

Graphical abstract

Keywords: Adenine phosphoribosyltransferase, African trypanosomes, Purine salvage, Aminopurinol
Highlights
-
•
African trypanosomes possess two distinct adenine phosphoribosyltransferases.
-
•
Trypanosoma brucei TbAPRT1 is cytosolic, TbAPRT2 localizes to the glycosome.
-
•
Aprt1,2 null mutants are viable but do not incorporate adenine into nucleotides.
-
•
Aprt1,2 null mutants are resistant to aminopurinol but still sensitive to adenine.
-
•
Aminopurinol is a trypanocide with submicromolar activity against T. brucei.
Abstract
African trypanosomes, like all obligate parasitic protozoa, cannot synthesize purines de novo and import purines from their hosts to build nucleic acids. The purine salvage pathways of Trypanosoma brucei being redundant, none of the involved enzymes is likely to be essential. Nevertheless they can be of pharmacological interest due to their role in activation of purine nucleobase or nucleoside analogues, which only become toxic when converted to nucleotides. Aminopurine antimetabolites, in particular, are potent trypanocides and even adenine itself is toxic to trypanosomes at elevated concentrations. Here we report on the T. brucei adenine phosphoribosyltransferases TbAPRT1 and TbAPRT2, encoded by the two genes Tb927.7.1780 and Tb927.7.1790, located in tandem on chromosome seven. The duplication is syntenic in all available Trypanosoma genomes but not in Leishmania. While TbAPRT1 is cytosolic, TbAPRT2 possesses a glycosomal targeting signal and co-localizes with the glycosomal marker aldolase. Interestingly, the distribution of glycosomal targeting signals among trypanosomatid adenine phosphoribosyltransferases is not consistent with their phylogeny, indicating that the acquisition of adenine salvage to the glycosome happened after the radiation of Trypanosoma. Double null mutant T. brucei Δtbaprt1,2 exhibited no growth phenotype but no longer incorporated exogenous adenine into the nucleotide pool. This, however, did not reduce their sensitivity to adenine. The Δtbaprt1,2 trypanosomes were resistant to the adenine isomer aminopurinol, indicating that it is activated by phosphoribosyl transfer. Aminopurinol was about 1000-fold more toxic to bloodstream-form T. brucei than the corresponding hypoxanthine isomer allopurinol. Aminopurinol uptake was not dependent on the aminopurine permease P2 that has been implicated in drug resistance.
1. Introduction
A common feature of most pathogens – viruses, bacteria, protozoa or tumor cells – is their fast proliferation and, consequently, high demand for nucleotides to replicate their genomes. Thus purine and pyrimidine analogues are widely used as chemotherapeutic agents. These are usually administered as nucleobase or nucleoside prodrugs that, once taken up by a target cell, need to be phosphorylated to the corresponding nucleotide in order to exert cytotoxic activity by inhibiting DNA or RNA polymerases. Knowledge on the molecular nature of a pathogen’s nucleotide salvage pathways will therefore promote the rational design of nucleobase or nucleoside antimetabolites that, ideally, are specifically phosphorylated by the target organism but not by human cells.
Like all obligate parasitic protozoa, Trypanosoma brucei ssp. do not synthesize purines de novo (Fish et al., 1982a,b), having lost the genes of the purine anabolic pathway, presumably in adaptation to parasitism. The trypanosomes therefore depend on salvage of purines from their hosts: the tsetse flies (Glossina spp.) and mammals. Trypanosoma brucei rhodesiense and T. b. gambiense respectively cause East- and West-African sleeping sickness in humans, also known as human African trypanosomiasis (HAT). The parasites proliferate extracellularly in the blood and eventually cross the blood–brain barrier, with fatal consequences for the patient. Purines are taken up from the blood by several transporters of overlapping substrate specificities (de Koning et al., 2005). The aminopurines adenine and adenosine are thought to be the favorite purine source of bloodstream-form T. brucei since of all physiological purines, these are taken up the fastest (Fish et al., 1982a). Adenosine is imported via P1- and P2-type transporters, adenine is taken up via P2, H2 and H3 (Carter and Fairlamb, 1993; de Koning and Jarvis, 1997). The pharmacological importance of trypanosomal purine permeases was underscored by the findings that P2 also transports trypanocidal drugs like melarsoprol, diminazene and pentamidine (Carter and Fairlamb, 1993; Carter et al., 1995), and that loss of the gene encoding P2, TbAT1, reduces the sensitivity to these drugs (Mäser et al., 1999; Matovu et al., 2003). Homozygous deletion of TbAT1 in T. brucei also resulted in resistance to adenosine analogs such as cordycepin (3′-deoxyadenosine) or tubercidin (7-deazaadenosine; Geiser et al., 2005; Lüscher et al., 2007).
Adenine at high (e.g. millimolar) concentrations is toxic to trypanosomes (Taliaferro and D’Alesandro, 1971; Geiser et al., 2005). Adenine toxicity is a phenomenon also known from Escherichia coli, where it is exacerbated by genetic disruption of hypoxanthine and guanine phosphoribosyltransferases (E.C. 2.4.2.8) and counteracted by addition of guanosine (Levine and Taylor, 1982). This clearly indicates that an excess of adenine may be cytotoxic to E. coli because it disturbs the cellular purine balance by depleting the guanine pool and increasing the ratio of [AMP] to [GMP]. However, it is unknown whether adenine has the same effect on T. brucei. The purine salvage machinery of bloodstream-form T. brucei is able to interconvert all the physiological purine nucleobases, nucleosides, and nucleotides (Fish et al., 1982a). Interestingly, some of the enzymes involved localize to the glycosomes, which are membrane-bound organelles of kinetoplastid parasites that are devoted mainly to glycolysis but are also involved in other metabolic pathways including purine salvage and pyrimidine biosynthesis (Opperdoes and Michels, 1993).
Glycosomal proteins carry targeting signals that are very similar to the known peroxisomal targeting signals (PTS), suggesting a common evolutionary origin of glycosomes and peroxisomes (Michels et al., 2005). Two types of PTS are known: serine-lysine-leucine (PTS1) or similar tripeptides at the C-terminus of glycosomal proteins, or a less conserved nonapeptide (PTS2) at the N-terminus of glycosomal proteins (Petriv et al., 2004). Hypoxanthine-guanine phosphoribosyltransferases identified from Leishmania spp., Trypanosoma cruzi and T. brucei all carried C-terminal PTS1 signals, and in T. brucei and Leishmania were localized to the glycosomes (Hassan et al., 1985; Shih et al., 1998a,b). Leishmanial HGPRTases are of high pharmacological interest since they accept allopurinol as a substrate (Hwang et al., 1996); human HGPRT is also able to phosphoribosylate allopurinol although it is a poor substrate (Krenitsky et al., 1969) but, unlike in hemoflagellates the resulting allopurinol riboside is not converted to aminopurinol riboside and consequently is not incorporated into nucleic acids (Hitchings, 1975; Shapiro et al., 1991). Allopurinol [4-hydroxypyrazolo(3,4-d)pyrimidine; HPP], used in humans primarily against hyperuricemia (e.g. gout) but also against leishmanioses, is a close structural isomer of hypoxanthine that carries the nitrogen at position 8 instead of 7 in the purine ring. Allopurinol is very active against Leishmania spp. and T. cruzi (Berens et al., 1982; Apt et al., 2003), but less so against T. brucei (Marr and Berens, 1983; Natto et al., 2005; de Koning, unpublished). The analogous N8 isomer of adenine, aminopurinol (4-aminopyrazolopyrimidine; APP) exhibits promising activity against T. brucei (Wallace et al., 2002; Natto et al., 2005). Here we identify and characterize the two APRT genes from T. b. brucei with particular respect to the subcellular localization of the gene products and their role in aminopurinol susceptibility.
