateful to all of the patients and their families and the control individuals described herein for their contributions to this study. We thank Dr. Lei Bu for critical reading and helpful discussions of this manuscript.
Funding Statement
This work is supported by the National Basic Research Program of China (No. 2007CB512103, 2011CB510100) (http://www.973.gov.cn/English/Index.aspx), the National Natural Science Foundation of China (No. 81030015) (http://www.nsfc.gov.cn/Portal0/default166.htm), and the E-Institutes of Shanghai Municipal Education Commission. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Pierpont ME, Basson CT, Benson DW Jr, Gelb BD, Giglia TM, et al. (2007) Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation 115: 3015–3038. [DOI] [PubMed] [Google Scholar]
- 2. Payne RM, Johnson MC, Grant JW, Strauss AW (1995) Toward a molecular understanding of congenital heart disease. Circulation 91: 494–504. [DOI] [PubMed] [Google Scholar]
- 3. Garg V (2006) Insights into the genetic basis of congenital heart disease. Cell Mol Life Sci 63: 1141–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Richards AA, Garg V (2010) Genetics of congenital heart disease. Curr Cardiol Rev 6: 91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Basson CT, Bachinsky DR, Lin RC, Levi T, Elkins JA, et al. (1997) Mutations in human TBX5 [corrected] cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet 15: 30–35. [DOI] [PubMed] [Google Scholar]
- 6. Li L, Krantz ID, Deng Y, Genin A, Banta AB, et al. (1997) Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet 16: 243–251. [DOI] [PubMed] [Google Scholar]
- 7. Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, et al. (1997) Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet 16: 235–242. [DOI] [PubMed] [Google Scholar]
- 8. Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, et al. (1998) Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 281: 108–111. [DOI] [PubMed] [Google Scholar]
- 9. Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, et al. (2003) GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 424: 443–447. [DOI] [PubMed] [Google Scholar]
- 10. Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, et al. (2005) Mutations in NOTCH1 cause aortic valve disease. Nature 437: 270–274. [DOI] [PubMed] [Google Scholar]
- 11. Wessels MW, Willems PJ (2010) Genetic factors in non-syndromic congenital heart malformations. Clin Genet 78: 103–123. [DOI] [PubMed] [Google Scholar]
- 12. Liao YC, Lo SH (2008) Deleted in liver cancer-1 (DLC-1): a tumor suppressor not just for liver. Int J Biochem Cell Biol 40: 843–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wong CM, Yam JW, Ching YP, Yau TO, Leung TH, et al. (2005) Rho GTPase-activating protein deleted in liver cancer suppresses cell proliferation and invasion in hepatocellular carcinoma. Cancer Res 65: 8861–8868. [DOI] [PubMed] [Google Scholar]
- 14. Ng IO, Liang ZD, Cao L, Lee TK (2000) DLC-1 is deleted in primary hepatocellular carcinoma and exerts inhibitory effects on the proliferation of hepatoma cell lines with deleted DLC-1. Cancer Res 60: 6581–6584. [PubMed] [Google Scholar]
- 15. Yuan BZ, Zhou X, Durkin ME, Zimonjic DB, Gumundsdottir K, et al. (2003) DLC-1 gene inhibits human breast cancer cell growth and in vivo tumorigenicity. Oncogene 22: 445–450. [DOI] [PubMed] [Google Scholar]
- 16. Guan M, Tripathi V, Zhou X, Popescu NC (2008) Adenovirus-mediated restoration of expression of the tumor suppressor gene DLC1 inhibits the proliferation and tumorigenicity of aggressive, androgen-independent human prostate cancer cell lines: prospects for gene therapy. Cancer Gene Ther 15: 371–381. [DOI] [PubMed] [Google Scholar]
- 17. Yuan BZ, Jefferson AM, Baldwin KT, Thorgeirsson SS, Popescu NC, et al. (2004) DLC-1 operates as a tumor suppressor gene in human non-small cell lung carcinomas. Oncogene 23: 1405–1411. [DOI] [PubMed] [Google Scholar]
