

Abstract
Compulsive drinking despite serious adverse medical, social and economic consequences is a characteristic of alcohol use disorders in humans. Although frontal cortical areas have been implicated in alcohol use disorders, little is known about the molecular mechanisms and pathways that sustain aversion-resistant intake. Here, we show that nucleus accumbens core (NAcore) NMDA-type glutamate receptors and medial prefrontal (mPFC) and insula glutamatergic inputs to the NAcore are necessary for aversion-resistant alcohol consumption in rats. Aversion-resistant intake was associated with a new type of NMDA receptor adaptation, in which hyperpolarization-active NMDA receptors were present at mPFC and insula but not amygdalar inputs in the NAcore. Accordingly, inhibition of Grin2c NMDA receptor subunits in the NAcore reduced aversion-resistant alcohol intake. None of these manipulations altered intake when alcohol was not paired with an aversive consequence. Our results identify a mechanism by which hyperpolarization-active NMDA receptors under mPFC- and insula-to-NAcore inputs sustain aversion-resistant alcohol intake.
Compulsive intake of alcoholic beverages despite negative economic, legal and physical consequences is a major obstacle to treatment of alcohol use disorders in humans1–4. To study aversion-resistant aspects of alcohol addiction, procedures have been developed whereby animals voluntary self-administer alcohol despite the presence of aversive stimuli, such as adulteration of alcohol with the bitter tastant quinine4–6. Although the compulsion to drink excessively is a major clinical problem1–4, very little is known about the neural or molecular underpinnings of this behavior.
Prefrontal cortical areas may mediate compulsive behavior in humans, as activity in these regions can correlate with craving and relapse1,3,4,7 and encode conflict in rats and humans8,9. Several groups2,3 have theorized that these cortical areas promote compulsive intake because of the presence of a conflict (that is, in the face of aversive consequences); this is in contrast to habitual intake, which is proposed to preferentially recruit striatal but not cortical areas in the absence of such challenges2,3. Thus, we predicted a priori that cortical areas would be more prominent in the regulation of aversion-resistant alcohol intake and would contribute much less to regulating alcohol intake not overtly paired with an aversive challenge (quinine or footshock). In rodents, the mPFC encodes aversiveness10 and regulates anxiety and fear8,11, in addition to regulating behavioral control, decision-making8,9 and drug seeking12–14 including aversion-resistant cocaine intake15. Aversive stimuli are also processed by the insula (INS), which encodes interoceptive cues that can promote addiction- and aversion-related behavior, including compulsive aspects of addiction1–3,7,16,17.
To identify neural circuits that regulate alcohol intake despite aversive consequences, we focused on glutamatergic mPFC and INS inputs to the nucleus accumbens (NAc) core, which integrates information about motivational drives and adaptive behavior9,12,15,17–21. The NAcore also encodes aversiveness10,21 and mediates some aversion-related behaviors21,22 (but see ref. 23). The NAcore receives inputs from mPFC and INS20, and glutamate release from mPFC-to-NAcore terminals promotes cocaine seeking24. In addition, repeated alcohol exposure produces a hyperglutamatergic state, including upregulation of NMDA receptor (NMDAR) function4. Also, NAc NMDARs contribute to operant alcohol self-administration25, alcohol conditioned place preference26 and the discriminative stimulus properties of alcohol27.
Here, we examined the hypothesis that excitatory cortical inputs that activate NAcore NMDARs drive aversion-resistant alcohol consumption. We used an intermittent alcohol drinking model that produces aversion-resistant intake6 and probed circuits and NMDARs using in vitro and in vivo optogenetics, pharmacology and RNA interference. Our findings suggest that excitatory mPFC and INS inputs onto Grin2c-containing NMDARs in the NAcore mediate aversion-resistant intake of alcohol.
Results
NAcore NMDARs promoted quinine-resistant alcohol intake
Pairing alcohol drinking with an aversive stimulus, the bitter tastant quinine, has been used to model aversion-resistant intake in rodents4–6. After 3–4 months of intermittent access to 20% alcohol, rats continue drinking despite adulteration of alcohol with quinine (10 or 30 mg l−1)6. We used this procedure to determine whether NAcore NMDARs mediate alcohol intake with or without quinine adulteration, using a within-subjects design. Rats were allowed ∼2.5 months of intermittent overnight access to alcohol, then ∼4 weeks of 20 min d−1, 5 d week−1 intake before beginning intake experiments. Histology examples for these and all other experiments are shown in Supplementary Figure 1.
Injecting the NMDAR blocker d(−)-2-amino-5-phosphonovaleric acid (AP5; 1 μg μl−1, 0.5 μl per side) into the NAcore significantly reduced alcohol intake in the 20-min intake session when alcohol was adulterated with 30 mg l−1 quinine (Fig. 1a), with no effect on quinine-free alcohol consumption (n = 11; Fquinine(1,10) = 9.803, P = 0.011; FAP5(1,10) = 20.086, P = 0.001; Finteraction(1,10) = 5.682, P = 0.038). The selective effect of AP5 on quinine-adulterated alcohol intake was also observed during the first 30 min of an intermittent overnight drinking session in a separate set of rats (Fig. 1b; n = 6; Fquinine(1,5) = 1.541, P = 0.270; FAP5(1,5) = 18.257, P = 0.008; Finteraction(1,5) = 9.831, P = 0.026). These results suggest that NAcore NMDARs mediated aversion-resistant alcohol intake.
Figure 1.

Inhibition of NMDARs in the NAcore reduced aversion-resistant alcohol intake. (a) Intra-NAcore infusion of AP5 (0.5 μl per side of 1 μg μl−1) reduced intake, relative to that in saline-infused rats, of quinine-adulterated alcohol but not quinine-free alcohol in rats drinking alcohol 20 min d−1, 5 d week−1. (b) Similar results were observed when examining the impact of intra-NAcore AP5 on alcohol intake during the first 30 min of an intermittent overnight alcohol intake session (see Online Methods). Tukey post hoc *P < 0.05. Error bars indicate s.e.m.
Cortical-NAcore inputs promoted aversion-resistant intake
To identify glutamatergic inputs to the NAcore that regulate aversion-resistant alcohol intake, we used halorhodopsin (eNpHR3.0)28–31 to selectively inhibit inputs from the mPFC or INS. We targeted a chronically implanted fiber-optic cable at the NAcore to activate halorhodopsin and reduce glutamate release. Laser stimulation to activate halorhodopsin in mPFC-to-NAcore inputs reduced intake of alcohol adulterated with 10 or 30 mg l−1 quinine but not of quinine-free alcohol (Fig. 2a; n = 11; Fquinine(1,10) = 11.233, P = 0.007; Flaser-stimulation(1,10) = 10.864, P < 0.001; Finteraction(1,10) = 5.576, P = 0.012). Laser stimulation did not alter intake in control rats expressing EYFP in mPFC-to-NAcore inputs (Fig. 2b; n = 9; Fquinine(1,8) = 1.622, P = 0.228; Flaser-stimulation(1,8) = 0.266, P = 0.620; Finteraction(1,8) = 0.062, P = 0.940). To compare drinking in rats with mPFC halorhodopsin versus EYFP, we measured the difference in intake between laser stimulation and no stimulation under six conditions: halorhodopsin or EYFP, each with 0, 10 or 30 mg l−1 quinine. Laser stimulation of halorhodopsin, but not EYFP, decreased alcohol intake in the presence of quinine and had no effect without quinine (Fig. 2c; Fquinine(2,18) = 2.785, P = 0.075; Fhalorhodopsin/EYFP(1,18) = 8.470, P = 0.009; Finteraction(2,18) = 3.727, P = 0.034). In addition, inhibiting mPFC-to-NAcore inputs had no effect on saccharin intake with or without quinine (Fig. 2d; n = 9; Fquinine(1,8) = 6.368, P = 0.036; Flaser-stimulation(1,8) = 0.230, P = 0.644; Finteraction(1,8) = 3.486, P = 0.099), suggesting that halorhodopsin inhibition of quinine-adulterated alcohol intake did not reflect changes in taste reactivity.
Figure 2.