2. Materials and methods
2.1. Cultivation of trypanosomes, transfection, drug sensitivity tests
Bloodstream-form T. b. brucei 221 (MITat 1.2) were cultivated in HMI-9 medium (Hirumi and Hirumi, 1989) and 10% FCS at 37 °C in a humidified atmosphere at 5% CO2. Procyclic T. b. brucei 427 were cultivated at 27 °C in SDM-79 (Brun and Schönenberger, 1979) supplemented with 5% fetal calf serum and 1 μg ml−1 hemin. For stable transfection, 108 procyclic or 2 × 107 bloodstream-form trypanosomes were washed and resuspended in 450 μl of electroporation buffer (120 mM KCl, 150 mM CaCl2, 4 mM MgCl2, 8.7 mM K2HPO4, 1.3 mM KH2PO4, 25 mM HEPES, 2 mM EDTA), mixed with 10 μg linearized plasmid DNA, and electroporated at 1.5 kV; antibiotic selection started 24 h after transformation. Positive transformants were cloned by limiting dilution and verified by PCR as well as Southern blots. Drug sensitivities were determined with the AlamarBlue® assay at inocula of 103 (bloodstream forms) or 5 × 104 (procyclics) and 72 h of incubation (Räz et al., 1997). The read-out was fluorescence, measured at wavelengths 530 nm for excitation and 590 nm for emission (Spectromax Gemini). IC50 values were calculated by non-linear fitting of the data to a sigmoidal dose-response curve with variable slope (Prism, GraphPad Software). All compounds were purchased from Sigma except 7-deazaadenine (Berry&Associates). Isothermal microcalorimetry was performed as described (Wenzler et al., 2012). Bloodstream-form cells, 105 in 2 ml HMI-9 medium and 10% horse serum, were inoculated to calorimetry ampoules. Drugs were added to the desired concentration and the ampoules were immediately sealed and placed into the microcalorimeter (TAM III, TA Instruments, New Castle DE, USA). Ampoules containing medium without trypanosomes served as negative controls, ampoules containing trypanosomes without drugs served as positive controls. All experiments were carried out twice, each in triplicate.
2.2. TbAPRT constructs and primers
The constructs for replacement by homologous recombination of the TbAPRT1,2 locus with selectable markers were made by ligating 160 bp of the TbAPRT1 5′-UTR, amplified by PCR from genomic DNA with primers KO180for (5′-ccggtaccgtcacttgtggaggttttgc-3′) and KO180rev (5′-ccaagcttgcgcatcacttcaggatttt-3′), and 160 bp of the TbAPRT2 3′-UTR, amplified with KO200for (5′-ccggtaccgtcacttgtggaggttttgc-3′) and KO200rev (5′-gctctagacacagtcgtgacctggtaatg-3′), to either side of hygromycin (first round) or neomycin (second round) resistance genes cloned into pBluescript II SK+. Verification of positive transformants by Southern blot was done using as probes cloned PCR products specific to TbAPRT1, amplified with primers Fw180 (5′-aatcttcagcagcccatcac-3′) and Rev180 (5′-gcgttcctttgaggaaagtg-3′), or TbAPRT2, amplified with Fw200 (5′-atgccaattgtgctcacgta-3′) and Rev200 (5′-gccacatcacagacggtaag-3′). The same probes were used for Northern blots on total RNA isolated with the hot phenol method (Roditi et al., 1989). The constructs for C-terminal in situ tagging were created by PCR using as template plasmid pMOTag3H containing a triple hemagglutinin tag (100 bp) plus neomycin resistance gene (800 bp) for TbAPRT1, and pMOTag3xM4 containing a triple Myc tag (520 bp) plus hygromycin resistance gene (1 kb) for TbAPRT2. Chimeric primers were used which consisted of the last 80 nt of the target TbAPRT coding region (forward) – respectively the first 80 nt of the target TbAPRT 3′-UTR (reverse) – plus 20 nt specific to the tagging cassette on the template plasmid (Oberholzer et al., 2006). PCR products of the expected size for directly electroporated into trypanosomes and positive transformants were verified by PCR and sequencing for in-frame insertion of the tag.
2.3. Immunofluorescence
For immunofluorescence, 2 × 106 bloodstream-form or 107 procyclic trypanosomes were washed with PBS supplemented with 10 g/l glucose, and spread onto Diagnostic Microscope Slides (Erie Scientific) which had been coated for 30 min with polylysin (100 μg/ml; Sigma). The cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS), washed three times with PBS (for procyclic forms supplemented with 0.1 M glycine), and permeabilized with 0.1% tritonX-100 (bloodstream forms) or 0.05% Tween-20 (procyclics) in PBS. The cells were washed three times with PBS and (procyclics only) incubated for 10 min in methanol at −20 °C followed by rehydration in PBS. After blocking with 2% BSA, the slides were incubated for 45 min in PBS plus 2% BSA with the primary antibody: for TbAPRT1 monoclonal mouse anti-HA diluted 1:300 (Roche), for TbAPRT2 monoclonal mouse anti-myc at 1:100 (Santa Cruz), for aldolase monoclonal rabbit anti-aldolase at 1:10,000 (kind gift of Paul Michels, Université Catholique de Louvain, Brussels). After three washes, the slides were incubated in the dark for 45 min with the secondary antibody: goat anti-mouse Alexa Fluor® 488 (Invitrogen) or goat anti-rabbit 594 (Molecular Probes) at 1:1000. The slides were washed three times and mounted in Vectashield® containing 1.5 μg/ml DAPI (Vector laboratories, Inc.).
2.4. Cell fractionations
For Western blots, total cell lysates of 107 cells were run on 10% SDS-polyacrylamide gels and transferred to nitrocellulose membranes (Immobilon-P, Millipore) in 3 g/l Tris, 14.4 g/l glycine, 20% methanol. After blocking in TBS (10 mM Tris–HCl pH 7.4, 150 mM NaCl) plus 5% milk powder, the blots were hybridized for 1.5 h with primary antibody (1:1000 for anti-HA and anti-Myc; 1:100,000 for anti-aldolase), washed three times in TBS 0.05% Tween-20, and incubated for 1.5 h with the secondary antibody (rabbit anti mouse-HRP; Dako). Signals were detected using ECL-plus (Amersham Biosciences). For digitonin extraction of proteins, 2.5 × 107 cells per assay were washed in SBG (22 mM glucose, 150 mM NaCl, 20 mM NaHPO4 pH 7.8), resuspended in 125 μl SoTE (0.6 M sorbitol, 20 mM Tris–HCl pH 8.0, 2 mM EDTA), and carefully mixed with an equal volume of digitonin at the indicated concentration. After 5 min on ice, soluble (supernatant of 6800 g) and insoluble (pellet of 20,000 g) fractions were separated by centrifugation.