- 18. Hankins GR, Sasaki T, Lieu AS, Saulle D, Karimi K, et al. (2008) Identification of the deleted in liver cancer 1 gene, DLC1, as a candidate meningioma tumor suppressor. Neurosurgery 63: 771–780 discussion 780–771. [DOI] [PubMed] [Google Scholar]
- 19. Goodison S, Yuan J, Sloan D, Kim R, Li C, et al. (2005) The RhoGAP protein DLC-1 functions as a metastasis suppressor in breast cancer cells. Cancer Res 65: 6042–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu PP, Jin YL, Shang YF, Jin Z, Wu P, et al. (2009) Restoration of DLC1 gene inhibits proliferation and migration of human colon cancer HT29 cells. Ann Clin Lab Sci 39: 263–269. [PubMed] [Google Scholar]
- 21. Zhang T, Zheng J, Jiang N, Wang G, Shi Q, et al. (2009) Overexpression of DLC-1 induces cell apoptosis and proliferation inhibition in the renal cell carcinoma. Cancer Lett 283: 59–67. [DOI] [PubMed] [Google Scholar]
- 22. Feng M, Huang B, Du Z, Xu X, Chen Z (2011) DLC-1 as a modulator of proliferation, apoptosis and migration in Burkitt's lymphoma cells. Mol Biol Rep 38: 1915–1920. [DOI] [PubMed] [Google Scholar]
- 23. Yam JW, Ko FC, Chan CY, Jin DY, Ng IO (2006) Interaction of deleted in liver cancer 1 with tensin2 in caveolae and implications in tumor suppression. Cancer Res 66: 8367–8372. [DOI] [PubMed] [Google Scholar]
- 24. Qian X, Li G, Asmussen HK, Asnaghi L, Vass WC, et al. (2007) Oncogenic inhibition by a deleted in liver cancer gene requires cooperation between tensin binding and Rho-specific GTPase-activating protein activities. Proc Natl Acad Sci U S A 104: 9012–9017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liao YC, Si L, deVere White RW, Lo SH (2007) The phosphotyrosine-independent interaction of DLC-1 and the SH2 domain of cten regulates focal adhesion localization and growth suppression activity of DLC-1. J Cell Biol 176: 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Durkin ME, Avner MR, Huh CG, Yuan BZ, Thorgeirsson SS, et al. (2005) DLC-1, a Rho GTPase-activating protein with tumor suppressor function, is essential for embryonic development. FEBS Lett 579: 1191–1196. [DOI] [PubMed] [Google Scholar]
- 27. Sabbir MG, Wigle N, Loewen S, Gu Y, Buse C, et al. (2010) Identification and characterization of Dlc1 isoforms in the mouse and study of the biological function of a single gene trapped isoform. BMC Biol 8: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ko FC, Yeung YS, Wong CM, Chan LK, Poon RT, et al. (2010) Deleted in liver cancer 1 isoforms are distinctly expressed in human tissues, functionally different and under differential transcriptional regulation in hepatocellular carcinoma. Liver Int 30: 139–148. [DOI] [PubMed] [Google Scholar]
- 29. O'Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, et al. (2012) Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338: 1619–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lynch M (2010) Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci U S A 107: 961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tennessen JA, Bigham AW, O'Connor TD, Fu W, Kenny EE, et al. (2012) Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337: 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hu G, Chong RA, Yang Q, Wei Y, Blanco MA, et al. (2009) MTDH activation by 8q22 genomic gain promotes chemoresistance and metastasis of poor-prognosis breast cancer. Cancer Cell 15: 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Theisen CS, Wahl JK 3rd, Johnson KR, Wheelock MJ (2007) NHERF links the N-cadherin/catenin complex to the platelet-derived growth factor receptor to modulate the actin cytoskeleton and regulate cell motility. Mol Biol Cell 18: 1220–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leung KW, Cheung LW, Pon YL, Wong RN, Mak NK, et al. (2007) Ginsenoside Rb1 inhibits tube-like structure formation of endothelial cells by regulating pigment epithelium-derived factor through the oestrogen beta receptor. Br J Pharmacol 152: 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Consortium TGP (2010) A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Consortium U (2012) Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Research 40: D71–D75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4: 1073–1082. [DOI] [PubMed] [Google Scholar]