Halorhodopsin inhibition of mPFC- and INS-to-NAcore inputs reduced quinine-resistant alcohol intake. (a) Halorhodopsin (eNpHr3.0) activation in mPFC-to-NAcore terminals significantly reduced intake, relative to that in the absence of laser stimulation, of alcohol adulterated with 10 or 30 mg l−1 quinine but not quinine-free alcohol. (b) Laser stimulation had no effect on alcohol intake in control rats infected with EYFP in mPFC-to-NAcore terminals. (c) To compare laser stimulation of halorhodopsin versus EYFP in mPFC terminals, we calculated the difference in intake between laser stimulation and no stimulation under six conditions: halorhodopsin + 10 or 30 mg l−1 quinine, halorhodopsin + no quinine, EYFP + 10 or 30 mg l−1 quinine, EYFP + no quinine. Laser stimulation only decreased alcohol intake with halorhodopsin + quinine. (d) Inhibiting mPFC-to-NAcore terminals did not alter intake of saccharin (sacc) ± quinine (quin). (e) Halorhodopsin inhibition of INS-to-NAcore terminals significantly reduced intake of quinine-adulterated alcohol (30 mg l−1 quinine) but not quinine-free alcohol. (f) Laser stimulation of INS-to-NAcore terminals had no effect in EYFP-infected rats. (g) Difference scores. Laser stimulation of INS-to-NAcore terminals decreased alcohol intake only with halorhodopsin + quinine. Tukey post hoc *P < 0.05. Error bars indicate s.e.m.
Optogenetic inhibition of INS-to-NAcore terminals also reduced consumption of quinine-adulterated (30 mg l−1) alcohol but not quinine-free alcohol (Fig. 2e; n = 11; Fquinine(1,10) = 11.233, P = 0.007; Flaser stimulation(1,10) = 10.864, P < 0.001; Finteraction(1,10) = 5.576, P = 0.012). Laser stimulation of EYFP in INS-to-NAcore terminals did not alter intake (Fig. 2f; n = 8; Fquinine(1,7) = 0.866, P = 0.383; Flaser-stimulation(1,7) = 0.088, P = 0.775; Finteraction(1,7) = 0.018, P = 0.897). We also analyzed the difference between intake with and without laser stimulation. Laser stimulation of halorhodopsin but not EYFP decreased alcohol intake in the presence of quinine, with no effect in the absence of quinine (Fig. 2g; Fquinine(1,17) = 5.026, P = 0.039; Fhalorhodopsin/EYFP(1,17) = 4.949, P = 0.040; Finteraction(1,17) = 4.478, P = 0.049). Halorhodopsin activation reduced glutamate release from INS- and mPFC-to-NAcore terminals in vitro (Supplementary Fig. 2), as seen in studies of mPFC32 and other terminals29,30. Thus, both mPFC and INS inputs to the NAcore promote aversion-resistant alcohol intake. Simultaneous stimulation of halorhodopsin in both INS- and mPFC-to-NAcore terminals also inhibited quinine-adulterated alcohol intake (Supplementary Fig. 3).
To investigate whether cortical inputs to the NAcore promote alcohol consumption paired with a different aversive stimulus, we examined footshock–resistant intake. After ∼2.5 months of two-bottle choice and 7–8 weeks of operant training, rats received intermittent footshocks during daily operant sessions. On the seventh or eighth footshock-paired session, we examined whether halorhodopsin inhibition of mPFC- or INS-to-NAcore terminals would reduce operant responding.
Inhibition of mPFC-to-NAcore inputs significantly reduced footshock-resistant responding for alcohol, with no effect during the baseline pre-shock period before the footshock-paired sessions began (n = 10; Fig. 3a; Fpre-shock/shock(1,9) = 11.941, P = 0.007; Flaser-stimulation(1,9) = 9.815, P = 0.012, Finteraction (1,9) = 5.849, P = 0.039), and reduced alcohol intake during footshock but not pre-shock sessions (Fig. 3b; Fpre-shock/shock(1,9) = 10.078, P = 0.011; Flaser-stimulation(1,9) = 16.975, P = 0.003, Finteraction(1,9) = 6.774, P = 0.029). Inhibiting INS-to-NAcore terminals also reduced footshock-resistant responding but not baseline responding (n = 10; Fig. 3c; Fpre-shock/shock(1,9) = 5.668, P = 0.041; Flaser-stimulation(1,9) = 6.701, P = 0.029, Finteraction(1,9) = 10.027, P = 0.011), with a similar pattern—effect on footshock-resistant but not basal responding—for alcohol intake (Fig. 3d; Fpre-shock/shock(1,9) = 5.735, P = 0.040; Flaser-stimulation(1,9) = 7.444, P = 0.023, Finteraction(1,9) = 9.645, P = 0.013). We observed individual differences in responding under footshock: some rats responded near baseline, while others greatly reduced responding, and halorhodopsin reduced footshock-resistant intake in the shock-resistant rats (Supplementary Fig. 4). Also, when examined across all mPFC-injected rats, inhibiting mPFC-to-NAcore inputs produced a small but significant reduction in baseline responding (no halorhodopsin, 83.9 ± 12.8 presses; halorhodopsin, 67.0 ± 9.7 presses; n = 17, t = 3.176, P = 0.006, paired t-test), which was not observed for INS-to-NAcore inputs (no halorhodopsin, 78.9 ± 12.2 presses; halorhodopsin, 76.1 ± 9.2 presses; n = 12, t = 0.251, P = 0.807, paired t-test). Thus, mPFC-and INS-to-NAcore inputs regulated alcohol intake paired with aversive stimuli involving different sensory modalities (somatosensory versus gustatory) and different manners of intake (operant versus two-bottle choice drinking).
Figure 3.

Halorhodopsin inhibition of mPFC- and INS-to-NAcore inputs reduced footshock-resistant alcohol intake. Rats underwent 7–8 weeks of fixed ratio 3 operant responding for alcohol, 20 min d−1, 5 d week−1. Thereafter, one in eight FR3 responses were paired with a footshock (0.25 mA, 0.5 s). By the sixth footshock session, alcohol intake could be considered shock-resistant in some rats (Supplementary Fig. 4a–c). We examined only those rats that gave more than 15 lever presses by the sixth footshock-paired session (10 of 17 rats for mPFC, 10 of 12 rats for INS). (a,b) Inhibiting mPFC-to-NAcore inputs significantly reduced responding for alcohol (a) and alcohol intake paired with intermittent footshock (b). Halorhodopsin (eNpHr3.0) had no effect on baseline, pre-shock sessions in this data set; however, when tested across all mPFC-injected rats, there was a small but significant reduction in baseline responding. (c,d) Inhibiting INS-to-NAcore inputs significantly reduced responding for alcohol (c) and alcohol intake paired with intermittent footshock (d). Halorhodopsin had no effect during baseline, pre-shock sessions. In addition, responding under footshock recovered in the 2 d after halorhodopsin exposure, suggesting no lasting impairments. “No laser” indicates average responding in the 2 d before halorhodopsin exposure under footshock. Tukey post hoc *P < 0.05. Error bars indicate s.e.m.
Hyperpolarization-active NAcore NMDARs in alcohol drinkers
Because NAcore NMDARs mediated aversion-resistant alcohol intake (Fig. 1) and repeated alcohol exposure can upregulate NMDARs4, we hypothesized that NMDAR function in the NAcore was elevated in rats with aversion-resistant intake6. We thus activated NMDARs using channelrhodopsin (ChR2) expressed in mPFC-to-NAcore inputs, with 6,7-dinitroquinoxaline-2,3-dione (DNQX; 10 μM) to block AMPA-type glutamate receptors. As expected, we were able to evoke NMDAR currents at potentials greater than 0 mV (ref. 33). In addition, however, we detected excitatory currents in neurons from alcohol-drinking rats at hyperpolarized potentials (Fig. 4a, n = 11 naive, 12 alcohol), where NMDARs are normally blocked by intracellular Mg2+ (ref. 33). AP5 (50 μM) inhibited currents evoked at −50 mV (Fig. 4b, 92.5 ± 0.5% inhibition, n = 5, paired t-test t = 14.35, P < 0.001), indicating that the currents were mediated by NMDARs. We next compared the magnitude of mPFC-evoked currents at −50 mV and +40 mV. At the hyperpolarized potential, NMDAR currents were significantly greater in alcohol-drinking rats than in alcohol-naive, age-matched control rats; in contrast, evoked currents at +40 mV were not different between control and alcohol-drinking rats (n = 11 each naive and alcohol; Fig. 4c,d; Fholding-potential(1,20) = 253.61, P < 0.001; Falcohol/naive(1,20) = 0.327, P = 0.574; Finteraction(1,20) = 6.664, P = 0.018). Thus, alcohol intake was associated with the appearance of NMDARs active at hyperpolarized potentials (HA-NMDARs) under mPFC-to-NAcore synaptic terminals, without changes in input resistance or cell capacitance (Supplementary Table 1).