2.5. Purine transport assays
Uptake of [3H]-hypoxanthine and [3H]-adenosine (both from GE Healthcare) was performed essentially as described previously (de Koning and Jarvis, 1997, 1999). Bloodstream trypanosomes T. b. brucei 221 were cultivated in HMI-9 medium for the adenosine uptake studies and isolated from adult female Wistar rats for the hypoxanthine uptake experiments, separating parasites from blood cells on DE52 anion exchange columns (Whatman) and washed into standard assay buffer (AB) as described (de Koning and Jarvis, 1999). Radiolabel concentrations for assaying H2, H3 and P2 activity were 0.1 μM [3H]-hypoxanthine, 1 μM [3H]-hypoxanthine and 0.05 μM [3H]-adenosine, respectively, and incubation time with radiolabel was 30 s. Assays were performed in AB (33 mM HEPES, 98 mM NaCl, 4.6 mM KCl, 0.55 mM CaCl2, 0.07 mM MgSO4, 5.8 mM NaH2PO4, 0.3 mM MgCl2, 23 mM NaHCO3, 14 mM glucose, pH 7.3) and incubations were terminated by the addition of 1 mM unlabelled permeant in ice-cold AB followed by centrifugation through oil (30 s, 12,000g). Radioactivity in the ensuing cell pellet was determined by liquid scintillation counting. Data was analyzed on GraphPad Prism 5.0 using non-linear regression after subtraction of non-specific radiolabel binding. When inhibition with aminopurionol was between 50% and 100% of uptake, incomplete inhibition curves were extrapolated to yield IC50 value based on the assumption of full inhibition and a Hill slope of −1. To measure incorporation of adenine into the nucleotide pool, procyclic trypanosomes were incubated at 107 cells ml−1 in AB plus 1 μM [3H]-adenine at 27 °C for a maximum of 10 min, washed with ice-cold buffer, and extracted in 0.1 M HCl by freeze-thawing. After centrifugation at 12,000 g, samples of the supernatant were spotted onto silica thin-layer chromatography plates (Merck LuxPLate 60F254) and run with a mixture of n-butanol, ethyl acetate, methanol and ammonia of 7:4:3:4 (vol). Nucleotide and nucleobase standards were run alongside the samples. Radiolabel was quantified by scanning the air-dried plates in a radioisotope detector (Berthold Technologies, Switzerland).
3. Results and discussion
3.1. Trypanosoma brucei possesses two adenine phosphoribosyltransferases
The T. b. brucei genome (Berriman et al., 2005) was annotated to contain two putative APRT genes in tandem on chromosome VII: Tb927.7.1780 and Tb927.7.1790. The two predicted proteins return highly significant expectancy of 10−31 and 10−18, respectively, when run with HMMer (Eddy, 1998) against the phosphoribosyl-transferase profile PF00156 from Pfam (Finn et al., 2008). We screened the predicted T. brucei proteome (version 4; 9192 proteins) for further phosphoribosyltransferase domains using HMMer with Pfam profile PF00156. This search returned nine hits with E-values below 10−12. The predicted proteomes of T. cruzi and Leishmania major were searched as well, and the mammalian host Homo sapiens was included as a reference. All the hits with an expectancy below 10−10 were aligned with ClustalW (Thompson et al., 1994). In the resulting phylogenetic tree (Supplementary Figure S1), the two T. brucei proteins Tb927.7.1780 and Tb927.7.1790 clustered with the known APRTases from L. major and H. sapiens and were therefore named TbAPRT1 and TbAPRT2. The APRT clade is proximate to the orotate phosphoribosyltransferases (OPRT; EC 2.4.2.10) and clearly distinct from the HGPRT (2.4.2.8)/XPRT (2.4.2.22) clade (Figure S1).
The two predicted proteins TbAPRT1 and TbAPRT2 share only 24% identity at the level of amino acids, and a nucleotide dot plot of the region encompassing the two genes does not reveal any similarities at the level of genomic DNA (not shown). Thus if the tandem location of TbAPRT1 and TbAPRT2 resulted from the duplication of a primordial gene, it must have been an ancient event. This presumed duplication is older than the radiation of Trypanosoma spp. since the tandem APRT locus is syntenic in Trypanosoma congolense, Trypanosoma vivax and T. cruzi [tritrypdb.org]. In contrast, Leishmania spp. possess only one APRT gene per genome, indicating that the presumed APRT duplication in Trypanosoma happened after the divergence from Leishmania (or that APRT2 was lost from the common ancestor of the Leishmania spp.; Fig. 1). The situation is different regarding HGPRT genes, which form tandem duplications in Trypanosoma as well as in Leishmania (Fig. 1). The two leishmanial genes encode for hypoxanthine-guanine and xanthine phosphoribosyltransferases, respectively. Genetic disruption of either gene was viable in Leishmania donovani (Boitz and Ullman, 2006b), but Δhgprt/xprt double mutants were synthetic lethal (Boitz and Ullman, 2006a, 2010).
Fig. 1.

Phylogenetic tree of trypanosomatid predicted APRT and HGPRT proteins plus human APRT (UniProt P07741) and HGPRT (UniProt P00492) as outgroups. Proteins with a C-terminal PTS1 signal as predicted by the PTS1 Predictor (Neuberger et al., 2003) are labeled with an asterisk. Bootstrapping values for the major branches are given as percent positives of 1000 rounds. The scale bar indicated number of changes per site. For the Trypanosoma tandem genes, the one upstream was labeled as number one. GeneDB accession numbers are Tc00.1047053508207.74 (TcrAPRT1), Tc00.1047053508207.70 (TcrAPRT2), TcIL3000.0.32550 (TcoAPRT1), TcIL3000.7.1260 (TcoAPRT2), TvY486_0701640 (TviAPRT1), TvY486_0701650 (TviAPRT2), LmjF26.0140 (LmaAPRT), Tb927.10.1400 (TbrHGPRT1), Tb927.10.1390 (TbrHGPRT2), TcIL3000.10.1180 (TcoHGPRT1), TcIL3000.10.1170 (TcoHGPRT2), TvY486_1001390 (TviHGPRT1), TvY486_1001370 (TviHGPRT2), Tc00.1047053506457.30 (TcrHGPRT1), Tc00.1047053506457.40 (TcrHGPRT2), LmjF21.0845 (LmajHGPRT), LmjF21.0850 (LmajXPRT).
To experimentally test the predicted adenine phosphoribosyltransferase function, we generated homozygous T. b. brucei tbaprt1,tbaprt2 double knock-out mutants (procyclic forms). A region of 2150 bp encompassing the two TbAPRT genes was replaced, via homologous recombination, with selectable constructs containing hygromycin and neomycin resistance genes. Positive transformants were cloned by limiting dilution and correct integration of the constructs into the TbAPRT1,2 locus was verified by Southern blots (Supplementary Figure S2). The resulting Δtbaprt1,2 trypanosomes grew as fast as their ‘wild-type’ parents (Fig. 2a) in standard cultivation medium; the population doubling times were 14.2 h for ‘wild-type’ and 13.4 h for Δtbaprt1,2 trypanosomes. However, when incubated with [3H]-adenine the Δtbaprt1,2 trypanosomes did not incorporate radiolabel into the nucleotide pool to significant levels (Fig. 2b), demonstrating that Δtbaprt1,2 mutants lack APRTase activity. The requirement of APRT to incorporate adenine into the nucleotide pool is consistent with the absence of an adenine deaminase activity in T. b. brucei, which is believed to be present only in T. vivax (Ogbunude and Ikediobi, 1983) as well as in Leishmania spp. (LaFon et al., 1982). Salvage of [3H]-hypoxanthine was not impaired in Δtbaprt1,2 trypanosomes (not shown).