- 38. Kawai K, Yamaga M, Iwamae Y, Kiyota M, Kamata H, et al. (2004) A PLCdelta1-binding protein, p122RhoGAP, is localized in focal adhesions. Biochem Soc Trans 32: 1107–1109. [DOI] [PubMed] [Google Scholar]
- 39. Kawai K, Iwamae Y, Yamaga M, Kiyota M, Ishii H, et al. (2009) Focal adhesion-localization of START-GAP1/DLC1 is essential for cell motility and morphology. Genes Cells 14: 227–241. [DOI] [PubMed] [Google Scholar]
- 40. Cao X, Voss C, Zhao B, Kaneko T, Li SS (2012) Differential regulation of the activity of deleted in liver cancer 1 (DLC1) by tensins controls cell migration and transformation. Proc Natl Acad Sci U S A 109: 1455–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yuan BZ, Jefferson AM, Millecchia L, Popescu NC, Reynolds SH (2007) Morphological changes and nuclear translocation of DLC1 tumor suppressor protein precede apoptosis in human non-small cell lung carcinoma cells. Exp Cell Res 313: 3868–3880. [DOI] [PubMed] [Google Scholar]
- 42. Gibson G (2011) Rare and common variants: twenty arguments. Nat Rev Genet 13: 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cirulli ET, Goldstein DB (2010) Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet 11: 415–425. [DOI] [PubMed] [Google Scholar]
- 44. Kurosaka S, Kashina A (2008) Cell biology of embryonic migration. Birth Defects Res C Embryo Today 84: 102–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim TY, Vigil D, Der CJ, Juliano RL (2009) Role of DLC-1, a tumor suppressor protein with RhoGAP activity, in regulation of the cytoskeleton and cell motility. Cancer Metastasis Rev 28: 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sakabe M, Matsui H, Sakata H, Ando K, Yamagishi T, et al. (2005) Understanding heart development and congenital heart defects through developmental biology: a segmental approach. Congenit Anom (Kyoto) 45: 107–118. [DOI] [PubMed] [Google Scholar]
- 47. Keyte A, Hutson MR (2012) The neural crest in cardiac congenital anomalies. Differentiation 84: 25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhong D, Zhang J, Yang S, Soh UJ, Buschdorf JP, et al. (2009) The SAM domain of the RhoGAP DLC1 binds EF1A1 to regulate cell migration. J Cell Sci 122: 414–424. [DOI] [PubMed] [Google Scholar]
- 49. Coffin JD, Poole TJ (1991) Endothelial cell origin and migration in embryonic heart and cranial blood vessel development. Anat Rec 231: 383–395. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S4 and Figures S1–S4. Table S1. The statistics of phenotype information of 148 non-trisomy CHD patients; Table S2. The primers for PCR to amplify the exons and portions of 5′UTR and 3′UTR regions of DLC1 isoform 1; Table S3. Rare variants of DLC1 isoform 1 identified in The 1000 Genomes project and Exome sequencing project; Table S4. The effects of 13 rare variants identified in the CHD cohort were predicted using multiple prediction algorithms; Figure S1. Effect of wild-type DLC1 isoform 1 and mutants on HUVEC proliferation; Figure S2. The apoptosis analysis of wild-type DLC1 isoform 1 and mutants in HUVECs; Figure S3. Percentage of cells overexpressing wild-type DLC1 isoform 1 and mutants that exhibited stress fibers; Figure S4. Wild-type DLC1 isoform 1 and mutants had similar effects on angiogenesis.
(DOC)