Figure 4.

NAcore neurons from alcohol-drinking rats showed hyperpolarization-active NMDARs under mPFC-to-NAcore inputs. Currents were evoked by ChR2 stimulation of mPFC-to-NAcore terminals in alcohol-drinking and naive rats. (a) Example traces of NMDAR currents at different holding potentials. Dashed lines highlight the greater NMDAR current at −50 mV in a neuron from an alcohol-drinking rat. Not all voltages were determined for all neurons. (b) Example trace of AP5-sensitive NMDAR currents at −50 mV. (c) Grouped data for NMDAR currents under mPFC-to-NAcore inputs at different holding membrane potentials. For some data points the error bars fall within the symbol. The reversal potential for NMDARs was somewhat positive to zero mV without junction null correction, in line with adult mouse NAc shell18. (d) Alcohol-drinking rats showed significantly greater NMDAR currents at −50 mV than alcohol-naive control rats, with no difference in NMDAR currents evoked at +40 mV holding potential. Tukey post hoc *P < 0.05; n.s., not significant. Error bars indicate s.e.m.
To use an independent method to look for HA-NMDARs, we measured whether AP5 could reduce excitatory postsynaptic currents (EPSCs) generated at −70 mV in the absence of DNQX. We predicted that AP5 would have no effect in control neurons because NMDARs are largely blocked by intracellular Mg2+ at −70 mV, and the AMPA receptor contribution to the EPSC predominates33. AP5 (50 μM) had no effect on mPFC-evoked EPSCs at −70 mV in control rats, but significantly reduced these currents in alcohol-drinking rats (Fig. 5a,b; n = 5 alcohol, 5 naive; t = 2.418, P = 0.042), confirming that NMDARs were active at hyperpolarized membrane potentials under mPFC-to-NAcore inputs in alcohol-drinking rats (Fig. 4).
Figure 5.

NAcore neurons from alcohol-drinking rats showed hyperpolarization-active NMDARs under mPFC and INS but not BLA inputs to the NAcore. (a,b) AP5 (50 μM) significantly reduced mPFC-ChR2-evoked EPSCs at a −70 mV membrane potential in alcohol-drinking but not control naive rats. (c) AP5-sensitive NMDAR currents were apparent in INS-ChR2-evoked EPSCs at −70 mV from alcohol-drinking but not control rats. (d,e) AP5 had no effect on EPSCs evoked at −70 mV by (d) ChR2 stimulation of BLA-NAcore terminals or (e) electrical field stimulation of glutamatergic terminals within the NAcore. *P < 0.05. Error bars indicate s.e.m.
We next examined whether HA-NMDARs were present in other excitatory inputs to the NAcore. When ChR2 was expressed in INS-to-NAcore terminals, AP5 significantly reduced light-evoked EPSCs at −70 mV in alcohol-drinking but not naive control rats (Fig. 5c; n = 8 alcohol, 9 naive; t = 2.466, P = 0.026). In contrast, AP5 did not alter light-evoked EPSCs when ChR2 was expressed in terminals from basolateral amygdala (BLA) neurons (Fig. 5d; n = 5 alcohol, 5 naive; t = 0.499, P = 0.631). AP5 also had no effect on EPSCs evoked by field electrical stimulation, which activates all glutamatergic synapses18,20 (Fig. 5e; n = 6 alcohol, 7 naive; t = 0.791, P = 0.445). Thus, alcohol-drinking rats showed HA-NMDARs under glutamatergic inputs from the mPFC and INS but not BLA.
Having more HA-NMDARs (Figs. 4 and 5a–c) might enhance action potential generation, as HA-NMDARs increase firing in the reticular thalamus despite the small magnitude of currents34. Thus, we stimulated ChR2 in mPFC-to-NAcore terminals at 20 Hz (ten pulses) to evoke EPSP trains every 10 s. Neurons were depolarized to approximately −55 mV to allow EPSP-evoked firing. AP5 significantly reduced action potential generation by mPFC-ChR2-evoked EPSP trains in NAcore neurons from alcohol-drinking but not naive rats (Fig. 6; n = 7 alcohol, 8 naive; Falcohol/naive(1,13) = 0.991, P = 0.338; Fpre/post-AP5(1,13) = 45.529, P < 0.001; Finteraction(1,13) = 18.498, P < 0.001). Thus, HA-NMDARs can promote firing in the NAcore of alcohol-drinking rats.
Figure 6.

NMDARs regulate evoked action potential firing under mPFC-to-NAcore inputs from alcohol-drinking but not naive rats. A ten-pulse train (20 Hz) of mPFC-ChR2-evoked EPSPs was stimulated every 10 s to evoke action potential firing; neurons were depolarized to about −55 mV (alcohol, −56.7 ± 2.5 mV; naive, −54.1 ± 2.9 mV; P = 0.491) with the patch amplifier to allow EPSPs to evoke ∼5 action potentials. (a,b) Traces (a) and time courses (b) from individual neurons showing AP5 inhibition of firing evoked by mPFC-to-NAcore ChR2 stimulation in neurons from alcohol-drinking but not naive rats. Each point represents the average of the six sweeps in that minute. Tick marks under firing traces in a indicate ChR2 stimulation. Firing does not completely track the timing of ChR2 stimulation, which likely reflects the strong potassium channel–mediated after hyperpolarization in NAcore neurons. (c) AP5 decreased firing in neurons from alcohol-drinking but not naive rats. Because of variability in firing across time, we averaged the firing across 4 min before AP5 administration (“basal”) and averaged the firing across the minutes 4–7 of AP5 exposure (“AP5”), as firing tended to run down in all cells after this point. For b, firing was normalized to the basal firing value. Tukey post hoc *P < 0.05. Error bars indicate s.e.m.
Grin2c NMDARs mediated aversion-resistant alcohol intake
HA-NMDARs might reflect the presence of Grin2c, Grin2d or Grin3 subunits (also called NR2C, NR2D and NR3), all of which show reduced Mg2+ inhibition and thus are active at hyperpolarized potentials33–35. Because NMDARs containing Grin1 and Grin3 are not blocked by AP5 (ref. 35), we generated short hairpin RNAs targeting Grin2c and Grin2d subunits (Supplementary Fig. 5) and used AAVs to infect NAcore cells with one of these shRNAs or a control shRNA not targeting any known coding sequence in the genome36. Expression of Grin2c shRNA but not control or Grin2d shRNA in NAcore neurons prevented AP5 from reducing mPFC-evoked EPSCs at −70 mV in neurons from alcohol-drinking rats (Fig. 7a; n = 6 control and Grin2d, 7 Grin2c; one-way ANOVA, F(2,16) = 5.504, P = 0.015), suggesting a role for Grin2c subunits. There are also considerable amounts of Grin2b in the NAcore37, although these subunits are relatively inactive at hyperpolarized resting potentials33. As expected, the Grin2b inhibitor ifenprodil (2 μM) had no effect on mPFC-evoked EPSCs in alcohol-drinking rats (Fig. 7b; n = 5; t = 3.840, P = 0.005, effect of ifenprodil versus AP5 in Fig. 5b). Thus, Grin2c subunits rather than Grin2d or Grin2b mediated HA-NMDARs under mPFC-to-NAcore inputs in alcohol-drinking rats.
Figure 7.