Fig. 2.
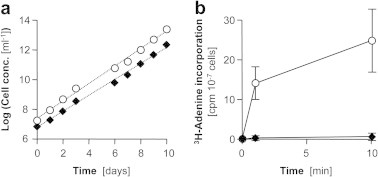
Homozygous disruption of TbAPRTs in procyclic T. brucei. (a) Double null mutant Δtbaprt1,2 trypanosomes (black diamonds) grew as fast as wild-type (white circles) in SDM-79. The cultures were diluted regularly during the experiment and the dilution factors accounted for in the virtual concentration given on the y-axis. (b) In contrast to wild-type trypanosomes, the Δtbaprt1,2 mutants did not incorporate [3H]adenine into the nucleotide pool as determined by thin layer chromatography of cells lysed at the indicated time points. Error bars represent standard deviation of three independent experiments.
3.2. Subcellular localization of TbAPRT1 and TbAPRT2 in trypanosomes
The genes TbAPRT1 and TbAPRT2 are expressed in bloodstream-form as well as in procyclic, tsetse fly midgut-stage trypanosomes as determined by Northern blot analysis (Fig. 3a), which is in agreement with existing proteomics data (Vertommen et al., 2008). Steady-state mRNA levels of TbAPRT2 appeared to be higher in procyclic than in bloodstream-form trypanosomes (Fig. 3a). While the predicted protein TbAPRT2 possesses a bona fide glycosomal targeting signal (serine-arginine-leucine), TbAPRT1 does not. To experimentally investigate their subcellular localization, the TbAPRT1 and the TbAPRT2 gene products were tagged C-terminally with haemagglutinin and c-myc, respectively, by homologous recombination in situ (Oberholzer et al., 2006). Heterozygous transformants carrying one tagged allele of either TbAPRT1 or TbAPRT2 were selected by means of the incorporated neomycin, respectively hygromycin, resistance markers and cloned by limiting dilution. Differential lysis of cells was performed with digitonin, which at lower concentration (0.1 mg digitonin per mg protein) only dissolves the plasma membrane whereas at higher concentration (0.6 mg per mg protein) it also dissolves organellar membranes. Differential lysis followed by Western blotting (Fig. 3b) or direct immunofluorescence of fixed cells (Fig. 4) demonstrated that TbAPRT1 localized to the cytosol while TbAPRT2 co-localized with the glycosomal marker aldolase. The result that a C-terminal myc tag apparently did not prevent TbAPRT2 from getting to the glycosome might be explained by the fact that APRTase is a dimeric enzyme. We speculate that a dimer containing one tagged subunit and one with a free C-terminal PTS1 could still be targeted to the glycosome. Peroxisomal proteins are imported folded and oligomeric (Leon et al., 2006). The presence of TbAPRT2 in the glycosomes implies that there ought to be an aminopurine permease in the glycosomal membrane. Genes encoding permeases for purine nucleobases, including adenine, have been identified from T. brucei (Mäser et al., 1999; Burchmore et al., 2003; Henriques et al., 2003; Ortiz et al., 2009). Several of these transporters were localized to the plasma membrane (Henriques et al., 2003; Ortiz et al., 2009) but no evidence exists for a glycosomal location.
Fig. 3.

Expression of TbAPRT1 and TbAPRT2 in T. brucei. (a) The genes are expressed in procyclic and in bloodstream-form trypanosomes as determined by Northern blot, TbAPRT2 more strongly in the procyclics (a probe of the 18S rRNA was used as a loading control). (b) Western blots of the in situ-tagged gene products after digitonin lysis of the cells. While TbAPRT1 was in the soluble fraction (S, supernatant) even at low digitonin concentration, TbAPRT2 and the glycosomal marker aldolase were in the insoluble fraction (P, pellet) at low digitonin concentration.
Fig. 4.

Immunofluorescence microscopy of procyclic T. brucei: wildtype (left), HA-tagged TbAPRT1 cells (middle), and Myc-tagged TbAPRT2 cells (right). (A) DAPI staining, (B) anti-TbAPRT stain (anti-HA for TbAPRT1 and wildtype, and anti-myc for TbAPRT2), (C) anti-aldolase stain, (D) overlay of B and C. TbAPRT2 but not TbAPRT1 appeared to co-localize with aldolase.
Leishmania have only one APRTase and it is a cytosolic enzyme (Zarella-Boitz et al., 2004). The South-American T. cruzi and the African T. brucei each possess two APRTase paralogues. An interesting difference between T. brucei and T. cruzi emerged when the presence of predicted glycosomal targeting signals was overlaid to the phylogeny of phosphoribosyl transferases (Fig. 1). While TbAPRT2 carries a PTS1 and TbAPRT1 does not, the situation is reversed in T. cruzi where TcAPRT1 has a PTS1 and TcAPRT2 does not (Fig. 1). The distribution of glycosomal targeting signals among trypanosomatid phosphoribosyltransferases is generally not consistent with their phylogeny (Figs. 1 and S1). An interesting case is presented by the phosphoribosyl-pyrophosphate synthetases (PRS; EC 2.7.6.1): T. brucei, T. cruzi and L. major each possess three PRS paralogues, of which in every species a different one is predicted to be targeted to the glycosome (Figure S1). In summary, this argues that the recruitment of adenine salvage enzymes to the glycosome happened independently, after the radiation of trypanosomatids. The physiological role of purine salvage enzymes in the glycosomes is unclear, since replacement of glycosomal XPRTase in L. donovani with a truncated, cytosolic form did not impair purine salvage (Zarella-Boitz et al., 2004).
3.3. Antitrypanosomal activity of purine nucleobase analogues
While adenosine antimetabolites such as cordycepin, tubercidin, or vidarabine are known as potent trypanocides (Bacchi et al., 1992; Kaminsky et al., 1996; Bressi et al., 2001; Drew et al., 2003; Geiser et al., 2005; Rodenko et al., 2007; Vodnala et al., 2008), adenine antimetabolites have so far received little attention (Wallace et al., 2002, 2004). Yet, adenine analogues are attractive trypanocides for many reasons. As mentioned above, trypanosomes lack adenine and adenosine deaminase (Ogbunude and Ikediobi, 1983), making adenine analogues highly stable. Secondly, T. brucei express at least three separate adenine transporters in bloodstream forms: the aminopurine transporter P2 (de Koning et al., 2005), and the broad specificity nucleobase transporters H2 and H3 (de Koning and Jarvis, 1997). Uptake by multiple transporters greatly reduces the chances of resistance through loss of transporter function, as we previously demonstrated for allopurinol (Natto et al., 2005) and pentamidine (de Koning, 2008). Moreover, adenines have a higher rate of membrane diffusion than hypoxanthine analogues or nucleosides; in Toxoplasma gondii adenine salvage was found to be non-saturable and consistent with simple diffusion (De Koning et al., 2003), made possible by a favourable octanol:water partition coefficient XLogP3 (Cheng et al., 2007) of just −0.1.
We determined the sensitivity of different T. brucei lines to adenine and analogues with an in vitro test using the fluorescent dye AlamarBlue® as an indicator of cell viability (Räz et al., 1997). As observed previously (Taliaferro and D’Alesandro, 1971; Geiser et al., 2005), adenine itself was toxic to trypanosomes with an IC50 value around 300 μM (Fig. 5); bloodstream-form and procyclic T. brucei were equally sensitive. Adenine toxicity in E. coli is thought to be linked to an imbalance between cellular GTP and ATP levels (Levine and Taylor, 1982). In T. brucei, however, deletion of TbAPRT did not alleviate adenine toxicity (Fig. 5). As the adenine was no longer incorporated into the nucleotide pool (Fig. 2b) this means that in trypanosomes adenine toxicity is not caused by an imbalance between [ATP] and [GTP]. Furthermore, adenine toxicity in E. coli could be counteracted by addition of excess guanosine (Levine and Taylor, 1982), which is not the case in T. brucei (data not shown). Our conclusion that adenine toxicity in trypanosomes is not linked to increased adenine nucleotide levels is in agreement with the fact that adenosine, which gets incorporated to the nucleotide pool by adenosine kinase (Lüscher et al., 2007), has no effect on T. b. brucei growth rates up to millimolar concentrations. Thus, the mechanism of adenine toxicity in trypanosomes remains elusive.
Fig. 5.

Structure of adenine and aminopurinol and activity against bloodstream-form (BSF) T. b. brucei, procyclic forms (PCF), and procyclic forms carrying a homozygous deletion of the complete TbAPRT1 and TbAPRT2 locus (Δaprt1,2). Activity was determined in vitro with the Alamar blue assay Hirumi and Hirumi, 1989.
In Leishmania, allopurinol is converted to allopurinol riboside monophosphate by HGPRT, aminated to aminopurinol riboside monophosphate in two steps by adenylosuccinate synthetase and -lyase, and then phosphorylated to the triphosphate which gets incorporated into RNA (Marr et al., 1978). Phosphoribosyl transfer to allopurinol is very inefficient in mammalian cells (Krenitsky et al., 1969) with most being rapidly metabolized to oxipurinol (Hitchings, 1975). The approximately 10% of allopurinol converted to allopurinol riboside monophosphate is not accepted by human adenylosuccinate synthetase whereas it can substitute for IMP as a substrate for the corresponding enzyme of Leishmania (Spector et al., 1984). Similarly, allopurinol riboside monophosphate is an inhibitor of L. donovani GMP reductase with 100-fold weaker activity against the human enzyme (Spector et al., 1984).
While these observations explain the therapeutic window of allopurinol for the treatment of leishmanioses and Chagas’ disease, allopurinol was not potent against T. brucei (IC50 against bloodstream forms of 170 μM; not shown). In contrast, aminopurinol exhibited submicromolar activity against T. brucei bloodstream forms with an IC50 value of 190 nM (Fig. 5). To determine whether amimopurinol acts in a cytostatic or a cytotoxic way, bloodstream-form trypanosomes were subjected to isothermal microcalorimetry. This technique allows to non-invasively monitor cell proliferation in real time by measuring the produced heat flow (Wenzler et al., 2012). In contrast to the cytotoxic reference drug pentamidine, aminopurinol did not kill the trypanosomes but merely inhibited their growth (Fig. 6).
Fig. 6.

Real-time isothermal microcalorimetry with bloodstream-form T. b. brucei exposed to aminopurinol (green, 7.5 μM; blue, 15 μM; magenta, 25 μM; dark blue, 45 μM), pentamidine (red, 15 nM), or no drug at all (black). The curves are averages of triplicates, except for the negative control without trypanosomes (grey). The experiment was performed as described (Wenzler et al., 2012). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.4. Uptake of allopurinol and aminopurinol by T. brucei bloodstream forms
An alternative explanation for the much greater efficacy of aminopurinol could lie in more efficient uptake of this prodrug. We previously reported allopurinol inhibition constants for the T. b. brucei nucleobase transporters H2 and H3 (Ki values of 4.0 and 194 μM, respectively; (de Koning and Jarvis, 1997)). We report here that these transporters in fact display lower affinity for aminopurinol (Fig. 7), with Ki values of 230 ± 35 and 105 ± 24 μM for H2 and H3, respectively. In addition, we determined the Ki value for the P2 aminopurine transporter at 75.3 ± 5.7 μM (n = 6; Fig. 7) – lower than for allopurinol (Ki = 260 μM) (de Koning and Jarvis, 1999), but much higher than for adenine (Ki = 0.3 μM). Thus, N8 appears to be unfavourable for binding to P2. We tested whether aminopurinol activity was dependent on P2 expression using a Δtbat1 strain (Matovu et al., 2003), but found no resistance to aminopurinol (Fig. 8) although we observed 18-fold resistance to diminazene, consistent with previous reports (de Koning et al., 2004). We thus conclude that the remarkable efficacy of aminopurinol versus allopurinol against T. b. brucei is highly unlikely to be related to more efficient uptake. Instead, it is most likely attributable to the much more efficient conversion of the prodrug to the active compound, aminopurinol riboside triphosphate. As allopurinol is a substrate for TbHGPRT (Allen and Ullman, 1993) it must be the adenylosuccinate synthetase/lyase step that is principally responsible for the striking difference.
Fig. 7.
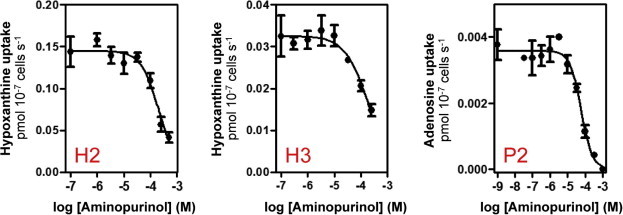
Inhibition of purine transporters of bloodstream-form T. brucei by aminopurinol. (A) inhibition of the H2 nucleobase transporter using 0.1 μM [3H]-hypoxanthine (de Koning and Jarvis, 1997), (B) inhibition of the H3 nucleobase transporter using 1 μM [3H]-hypoxanthine (de Koning and Jarvis, 1997), (C) inhibition of the P2 aminopurine transporter using 0.05 μM [3H]-adenosine in the presence of 1 mM inosine (to saturate P1; Carter and Fairlamb, 1993). All experiments were conducted in triplicate and standard errors are indicated. Experiments shown are representative of at least three independently conducted experiments. In all cases, incubation time with radiolabel was 30 s, well within the linear phase of uptake (de Koning and Jarvis, 1997, 1999).
Fig. 8.

Effect of adenine on aminopurinol sensitivity of wild-type and mutant T. brucei lines. IC50 values were determined with the Alamar blue assay Räz et al., 1997 in the presence (grey) or absence (white) of 100 μM adenine and are shown on a log scale. The statistical significance (asterisks) was determined on the untransformed data with two-tailed t-tests. Error bars represent standard deviations of at least three independent experiments.