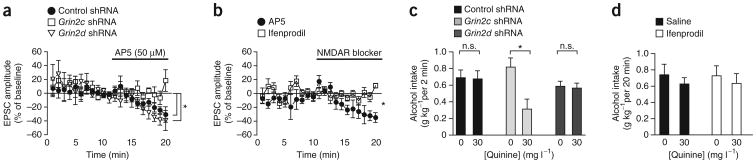
Grin2c but not Grin2d or Grin2b subunits mediate hyperpolarization-active NMDARs under mPFC-to-NAcore terminals and promote quinine-resistant alcohol intake. (a) Infection of NAcore neurons with Grin2c but not Grin2d or control36 shRNA prevented AP5 from inhibiting mPFC-evoked EPSCs at −70 mV. (b) The Grin2b inhibitor ifenprodil (2 μM) did not reduce mPFC-evoked EPSCs. Data for AP5 in b are the same as shown in Figure 5b. (c) Infecting NAcore neurons with Grin2c but not Grin2d or control34 shRNA reduced intake of quinine-adulterated alcohol. (d) Injecting the Grin2b inhibitor ifenprodil in the NAcore (1 μg μl−1) did not alter alcohol intake. *P < 0.05. Error bars indicate s.e.m.
We next used the NMDAR shRNAs to determine whether NAcore HA-NMDARs promote aversion-resistant alcohol intake. Only Grin2c shRNA significantly reduced intake of quinine-adulterated alcohol relative to quinine-free alcohol (Fig. 7c; n = 7 control, 6 Grin2c, 8 Grin2d; Fquinine(1,18) = 23.351, P < 0.001; FshRNA(2,18) = 0.621, P = 0.548; Finteraction(2,18) = 17.698, P < 0.001). In addition, intra- NAcore injection of the Grin2b inhibitor ifenprodil (1 μg μl−1, 0.5 μl per side, which reduces operant intake in dorsal striatum but not NAc38) did not alter consumption of quinine-adulterated alcohol (Fig. 7d, n = 11; Fquinine(1,10) = 1.184, P = 0.302; Fifenprodil(1,10) = 0.004, P = 0.952; Finteraction(1,10) = 0.054, P = 0.821). Thus, Grin2c but not Grin2d or Grin2b in the NAcore mediated quinine-resistant alcohol intake.
Discussion
Here, we found that NMDARs under inputs to the NAcore from the INS and mPFC mediated aversion-resistant alcohol intake. Both mPFC- and INS-to-NAcore inputs promoted quinine-resistant two-bottle-choice alcohol intake, as well as footshock-resistant operant alcohol intake, with little effect on intake not paired with a specific aversive consequence (quinine or footshock). In addition, alcohol-drinking rats showed an unexpected NMDAR adaptation in the NAcore, with NMDARs active at hyperpolarized membrane potentials under INS and mPFC but not BLA inputs. HA-NMDARs in alcohol-drinking rats promoted action potential firing. RNA interference studies demonstrated that Grin2c subunits contributed to HA-NMDARs and mediated quinine-resistant alcohol intake. Thus, our studies reveal an alcohol-related adaptation in NAcore NMDARs, whereby the appearance of Grin2c subunits under several cortical inputs promotes aversion-resistant alcohol intake.
Several studies implicate NAc NMDARs in alcohol reward25–27, but we found that blocking NAcore NMDARs with AP5 did not reduce quinine-free alcohol intake. There are several possible explanations for our seemingly paradoxical result. A noncompetitive NMDAR blocker substitutes for the discriminative stimulus properties of alcohol, whereas a competitive NMDAR blocker does not27, and AP5 is a competitive NMDAR antagonist33. Furthermore, AP5 alters alcohol place preference when infused simultaneously into both the NAcore and NAc shell26, but it can have divergent effects when administered separately into these two subregions19,39,40. In addition, AP5 concentrations that reduce operant alcohol self-administration26 are higher (3–6 μg) than those used here and can suppress locomotor activity39–41. Given these caveats, we conclude that NAcore NMDARs supported quinine-resistant but not quinine-free alcohol intake.
Little is known about the molecular and circuit mechanisms that mediate compulsive, aversion-resistant alcohol intake. Here we used optogenetic inhibition of specific projections to demonstrate that both mPFC- and INS-to-NAcore inputs promote aversion-resistant intake with aversive stimuli of different sensory modalities (somatosensory versus gustatory) and different manners of intake (operant versus two-bottle-choice drinking). Thus, our results suggest that a common neural circuit involving mPFC and INS inputs to the NAcore is a critical mechanism that promotes aversion-resistant intake and that this circuit is related more to the choice to self-administer despite aversive consequences rather than to sensory or motivational aspects of each aversive consequence. In addition, this circuit had little impact on quinine- or footshock-free alcohol intake. In this regard, cortical regions have been proposed to predominate in the aversion-resistant, compulsive aspects of human addiction, where intake is accompanied by conflict or challenge1–3,7; in contrast, intake in the absence of conflict is considered to use more habit-based striatal systems, with much less of a role for cortical areas2,3. Ours is, to our knowledge, the first direct evidence supporting the hypothesis that cortical regions can act more prominently in aversion-resistant intake.
The mPFC mediates decision making and conflict processing8,9 and promotes expression of fear and aversion8,10,11. In this case, inhibiting mPFC-to-NAcore inputs did not alter consumption of quinine-free alcohol, saccharin or saccharin + quinine, suggesting that reduced intake of quinine-adulterated alcohol was not secondary to locomotor effects or changes in taste reactivity. However, the taste of alcohol + quinine may differ from these other substances, and effects on taste reactivity cannot be completely ruled out. Also, the broader mPFC role in behavioral control may explain the small decrease in basal operant responding after inhibiting mPFC-to-NAcore inputs. In addition, inhibiting mPFC cell bodies with halorhodopsin enhances compulsive cocaine seeking, whereas activating the mPFC with ChR2 reduces compulsive cocaine seeking15, perhaps suggesting differential regulation of aversion-resistant alcohol and cocaine intake by the mPFC. Our results also implicate the INS, a regulator of interoceptive stimuli2,16 and aversive and taste stimuli2,16,17,42 that can drive addictive behaviors2,16. The INS is activated by alcohol cues in alcoholics1,7,17, which could promote compulsive aspects of intake1–3,8. However, ours is, to our knowledge, the first study directly implicating the INS in aversion-resistant alcohol intake. The NAc also receives inputs from the hippocampus and amygdala, which can regulate many goal-directed behaviors, including alcohol intake, as does the NAc shell1,15,16,43–46. Future experiments will be able to examine the contribution of subcortical NAc inputs and the NAc shell to aversion-resistant intake.
One possible concern with optogenetics is a nonspecific effect during prolonged laser stimulation. Other groups use longer laser exposure without adverse side effects29,30,32, with some observing slower recovery after halorhodopsin activation29,32 (Supplementary Fig. 2). Here, laser stimulation did not change alcohol drinking in control EYFP-infected rats and did not reduce quinine- or footshock-free alcohol intake or saccharin ± quinine consumption, suggesting that halorhodopsin inhibition of aversion-resistant alcohol intake did not reflect nonspecific motor or motivational effects.
Alcohol exposure can induce a hyperglutamatergic state, including increased NMDAR function4. In this study, alcohol-drinking rats showed NMDARs active at hyperpolarized potentials under mPFC-and INS- but not BLA-to-NAcore inputs, which might reflect activity of Grin2c, Grin2d or Grin3 subunits33–35. Grin2c but not Grin2d or Grin2b subunits mediated both mPFC-evoked HA-NMDAR currents in vitro and quinine-resistant alcohol intake. Little is otherwise known about the behavioral functions of Grin2c and Grin2d, although alcohol intake decreases Grin2c striatal mRNA47 and Grin2d is implicated in acute drug responses48 and spatial memory49. Adult striatum has very little Grin2c and Grin2d50, and these subunits have not been detectable in total brain tissue (data not shown). This would be predicted if only a small subset of glutamatergic terminals contained the alcohol-related adaptation. HA-NMDARs were not observed under BLA-to-NAcore inputs or in EPSCs evoked with field electrical stimulation, which activates all glutamate inputs onto a NAcore neuron20, and thus mPFC and INS synapses may represent a minority of glutamatergic synapses impinging on NAcore neurons. Although the magnitude of HA-NMDAR currents was relatively small, NAcore HA-NMDARs regulated action potential firing evoked by ChR2 stimulation of mPFC-to-NAcore inputs in alcohol-drinking but not naive rats. Together, these results support the possibility that HA-NMDARs regulate physiological activity in the NAcore in alcohol-drinking rats.