3.5. Phenotype of procyclic form APRT null mutants
Double null mutant Δtbaprt1,2 procyclics were significantly less sensitive to aminopurinol than their ‘wild-type’ parent (p < 0.001, one-way Anova followed by Tukey’s multiple comparison test) even though procyclic cells were already much less sensitive than bloodstream forms (Fig. 5), confirming that the drug is activated by phosphoribosyl transfer. This is in agreement with results from L. donovani, where aminopurinol was used to select for Δaprt null mutants (Hwang et al., 1996). Thus, aminopurinol is likely to have the same mechanism of action as allopurinol, but requires only a one-step activation by APRT to aminopurinol riboside monophosphate. Somewhat surprisingly, given that the purine salvage pathways are similar in the two life stages (Fish et al., 1982a,b), aminopurinol was over two orders of magnitude more active against bloodstream-form than against procyclic trypanosomes (Fig. 5). One difference in purine salvage between procyclic and bloodstream-form trypanosomes is that the aminopurine permease P2 is only active in the latter (de Koning et al., 1998; de Koning and Jarvis, 1999), but the relatively low affinity of the compound for this transporter, and the lack of resistance to aminopurinol in the Δtbat1 line, show this is not a crucial difference in this case. Addition of adenine at 100 μM decreased aminopurinol susceptibility in bloodstream-form as well as in procyclic T. brucei (Fig. 8), as observed previously for T. cruzi (Marr et al., 1978). The two possible explanations for this observation are that adenine could either be competing with aminopurinol for transport into the cell, or alternatively inside the cell at the APRT binding site. Since 100 μM adenine had no effect on aminopurinol susceptibility in Δtbaprt1,2 trypanosomes (Fig. 8), the counter-acting effect of adenine must be caused at the APRT site rather than at the level of uptake. We conclude that aminopurinol uptake is largely independent of any adenine-sensitive transporters and may to a significant degree involve simple diffusion. Its physical characteristics, which are highly similar to adenine with a XLogP3 of −0.2 (compare −1.1 for hypoxanthine and adenosine (source: pubchem.ncbi.nlm.nih.gov)) would allow a sufficient penetration into the trypanosome considering the duration of the standard drug sensitivity protocol with Alamar blue (72 h of exposure in vitro).
4. Conclusion
T. brucei possess two adenine phosphoribosyltransferase genes located in tandem on chromosome VII: TbAPRT1 (Tb927.7.1780) and TbAPRT2 (Tb927.7.1790). The duplication is syntenic in T. congolense, T. vivax and T. cruzi, but absent in Leishmania spp. (Fig. 1). The two genes are expressed in bloodstream-form as well as procyclic trypanosomes (Fig. 3a). While TbAPRT1 is cytosolic, TbAPRT2 carries a C-terminal PTS1 signal and co-localizes with the glycosomal marker aldolase (Figs. 3b and 4). As expected given the high degree of redundancy of the purine salvage pathways in T. brucei, TbAPRT1 and TbAPRT2 are not essential genes in vitro and there was no growth phenotype (Fig. 2a). Double null mutant Δtbaprt1,2 trypanosomes lack APRTase activity (Fig. 2b) which, however, does not protect them from the toxic effect of high adenine concentrations. Thus, in contrast to the situation in E. coli, adenine toxicity in T. brucei does not seem to be caused at the level of purine nucleotide imbalance and remains an unresolved phenomenon – which nonetheless demonstrates that trypanosomal purine salvage harbors vulnerable targets. The pharmacological importance of purine salvage was further underscored by the finding that the Δtbaprt1,2 trypanosomes were resistant to aminopurinol (Fig. 5). With an IC50 of 190 nM (Fig. 5), the adenine isomer aminopurinol is 1000-fold more active against T. brucei bloodstream forms than the corresponding hypoxanthine isomer allopurinol, consistent with the model that the aminopurines adenine and adenosine are the preferred purine sources for bloodstream-form trypanosomes and analogues thereof the most potent among the trypanocidal purine antimetabolites. A potential advantage of aminopurinol over adenosine analogues is that Δtbat1 trypanosomes are fully sensitive (Fig. 8). Loss-of-function mutations in TbAT1, the gene encoding the aminopurine permease P2, have been reported from T. brucei field isolates and implicated in resistance to melaminophenyl arsenicals, diamidines, and adenosine antimetabolites. Allopurinol not being suitable against African trypanosomes for lack of efficacy, aminopurinol may be an interesting alternative for further studies. However, the finding that aminopurinol exerts a cytostatic rather than a cytotoxic effect on bloodstream-form T. brucei (Fig. 6) limits its potential usefulness.
Acknowledgements
We are grateful to Paul Michels for the aldolase antiserum, Patrick Bregy for help with immunofluorescence, Tom Seebeck for help with thin layer chromatography, Peter Bütikofer for help with tritium-scanning, and Juma Ali and Anthonius Eze for assistance with the determination of the Ki value of aminopurinol on T. brucei purine transporters. This work was funded by the Swiss National Science Foundation.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supplementary data
Phylogenetic tree of all the hits in the predicted proteomes of Trypanosoma brucei (red), Trypanosoma cruzi (blue), Leishmania major (green) and Homo sapiens (black) with an E-value below 10−10 against Pfam profile PF00156 (‘Phosphoribosyl transferase domain’) as calulated by HMMer. Predicted glyocosomal location is indicated with one (PTS1), respectively two asterisks (PTS2). Bootstrap values are in percent positives of 1000 rounds. The scale bar indicates number of amino acid substitutions per site. APRT, adenine phosphoribosyltransferases (EC 2.4.2.7); OPRT, orotate phosphoribosyltransferases (EC 2.4.2.10); PRS, phosphoribosyl pyrophosphate synthetases (ribose-phosphate pyrophosphokinases, EC 2.7.6.1); HGXPRT, hypoxanthine/guanine/xanthine phosphoribosyltransferases (EC 2.4.2.8, 2.4.2.22).
Supplementary Figure S2.

Southern blots of parental Trypanosoma brucei (lane 0) and four homozygous Δtbaprt1,2 double knock-out mutants (lanes 1–4). The T. brucei genomic DNA was digested with the indicated enzymes and hybridized to probes for the two APRT genes and the antibiotic resistance markers used for selection. The four transgenic lines are negative for either APRT paralogue and positive for the two antibiotic resistance markers, indicating correct integration of the deletion constructs.