In conclusion, mPFC- and INS-to-NAcore inputs and NAcore HA-NMDARs all supported aversion-resistant alcohol intake, with little contribution to alcohol intake not paired with the aversive stimuli footshock or quinine. Our results are the first experimental demonstration of the proposed2,3 critical role for frontal cortical regions in regulating aversion-resistant intake. mPFC- and INS-to-NAcore inputs promote both quinine- and footshock-resistant intake. In addition, Grin2c subunits mediated a newly identified HA-NMDAR neuroadaptation under cortical inputs to the NAcore in alcohol-drinking rats, and this NMDAR change promoted aversion-resistant intake, the first identified behavioral role for Grin2c. Thus, NMDAR blockers or inhibitory allosteric modulators might decrease compulsive aspects of alcohol use disorders4.
Online Methods
Animal handling
All procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals, as adopted by the NIH, and approval of an Institutional Animal Care and Use Committee of the Ernest Gallo Clinic and Research Center. Adult male Wistar rats (∼450–550 g, ∼5–6 months) at the time of the surgery were individually housed and maintained on a 12-h light cycle with ad libitum access to food and water except for experiments in the operant chamber. Experiments were performed in the light cycle.
Two-bottle choice alcohol intake
Rats were trained to voluntarily consume alcohol using a two-bottle, home-cage intermittent access to 20% alcohol procedure (IAA)51. Rats had 24 h of continuous access to two bottles, one containing alcohol (20% vol/vol with water) and the other water, each Monday, Wednesday and Friday evening. The amount of intake was calculated by bottle weight. For some in vitro experiments, patch-clamp in NAcore neurons was performed after 3–4 months IAA (Fig. 5). For other in vitro experiments (Figs. 4 and 7) and all in vivo experiments, rats were allowed ∼2.5 months of overnight IAA intake, then were switched to two-bottle choice access to alcohol or water for 20 min d−1, 5 d week−1 for at least 4 weeks. For two-bottle choice halorhodopsin or EYFP experiments, rats were then switched to drink alcohol for 2 min d−1, 5 d week−1 in an optogenetic chamber (an operant chamber modified to allow two-bottle choice intake). Intakes of alcohol in 2 min and 20 min were similar (for example, compare Figs. 1a and 7c with Fig. 2a,e), indicating that most alcohol was consumed in the first 2 min, representing what has been previously described as the initial “loading-up” period52. Rats had at least 2 weeks of daily intake with dummy cables attached to their chronically implanted fiber optic cables (see below for details) before beginning intake experiments. On each experimental day, rats were placed in the optogenetic chamber with dummy cables attached for 5 min before we provided the bottles. Blood ethanol concentrations at the end of a 20 min two-bottle intake session (determined as in ref. 51) were 42.3 ± 10.7 mg/dl for n = 16 rats that drank 1.01 ± 0.10 g alcohol per kilogram body weight, in agreement with a previous study of drinking in intermittent-access rats51. Although this does not reach what is considered binge drinking as defined by BAC in humans, intake under the intermittent access model used here is associated with several factors considered to model aspects of human alcohol use disorders, including escalation of intake51, sensitivity to compounds that reduce intake in human alcoholics51 and moderate signs of dependence53, as well as continued intake despite pairing with aversive consequences.
Alcohol intake with and without quinine
Rats first had 2–3 d of habituation to alcohol + quinine. Alcohol intake was tested using a within-rat design for drinking alcohol ± quinine and with treatment versus control (for example, for AP5 experiments, the four groups were AP5 + quinine, AP5 + no quinine, saline + quinine, saline + no quinine). The different conditions were randomized across and within rats using a Latin-square design. Rats were given at least 1 d of drinking alcohol without quinine between each treatment day. In addition, for all experiments except AP5 and ifenprodil, rats were subject to each of the four experimental conditions twice. The two sessions were then averaged, giving a single intake value for each rat for each of the four experimental conditions.
For shRNA experiments, we could not examine alcohol intake with and without shRNA in the same randomized way. Thus, alcohol intakes with or without quinine were each averaged from two intake sessions ∼6–7 weeks after shRNA infection.
Saccharin intake with and without quinine
Two-bottle choice saccharin ± quinine intake was measured using methods similar to those described for alcohol intake with and without quinine, except examining drinking of 300 mg l−1 saccharin or 1,300 mg l−1 saccharin plus 30 mg l−1 quinine. Rats for these studies were previously used to examine mPFC-to-NAcore inhibition and two-bottle-choice alcohol intake. Preliminary experiments identified two concentrations that gave relatively low volumes of intake that were relatively similar to the alcohol volume consumed. Rats were then allowed to drink one of these two solutions in the late morning, 20 min d−1, 5 d week−1, under two-bottle choice conditions. The other bottle contained only water. Rats also drank the usual 20% alcohol under two-bottle choice in the late afternoon each day, 20 min d−1, 5 d week−1. After 1–2 weeks of saccharin ± quinine intake, rats began drinking the saccharin for 8 min d−1, 5 d week−1 in the optogenetic chamber each late morning. Laser stimulation experiments began after 1–2 weeks habituation to having dummy cables attached. On each day, rats were placed in the optogenetic chamber for 5 min before we provided the bottles. Exposure to the two saccharin solutions, with or without laser stimulation in the NAcore, was randomized using a within-rat, Latin-squares design. Unlike for alcohol, rats did not show a rapid load-up period of intake of saccharin ± quinine; thus, we allowed rats 8 min for intake during saccharin optogenetic experiments.
One concern is that laser stimulation of halorhodopsin could have nonspecific locomotor effects, but that giving rats 8 min to drink saccharin ± quinine but 2 min to drink alcohol might allow rats to overcome the locomotor deficit in 8 min. This would give a false negative result for saccharin ± quinine intake. However, we believe this not to be the case. First, halorhodopsin stimulation did not alter intake of quinine-free alcohol with 2 min access, suggesting no locomotor effects per se. Second, rats were allowed 20 min to drink alcohol in the intra-NAcore AP5 and shRNA experiments and operant footshock experiments. In these cases, manipulations had no effect on quinine- or footshock-free alcohol intake, even though there would have plenty of time to overcome locomotor deficits.
Operant self-administration methods
After ∼2.5 months of overnight IAA intake, rats were trained to operantly respond for alcohol54. After two overnight sessions, rats were switched to respond for alcohol 20 min d−1, 5 d week−1, all under a fixed ratio 3 (FR3) schedule. Operant sessions were initiated by presentation of a tone and light (which were also paired with each FR3 response) and raising of the dipper containing 100 μl alcohol; licking the dipper resulted in lowering the dipper and extension of two levers. Thereafter, each FR3 response on the active lever resulted in raising the alcohol-containing dipper for 5 s and presenting the light/tone cue for the first 2 s access to alcohol. Surgery was performed after 2 weeks of operant training. After a further 7–8 weeks, rats were tested for the impact of halorhodopsin activation on baseline operant responding for alcohol. We then introduced daily intermittent footshock, where 1 in 8 FR3 responses paired with a footshock (0.25 mA, 0.5 s) just before raising the dipper containing alcohol.
We observed shock-resistant and shock-sensitive subpopulations of rats, as observed in some studies55,56 of aversion-resistant cocaine intake. We should note that aversive challenges of different qualities could have different impact on alcohol intake, for example, where the majority of rats tolerated quinine while only ∼50% tolerated footshock. In fact, studies of cocaine self-administration have found that all rats will give up responding for cocaine when the shock intensity is high enough57. In addition, compulsive-like cocaine self-administration is observed in a subset of rats responding for cocaine with footshock55,56 but is seen all rats responding for cocaine when paired with a cue predicting footshock58. Thus, rats may differ in the threshold of adversity that would be tolerated to receive a given reward.
In vivo optogenetics
Surgery was performed 6–8 weeks before onset of experiments. Rats were infected in the mPFC (AP +3.20, ML ±1.20, DV −3.00, 10 degrees tilt) or INS (AP +2.80, ML ±4.80 DV −4.70) with 0.6 μl per side of an adeno-associated virus (AAV) encoding halorhodopsin (eNpHR3.0), a chloride pump that silences neurons when activated by a 532-nm green laser. Halorhodopsin was under control of a Camk2a promoter and tethered to EYFP to allow visualization for histology. Control rats were infected with an AAV encoding EYFP alone under control of a Camk2a promoter. During the same surgery, rats were implanted bilaterally with chronically implanted fiber-optic ferrules (CFOs) targeting the medial NAcore (AP +2.20, ML ±2.70, DV −7.18, 8 degrees tilt). These were custom made by inserting a 200 μm optical fiber into a ferrule (a hollow metal cylinder 12 mm long, 2 mm OD, 260 μm ID, Fiber Optics for Sale Co., part F1-0064F-25) and gluing it, so that the fiber extended beyond the ferrule ∼7.5 mm to reach the NAcore. For some INS-virally-injection rats, viral infection spread into the adjacent somatosensory or orbito-frontal cortices which do not project to the medial NAcore59; however, the majority did not exhibit such spread. ChR2 distribution in terminals (Supplementary Fig. 1c,d) was consistent with that reported for mPFC and INS59.
During in vivo optogenetic experiments, a 532 nm laser light (8–12 mW) was turned on during the entire intake session (2 min for two-bottle choice alcohol, 8 min for saccharin, 20 min for operant). The laser was attached by a single cable to a rotating uni-to-bilateral optical commutator (Doric Lenses, custom-made part FRJ-ID(50:50)_FC-sma(2)_v01); light then emerged into two fiber-optic cables with mini-SMA plugs (custom made by Doric) which attached to the bottom of the commutator. At the other end of each cable, the fiber-optic was mounted through a metal ferrule to which a zirconium sleeve (Fiber Instrument Sales, part F18300SSC25) was attached. This was then attached to the CFO in the rat's head during behavioral experiments.
In vitro optogenetics
Surgery was performed 6–8 weeks before onset of experiments. Rats were infected in the mPFC, INS, or BLA (AP: −2.80, ML: ±5.55, DV: −8.70, 10 degrees tilt) with 0.6 μl per side of an AAV encoding ChR2, a cation channel that excites neurons when activated by blue light. ChR2 was under control of a Camk2a promoter and tethered to a yellow fluorescent protein (EYFP) to allow visualization for histology. To generate ChR2-evoked EPSCs in vitro, we adjusted the LED intensity (Prizmatrix, 480 nm, 5ms) to get an EPSC of 130–160 pA at −70 mV or the maximum current that could be generated.
The reversal potential (ERev) for NMDARs in Figure 4c was somewhat positive to 0 mV without junction null correction, which would shift the ERev ∼10–15 mV hyperpolarized (based on the junction null calculator in Clampex), in line with observations in the adult mouse NAc shell. We should note that the NMDAR currents in these cells are small compared to the background noise except at +40 mV, making accurate measurement of ERev somewhat challenging.
For histology, the anterior cortex or BLA was sliced in 500 μm sections, and these living slices were placed into 4% paraformaldehyde just after cutting for assessment of viral injection placement.
shRnAs
Sequences for shRNA were identified using the iRNAi program (www.mekentosj.com). Identified sequences were additionally analyzed using BLAST to ensure subunit specificity. The shRNA targeting sequence for Grin2c was AACCCCCTACACCAAGCTCTGTT and for Grin2d was AAACGCCCTGGTGGCTCTACCTT. Assessment of shRNA knockdown of Grin2c or Grin2d was performed using plasmids for rat Grin2c or Grin2d receptors (a kind gift from Dr. John Woodward) (Supplementary Fig. 5), which were transfected into HEK293 cells along with the Grin2c, Grin2d or control shRNA, using described methods60. 24 h later, protein levels were analyzed with western blot60 (polyclonal anti-Grin2c, sc-1470 Santa Cruz, 1:2,000; polyclonal anti-Grin2d, sc-1471 Santa Cruz, 1:2000, anti-GAPDH, sc-25778 Santa Cruz, 1:50,000) The NMDAR subunit shRNAs were cloned into a pAAV modified to have multiple cloning sites, and with a U6 promoter to drive the shRNA expression and CMV to drive GFP expression. Infected neurons for in vitro electrophysiology could be identified by co-expression of EYFP from the same AAV vector containing the shRNA.
Surgery
Bilateral implantation of cannulas and fiber optic implants targeting the NAcore and other surgical details were as described54. The coordinates used for cannulas and fiber optic implants were previously used54 to show that injection of potassium channel modulators within the NAcore but not NAc shell altered alcohol intake; thus, these coordinates have been validated as targeting the NAcore. For viral infection, injection occurred over 10 min followed by an additional 10 min to allow diffusion of viral particles away from the injection site.
Patch-clamp electrophysiology
Slice preparation and electrophysiology methods were generally as described54, except for in vitro optogenetics, for which the methods are described above. All experiments were performed using whole-cell recording and visualized using infrared-DIC with 3.0 to 4.5 MW electrodes. The internal solution for EPSCs contained (in millimolar): 120 cesium methanesulfonate, 20 HEPES, 0.4 EGTA, 2.8 NaCl, 5 TEA chloride, 2.5 Mg-ATP and 0.25 Na-GTP, pH 7.2 to 7.3, 270 to 285 mOsm. Electrical signals were recorded using Clampex 9.2 or 10.1 and an Axon 700A or 700B patch amplifier (Axon Instruments, Foster City, CA). During recordings, EPSCs were filtered at 2 kHz, digitized at 10 kHz and collected online using Igor Pro software (Wavemetrics, Lake Oswego, OR). Series resistance (10–30 MΩ) and input resistance were monitored on-line with a 4-mV depolarizing step (50 ms) given after every EPSC. Electrically evoked currents were elicited using a bipolar stimulating electrode placed dorsal to the anterior commissure.
For in vitro experiments examining changes in EPSCs at −70 mV caused by NMDAR blockers, we first normalized the baseline EPSC values during the 4 min before drug exposure to 100%, then calculated the percent change in EPSC relative to baseline during the last 3 min of blocker exposure. We then compared blocker-induced changes in EPSCs between neurons from alcohol-drinking and control rats. For firing experiments, EPSPs were evoked by a train of 10 laser pulses at 20 Hz to excite ChR2. Neurons were brought to ∼ −55–60 mV with the patch amplifier to allow EPSPs to evoke action potentials. Measurements of eNpHR3.0 inhibition in vitro in Supplementary Figure 2 used sagittal slices which may enrich for prefrontal inputs to the NAcore.
Statistics
Unless otherwise indicated, results were analyzed with SigmaStat 3.1 using two-way repeated-measures ANOVA with a Tukey post hoc test for multiple comparisons, since the majority of results involve within-rat measurements of intake with and without aversive consequences. All t-tests were unpaired unless otherwise indicated and were two-sided. All data conform to the requirements for each statistical test (for example, normal distribution), and the variances were not different between groups that were compared statistically. Sample sizes were taken from similar previous studies where statistical differences in alcohol intake or in vitro parameters after alcohol intake were observed54. For behavior experiments, n values represent number of individual rats. For in vitro experiments, n values represent number of individual cells, with no more than two cells for a given condition from a particular rat. All data in manuscript and figures are shown as mean ± s.e.m.
For in vitro and in vivo experiments, rats were randomized before surgery so that the mean and s.e.m. of alcohol intake was approximately equal in each group (for example, see Supplementary Fig. 5d). Rats with intake less than 0.2 g alcohol kg−1 under two-bottle choice were not included for analysis. Researchers were not blind to the experimental condition; however, in vitro experiments were typically conducted by two researchers working at the same time on different electrophysiology rigs.