References
- Allen T.E., Ullman B. Cloning and expression of the hypoxanthine-guanine phosphoribosyltransferase gene from Trypanosoma brucei. Nucleic Acids Res. 1993;21:5431–5438. doi: 10.1093/nar/21.23.5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apt W., Arribada A., Zulantay I., Sanchez G., Vargas S.L., Rodriguez J. Itraconazole or allopurinol in the treatment of chronic American trypanosomiasis: the regression and prevention of electrocardiographic abnormalities during 9 years of follow-up. Ann. Trop. Med. Parasitol. 2003;97:23–29. doi: 10.1179/000349803125002751. [DOI] [PubMed] [Google Scholar]
- Bacchi C.J., Nathan H.C., Yarlett N., Goldberg B., McCann P.P., Bitonti A.J., Sjoerdsma A. Cure of murine Trypanosoma brucei rhodesiense infections with an S-adenosylmethionine decarboxylase inhibitor. Antimicrob. Agents Chemother. 1992;36:2736–2740. doi: 10.1128/aac.36.12.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berens R.L., Marr J.J., Steele da Cruz F.S., Nelson D.J. Effect of allopurinol on Trypanosoma cruzi: metabolism and biological activity in intracellular and bloodstream forms. Antimicrob. Agents Chemother. 1982;22:657–661. doi: 10.1128/aac.22.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berriman M., Ghedin E., Hertz-Fowler C., Blandin G., Renauld H., Bartholomeu D.C., Lennard N.J., Caler E., Hamlin N.E., Haas B., Bohme U., Hannick L., Aslett M.A., Shallom J., Marcello L., Hou L., Wickstead B., Alsmark U.C., Arrowsmith C., Atkin R.J., Barron A.J., Bringaud F., Brooks K., Carrington M., Cherevach I., Chillingworth T.J., Churcher C., Clark L.N., Corton C.H., Cronin A., Davies R.M., Doggett J., Djikeng A., Feldblyum T., Field M.C., Fraser A., Goodhead I., Hance Z., Harper D., Harris B.R., Hauser H., Hostetler J., Ivens A., Jagels K., Johnson D., Johnson J., Jones K., Kerhornou A.X., Koo H., Larke N., Landfear S., Larkin C., Leech V., Line A., Lord A., Macleod A., Mooney P.J., Moule S., Martin D.M., Morgan G.W., Mungall K., Norbertczak H., Ormond D., Pai G., Peacock C.S., Peterson J., Quail M.A., Rabbinowitsch E., Rajandream M.A., Reitter C., Salzberg S.L., Sanders M., Schobel S., Sharp S., Simmonds M., Simpson A.J., Tallon L., Turner C.M., Tait A., Tivey A.R., Van Aken S., Walker D., Wanless D., Wang S., White B., White O., Whitehead S., Woodward J., Wortman J., Adams M.D., Embley T.M., Gull K., Ullu E., Barry J.D., Fairlamb A.H., Opperdoes F., Barrell B.G., Donelson J.E., Hall N., Fraser C.M., Melville S.E., El-Sayed N.M. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- Boitz J.M., Ullman B. A conditional mutant deficient in hypoxanthine-guanine phosphoribosyltransferase and xanthine phosphoribosyltransferase validates the purine salvage pathway of Leishmania donovani. J. Biol. Chem. 2006;281:16084–16089. doi: 10.1074/jbc.M600188200. [DOI] [PubMed] [Google Scholar]
- Boitz J.M., Ullman B. Leishmania donovani singly deficient in HGPRT, APRT or XPRT are viable in vitro and within mammalian macrophages. Mol. Biochem. Parasitol. 2006;148:24–30. doi: 10.1016/j.molbiopara.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Boitz J.M., Ullman B. Amplification of adenine phosphoribosyltransferase suppresses the conditionally lethal growth and virulence phenotype of Leishmania donovani mutants lacking both hypoxanthine-guanine and xanthine phosphoribosyltransferases. J. Biol. Chem. 2010;285:18555–18564. doi: 10.1074/jbc.M110.125393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressi J.C., Verlinde C.L., Aronov A.M., Shaw M.L., Shin S.S., Nguyen L.N., Suresh S., Buckner F.S., Van Voorhis W.C., Kuntz I.D., Hol W.G., Gelb M.H. Adenosine analogues as selective inhibitors of glyceraldehyde-3-phosphate dehydrogenase of Trypanosomatidae via structure-based drug design. J. Med. Chem. 2001;44:2080–2093. doi: 10.1021/jm000472o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun R., Schönenberger M. Cultivation and in vivo cloning of procyclic culture forms of Trypanosoma brucei in a semi-defined medium. Acta Trop. 1979;36:289–292. [PubMed] [Google Scholar]
- Burchmore R.J., Wallace L.J., Candlish D., Al-Salabi M.I., Beal P.R., Barrett M.P., Baldwin S.A., de Koning H.P. Cloning, heterologous expression, and in situ characterization of the first high affinity nucleobase transporter from a protozoan. J. Biol. Chem. 2003;278:23502–23507. doi: 10.1074/jbc.M301252200. [DOI] [PubMed] [Google Scholar]
- Carter N.S., Fairlamb A.H. Arsenical-resistant trypanosomes lack an unusual adenosine transporter. Nature. 1993;361:173–175. doi: 10.1038/361173a0. [DOI] [PubMed] [Google Scholar]
- Carter N.S., Berger B.J., Fairlamb A.H. Uptake of diamidine drugs by the P2 nucleoside transporter in melarsen-sensitive and -resistant Trypanosoma brucei brucei. J. Biol. Chem. 1995;270:28153–28157. doi: 10.1074/jbc.270.47.28153. [DOI] [PubMed] [Google Scholar]
- Cheng T., Zhao Y., Li X., Lin F., Xu Y., Zhang X., Li Y., Wang R., Lai L. Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J. Chem. Inf. Model. 2007;47:2140–2148. doi: 10.1021/ci700257y. [DOI] [PubMed] [Google Scholar]
- de Koning H.P., Jarvis S.M. Purine nucleobase transport in bloodstream forms of Trypanosoma brucei brucei is mediated by two novel transporters. Mol. Biochem. Parasitol. 1997;89:245–258. doi: 10.1016/s0166-6851(97)00129-1. [DOI] [PubMed] [Google Scholar]
- de Koning H.P., Watson C.J., Jarvis S.M. Characterization of a nucleoside/proton symporter in procyclic Trypanosoma brucei brucei. J. Biol. Chem. 1998;273:9486–9494. doi: 10.1074/jbc.273.16.9486. [DOI] [PubMed] [Google Scholar]
- de Koning H.P., Jarvis S.M. Adenosine transporters in bloodstream forms of Trypanosoma brucei brucei: substrate recognition motifs and affinity for trypanocidal drugs. Mol. Pharmacol. 1999;56:1162–1170. doi: 10.1124/mol.56.6.1162. [DOI] [PubMed] [Google Scholar]
- de Koning H.P., Al-Salabi M.I., Cohen A.M., Coombs G.H., Wastling J.M. Identification and characterisation of high affinity nucleoside and nucleobase transporters in Toxoplasma gondii. Int. J. Parasitol. 2003;33:821–831. doi: 10.1016/s0020-7519(03)00091-2. [DOI] [PubMed] [Google Scholar]
- de Koning H.P., Anderson L.F., Stewart M., Burchmore R.J., Wallace L.J., Barrett M.P. The trypanocide diminazene aceturate is accumulated predominantly through the TbAT1 purine transporter: additional insights on diamidine resistance in african trypanosomes. Antimicrob. Agents Chemother. 2004;48:1515–1519. doi: 10.1128/AAC.48.5.1515-1519.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Koning H.P., Bridges D.J., Burchmore R.J. Purine and pyrimidine transport in pathogenic protozoa: from biology to therapy. FEMS Microbiol. Rev. 2005;29:987–1020. doi: 10.1016/j.femsre.2005.03.004. [DOI] [PubMed] [Google Scholar]