Reagents
AAV-ChR2, AAV-eNpHR3.0 and AAV-EYFP were from the University of North Carolina, Chapel Hill Vector Core. AAVs containing shRNAs for Grin2c and Grin2d were packaged at the NIDA Optogenetics and Transgenic Technology Core facility.
Supplementary Material
Acknowledgments
We thank V. Kharazia and the Gallo Center P50 National Institute on Alcohol Abuse and Alcoholism (NIAAA) AA017072 Histology Core for assistance with histology and R. Maiya for shRNA assistance. Supported by NIAAA RO1AA015358 (F.W.H.), National Institute on Drug Abuse F32DA028065 (T.S.), NIAAA/NIH RO1A/MH13438 (D.R.) and funds provided by the State of California for medical research for alcohol substance abuse through the University of California San Francisco (D.R., A.B., R.O.M.).
Footnotes
Author Contributions: T.S., B.T.C., D.R., R.O.M., A.B. and F.W.H. designed experiments. T.S., S.-J.C., J.A.S., S.L.G. and F.W.H. collected and analyzed data. S.L.G., J.D., B.T.C., B.K.H., D.R. and R.O.M. generated and tested NMDAR shRNAs. T.S., F.W.H. and R.O.M. wrote the paper.
Competing Financial Interests: The authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
Note: Supplementary information is available in the online version of the paper.
References
- 1.Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naqvi NH, Bechara A. The insula and drug addiction: an interoceptive view of pleasure, urges, and decision-making. Brain Struct Funct. 2010;214:435–450. doi: 10.1007/s00429-010-0268-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tiffany ST, Conklin CA. A cognitive processing model of alcohol craving and compulsive alcohol use. Addiction. 2000;95(suppl. 2):S145–S153. doi: 10.1080/09652140050111717. [DOI] [PubMed] [Google Scholar]
- 4.Spanagel R. Alcoholism: a systems approach from molecular physiology to addictive behavior. Physiol Rev. 2009;89:649–705. doi: 10.1152/physrev.00013.2008. [DOI] [PubMed] [Google Scholar]
- 5.Wolffgramm J, Galli G, Thimm F, Heyne A. Animal models of addiction: models for therapeutic strategies? J Neural Transm. 2000;107:649–668. doi: 10.1007/s007020070067. [DOI] [PubMed] [Google Scholar]
- 6.Hopf FW, Chang SJ, Sparta DR, Bowers MS, Bonci A. Motivation for alcohol becomes resistant to quinine adulteration after 3–4 months of intermittent alcohol self-administration. Alcohol Clin Exp Res. 2010;34:1565–1573. doi: 10.1111/j.1530-0277.2010.01241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Breese GR, Sinha R, Heilig M. Chronic alcohol neuroadaptation and stress contribute to susceptibility for alcohol craving and relapse. Pharmacol Ther. 2011;129:149–171. doi: 10.1016/j.pharmthera.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Visser L, Baars AM, van 't Klooster J, van den Bos R. Transient inactivation of the medial prefrontal cortex affects both anxiety and decision-making in male Wistar rats. Front Neurosci. 2011;5:102. doi: 10.3389/fnins.2011.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balleine BW, O'Doherty JP. Human and rodent homologies in action control: corticostriatal determinants of goal-directed and habitual action. Neuropsychopharmacology. 2010;35:48–69. doi: 10.1038/npp.2009.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bassareo V, De Luca MA, Di Chiara G. Differential expression of motivational stimulus properties by dopamine in nucleus accumbens shell versus core and prefrontal cortex. J Neurosci. 2002;22:4709–4719. doi: 10.1523/JNEUROSCI.22-11-04709.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peters J, Kalivas PW, Quirk GJ. Extinction circuits for fear and addiction overlap in prefrontal cortex. Learn Mem. 2009;16:279–288. doi: 10.1101/lm.1041309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LaLumiere RT, Kalivas PW. Glutamate release in the nucleus accumbens core is necessary for heroin seeking. J Neurosci. 2008;28:3170–3177. doi: 10.1523/JNEUROSCI.5129-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Pietro NC, Black YD, Kantak KM. Context-dependent prefrontal cortex regulation of cocaine self-administration and reinstatement behaviors in rats. Eur J Neurosci. 2006;24:3285–3298. doi: 10.1111/j.1460-9568.2006.05193.x. [DOI] [PubMed] [Google Scholar]
- 14.Sesack SR, Grace AA. Cortico-basal ganglia reward network: microcircuitry. Neuropsychopharmacology. 2010;35:27–47. doi: 10.1038/npp.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen BT, et al. Rescuing cocaine-induced prefrontal cortex hypoactivity prevents compulsive cocaine seeking. Nature. 2013;496:359–362. doi: 10.1038/nature12024. [DOI] [PubMed] [Google Scholar]
- 16.Craig AD. How do you feel—now? The anterior insula and human awareness. Nat Rev Neurosci. 2009;10:59–70. doi: 10.1038/nrn2555. [DOI] [PubMed] [Google Scholar]
- 17.Filbey FM, et al. Exposure to the taste of alcohol elicits activation of the mesocorticolimbic neurocircuitry. Neuropsychopharmacology. 2008;33:1391–1401. doi: 10.1038/sj.npp.1301513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Britt JP, et al. Synaptic and behavioral profile of multiple glutamatergic inputs to the nucleus accumbens. Neuron. 2012;76:790–803. doi: 10.1016/j.neuron.2012.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zahm DS. Functional-anatomical implications of the nucleus accumbens core and shell subterritories. Ann NY Acad Sci. 1999;877:113–128. doi: 10.1111/j.1749-6632.1999.tb09264.x. [DOI] [PubMed] [Google Scholar]
- 20.Gerfen CR. Basal ganglia. In: Paxinos G, editor. The Rat Nervous System. 3rd. Elsevier Academic; 2004. pp. 455–508. [Google Scholar]
- 21.Roitman MF, Wheeler RA, Carelli RM. Nucleus accumbens neurons are innately tuned for rewarding and aversive taste stimuli, encode their predictors, and are linked to motor output. Neuron. 2005;45:587–597. doi: 10.1016/j.neuron.2004.12.055. [DOI] [PubMed] [Google Scholar]
- 22.Singh T, McDannald MA, Haney RZ, Cerri DH, Schoenbaum G. Nucleus accumbens core and shell are necessary for reinforcer devaluation effects on pavlovian conditioned responding. Front Integr Neurosci. 2010;4:126. doi: 10.3389/fnint.2010.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.da Cunha IC, et al. The microinjection of AMPA receptor antagonist into the accumbens shell, but not into the accumbens core, induces anxiolysis in an animal model of anxiety. Behav Brain Res. 2008;188:91–99. doi: 10.1016/j.bbr.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 24.McFarland K, Davidge SB, Lapish CC, Kalivas PW. Limbic and motor circuitry underlying footshock-induced reinstatement of cocaine-seeking behavior. J Neurosci. 2004;24:1551–1560. doi: 10.1523/JNEUROSCI.4177-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rassnick S, Pulvirenti L, Koob GF. Oral ethanol self-administration in rats is reduced by the administration of dopamine and glutamate receptor antagonists into the nucleus accumbens. Psychopharmacology (Berl) 1992;109:92–98. doi: 10.1007/BF02245485. [DOI] [PubMed] [Google Scholar]
- 26.Gremel CM, Cunningham CL. Involvement of amygdala dopamine and nucleus accumbens NMDA receptors in ethanol-seeking behavior in mice. Neuropsychopharmacology. 2009;34:1443–1453. doi: 10.1038/npp.2008.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hodge CW, Cox AA. The discriminative stimulus effects of ethanol are mediated by NMDA and GABAA receptors in specifc limbic brain regions. Psychopharmacology (Berl) 1998;139:95–107. doi: 10.1007/s002130050694. [DOI] [PubMed] [Google Scholar]
- 28.Stuber GD, et al. Excitatory transmission from the amygdala to nucleus accumbens facilitates reward seeking. Nature. 2011;475:377–380. doi: 10.1038/nature10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tye KM, et al. Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature. 2011;471:358–362. doi: 10.1038/nature09820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Witten IB, et al. Cholinergic interneurons control local circuit activity and cocaine conditioning. Science. 2010;330:1677–1681. doi: 10.1126/science.1193771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sparta DR, et al. Construction of implantable optical fibers for long-term optogenetic manipulation of neural circuits. Nat Protoc. 2012;7:12–23. doi: 10.1038/nprot.2011.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stefanik MT, et al. Optogenetic inhibition of cocaine seeking in rats. Addict Biol. 2013;18:50–53. doi: 10.1111/j.1369-1600.2012.00479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cull-Candy SG, Leszkiewicz DN. Role of distinct NMDA receptor subtypes at central synapses. Sci STKE. 2004;2004:re16. doi: 10.1126/stke.2552004re16. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, Llinas RR, Lisman JE. Inhibition of NMDARs in the nucleus reticularis of the thalamus produces delta frequency bursting. Front Neural Circuits. 2009;3:20. doi: 10.3389/neuro.04.020.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Low CM, Wee KS. New insights into the not-so-new NR3 subunits of N-methyl-D-aspartate receptor: localization, structure, and function. Mol Pharmacol. 2010;78:1–11. doi: 10.1124/mol.110.064006. [DOI] [PubMed] [Google Scholar]
- 36.Lasek AW, Janak PH, He L, Whistler JL, Heberlein U. Downregulation of mu opioid receptor by RNA interference in the ventral tegmental area reduces ethanol consumption in mice. Genes Brain Behav. 2007;6:728–735. doi: 10.1111/j.1601-183X.2007.00303.x. [DOI] [PubMed] [Google Scholar]
- 37.Shi M, et al. Effects of NR2A and NR2B-containing N-methyl-D-aspartate receptors on neuronal-fring properties. Neuroreport. 2011;22:762–766. doi: 10.1097/WNR.0b013e32834ae32e. [DOI] [PubMed] [Google Scholar]
- 38.Wang J, et al. Ethanol induces long-term facilitation of NR2B-NMDA receptor activity in the dorsal striatum: implications for alcohol drinking behavior. J Neurosci. 2007;27:3593–3602. doi: 10.1523/JNEUROSCI.4749-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maldonado-Irizarry CS, Kelley AE. Differential behavioral effects following microinjection of an NMDA antagonist into nucleus accumbens subregions. Psychopharmacology (Berl) 1994;116:65–72. doi: 10.1007/BF02244872. [DOI] [PubMed] [Google Scholar]
- 40.Di Ciano P, Everitt BJ. Dissociable effects of antagonism of NMDA and AMPA/KA receptors in the nucleus accumbens core and shell on cocaine-seeking behavior. Neuropsychopharmacology. 2001;25:341–360. doi: 10.1016/S0893-133X(01)00235-4. [DOI] [PubMed] [Google Scholar]
- 41.Backström P, Hyytia P. Involvement of AMPA/kainate, NMDA, and mGlu5 receptors in the nucleus accumbens core in cue-induced reinstatement of cocaine seeking in rats. Psychopharmacology (Berl) 2007;192:571–580. doi: 10.1007/s00213-007-0753-8. [DOI] [PubMed] [Google Scholar]
- 42.Yamamoto T. Neural substrates for the processing of cognitive and affective aspects of taste in the brain. Arch Histol Cytol. 2006;69:243–255. doi: 10.1679/aohc.69.243. [DOI] [PubMed] [Google Scholar]
- 43.Sinclair CM, Cleva RM, Hood LE, Olive MF, Gass JT. mGluR5 receptors in the basolateral amygdala and nucleus accumbens regulate cue-induced reinstatement of ethanol-seeking behavior. Pharmacol Biochem Behav. 2012;101:329–335. doi: 10.1016/j.pbb.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.June HL, et al. GABA(A) receptors containing α5 subunits in the CA1 and CA3 hippocampal fields regulate ethanol-motivated behaviors: an extended ethanol reward circuitry. J Neurosci. 2001;21:2166–2177. doi: 10.1523/JNEUROSCI.21-06-02166.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roberts AJ, Cole M, Koob GF. Intra-amygdala muscimol decreases operant ethanol self-administration in dependent rats. Alcohol Clin Exp Res. 1996;20:1289–1298. doi: 10.1111/j.1530-0277.1996.tb01125.x. [DOI] [PubMed] [Google Scholar]
- 46.Chaudhri N, Sahuque LL, Janak PH. Ethanol seeking triggered by environmental context is attenuated by blocking dopamine D1 receptors in the nucleus accumbens core and shell in rats. Psychopharmacology (Berl) 2009;207:303–314. doi: 10.1007/s00213-009-1657-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raeder H, et al. Expression of N-methyl-d-aspartate (NMDA) receptor subunits and splice variants in an animal model of long-term voluntary alcohol self-administration. Drug Alcohol Depend. 2008;96:16–21. doi: 10.1016/j.drugalcdep.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 48.Hagino Y, et al. Essential role of NMDA receptor channel epsilon4 subunit (GluN2D) in the effects of phencyclidine, but not methamphetamine. PLoS ONE. 2010;5:e13722. doi: 10.1371/journal.pone.0013722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Obiang P, et al. GluN2D subunit-containing NMDA receptors control tissue plasminogen activator-mediated spatial memory. J Neurosci. 2012;32:12726–12734. doi: 10.1523/JNEUROSCI.6202-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wenzel A, et al. Distribution of NMDA receptor subunit proteins NR2A, 2B, 2C and 2D in rat brain. Neuroreport. 1995;7:45–48. [PubMed] [Google Scholar]
- 51.Simms JA, et al. Intermittent access to 20% ethanol induces high ethanol consumption in Long-Evans and Wistar rats. Alcohol Clin Exp Res. 2008;32:1816–1823. doi: 10.1111/j.1530-0277.2008.00753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rhodes JS, et al. Mouse inbred strain differences in ethanol drinking to intoxication. Genes Brain Behav. 2007;6:1–18. doi: 10.1111/j.1601-183X.2006.00210.x. [DOI] [PubMed] [Google Scholar]
- 53.Li J, Bian W, Dave V, Ye JH. Blockade of GABAA receptors in the paraventricular nucleus of the hypothalamus attenuates voluntary ethanol intake and activates the hypothalamic-pituitary-adrenocortical axis. Addict Biol. 2011;16:600–614. doi: 10.1111/j.1369-1600.2011.00344.x. [DOI] [PubMed] [Google Scholar]
- 54.Hopf FW, et al. Reduced nucleus accumbens SK channel activity enhances alcohol seeking during abstinence. Neuron. 2010;65:682–694. doi: 10.1016/j.neuron.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deroche-Gamonet V, Belin D, Piazza PV. Evidence for addiction-like behavior in the rat. Science. 2004;305:1014–1017. doi: 10.1126/science.1099020. [DOI] [PubMed] [Google Scholar]
- 56.Belin D, Berson N, Balado E, Piazza PV, Deroche-Gamonet V. High-novelty-preference rats are predisposed to compulsive cocaine self-administration. Neuropsychopharmacology. 2011;36:569–579. doi: 10.1038/npp.2010.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cooper A, Barnea-Ygael N, Levy D, Shaham Y, Zangen A. A confict rat model of cue-induced relapse to cocaine seeking. Psychopharmacology (Berl) 2007;194:117–125. doi: 10.1007/s00213-007-0827-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vanderschuren LJ, Everitt BJ. Drug seeking becomes compulsive after prolonged cocaine self-administration. Science. 2004;305:1017–1019. doi: 10.1126/science.1098975. [DOI] [PubMed] [Google Scholar]
- 59.Berendse HW, Galis-de Graaf Y, Groenewegen HJ. Topographical organization and relationship with ventral striatal compartments of prefrontal corticostriatal projections in the rat. J Comp Neurol. 1992;316:314–347. doi: 10.1002/cne.903160305. [DOI] [PubMed] [Google Scholar]
- 60.Gibb SL, et al. Lyn kinase regulates mesolimbic dopamine release: implication for alcohol reward. J Neurosci. 2011;31:2180–2187. doi: 10.1523/JNEUROSCI.5540-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.