- de Koning H.P. The ever-increasing complexities of arsenical-diamidine cross-resistance in African trypanosomes. Trends Parasitol. 2008;24:345–349. doi: 10.1016/j.pt.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Drew M.E., Morris J.C., Wang Z., Wells L., Sanchez M., Landfear S.M., Englund P.T. The adenosine analog tubercidin inhibits glycolysis in Trypanosoma brucei as revealed by an RNA interference library. J. Biol. Chem. 2003;278:46596–46600. doi: 10.1074/jbc.M309320200. [DOI] [PubMed] [Google Scholar]
- Eddy S.R. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Finn R.D., Tate J., Mistry J., Coggill P.C., Sammut S.J., Hotz H.R., Ceric G., Forslund K., Eddy S.R., Sonnhammer E.L., Bateman A. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–D288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish W.R., Looker D.L., Marr J.J., Berens R.L. Purine metabolism in the bloodstream forms of Trypanosoma gambiense and Trypanosoma rhodesiense. Biochim. Biophys. Acta. 1982;719:223–231. doi: 10.1016/0304-4165(82)90092-7. [DOI] [PubMed] [Google Scholar]
- Fish W.R., Marr J.J., Berens R.L. Purine metabolism in Trypanosoma brucei gambiense. Biochim. Biophys. Acta. 1982;714:422–428. doi: 10.1016/0304-4165(82)90149-0. [DOI] [PubMed] [Google Scholar]
- Geiser F., Luscher A., de Koning H.P., Seebeck T., Mäser P. Molecular pharmacology of adenosine transport in Trypanosoma brucei: P1/P2 revisited. Mol. Pharmacol. 2005;68:589–595. doi: 10.1124/mol.104.010298. [DOI] [PubMed] [Google Scholar]
- Hassan H.F., Mottram J.C., Coombs G.H. Subcellular localisation of purine-metabolising enzymes in Leishmania mexicana mexicana. Comp. Biochem. Physiol. B. 1985;81:1037–1040. doi: 10.1016/0305-0491(85)90110-5. [DOI] [PubMed] [Google Scholar]
- Henriques C., Sanchez M.A., Tryon R., Landfear S.M. Molecular and functional characterization of the first nucleobase transporter gene from African trypanosomes. Mol. Biochem. Parasitol. 2003;130:101–110. doi: 10.1016/s0166-6851(03)00167-1. [DOI] [PubMed] [Google Scholar]
- Hirumi H., Hirumi K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 1989;75:985–989. [PubMed] [Google Scholar]
- Hitchings G.H. Pharmacology of allopurinol. Arthritis Rheum. 1975;18:863–870. doi: 10.1002/art.1780180733. [DOI] [PubMed] [Google Scholar]
- Hwang H.Y., Gilberts T., Jardim A., Shih S., Ullman B. Creation of homozygous mutants of Leishmania donovani with single targeting constructs. J. Biol. Chem. 1996;271:30840–30846. doi: 10.1074/jbc.271.48.30840. [DOI] [PubMed] [Google Scholar]
- Kaminsky R., Schmid C., Grether Y., Holy A., De Clercq E., Naesens L., Brun R. (S)-9-(3-hydroxy-2-phosphonylmethoxypropyl)adenine [(S)-HPMPA]: a purine analogue with trypanocidal activity in vitro and in vivo. Trop. Med. Int. Health. 1996;1:255–263. doi: 10.1111/j.1365-3156.1996.tb00036.x. [DOI] [PubMed] [Google Scholar]
- Krenitsky T.A., Papaioannou R., Elion G.B. Human hypoxanthine phosphoribosyltransferase. I. Purification, properties, and specificity. J. Biol. Chem. 1969;244:1263–1270. [PubMed] [Google Scholar]
- LaFon S.W., Nelson D.J., Berens R.L., Marr J.J. Purine and pyrimidine salvage pathways in Leishmania donovani. Biochem. Pharmacol. 1982;31:231–238. doi: 10.1016/0006-2952(82)90216-7. [DOI] [PubMed] [Google Scholar]
- Leon S., Goodman J.M., Subramani S. Uniqueness of the mechanism of protein import into the peroxisome matrix: transport of folded, co-factor-bound and oligomeric proteins by shuttling receptors. Biochim. Biophys. Acta. 2006;1763:1552–1564. doi: 10.1016/j.bbamcr.2006.08.037. [DOI] [PubMed] [Google Scholar]
- Levine R.A., Taylor M.W. Mechanism of adenine toxicity in Escherichia coli. J. Bacteriol. 1982;149:923–930. doi: 10.1128/jb.149.3.923-930.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher A., Onal P., Schweingruber A.M., Mäser P. Adenosine kinase of Trypanosoma brucei and its role in susceptibility to adenosine antimetabolites. Antimicrob. Agents Chemother. 2007;51:3895–3901. doi: 10.1128/AAC.00458-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marr J.J., Berens R.L., Nelson D.J. Antitrypanosomal effect of allopurinol: conversion in vivo to aminopyrazolopyrimidine nucleotides by Trypanosoma cruzi. Science. 1978;201:1018–1020. doi: 10.1126/science.356267. [DOI] [PubMed] [Google Scholar]
- Marr J.J., Berens R.L. Pyrazolopyrimidine metabolism in the pathogenic trypanosomatidae. Mol. Biochem. Parasitol. 1983;7:339–356. doi: 10.1016/0166-6851(83)90016-6. [DOI] [PubMed] [Google Scholar]
- Mäser P., Sütterlin C., Kralli A., Kaminsky R. A nucleoside transporter from Trypanosoma brucei involved in drug resistance. Science. 1999;285:242–244. doi: 10.1126/science.285.5425.242. [DOI] [PubMed] [Google Scholar]
- Matovu E., Stewart M.L., Geiser F., Brun R., Mäser P., Wallace L.J., Burchmore R.J., Enyaru J.C., Barrett M.P., Kaminsky R., Seebeck T., de Koning H.P. Mechanisms of arsenical and diamidine uptake and resistance in Trypanosoma brucei. Euk. Cell. 2003;2:1003–1008. doi: 10.1128/EC.2.5.1003-1008.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michels P.A., Moyersoen J., Krazy H., Galland N., Herman M., Hannaert V. Peroxisomes, glyoxysomes and glycosomes (review) Mol. Membr. Biol. 2005;22:133–145. doi: 10.1080/09687860400024186. [DOI] [PubMed] [Google Scholar]
- Natto M.J., Wallace L.J., Candlish D., Al-Salabi M.I., Coutts S.E., de Koning H.P. Trypanosoma brucei: expression of multiple purine transporters prevents the development of allopurinol resistance. Exp. Parasitol. 2005;109:80–86. doi: 10.1016/j.exppara.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Neuberger G., Maurer-Stroh S., Eisenhaber B., Hartig A., Eisenhaber F. Prediction of peroxisomal targeting signal 1 containing proteins from amino acid sequence. J. Mol. Biol. 2003;328:581–592. doi: 10.1016/s0022-2836(03)00319-x. [DOI] [PubMed] [Google Scholar]
- Oberholzer M., Morand S., Kunz S., Seebeck T. A vector series for rapid PCR-mediated C-terminal in situ tagging of Trypanosoma brucei genes. Mol. Biochem. Parasitol. 2006;145:117–120. doi: 10.1016/j.molbiopara.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Ogbunude P.O., Ikediobi C.O. Comparative aspects of purine metabolism in some African trypanosomes. Mol. Biochem. Parasitol. 1983;9:279–287. doi: 10.1016/0166-6851(83)90084-1. [DOI] [PubMed] [Google Scholar]
- Opperdoes F.R., Michels P.A. The glycosomes of the Kinetoplastida. Biochimie. 1993;75:231–234. doi: 10.1016/0300-9084(93)90081-3. [DOI] [PubMed] [Google Scholar]
- Ortiz D., Sanchez M.A., Quecke P., Landfear S.M. Two novel nucleobase/pentamidine transporters from Trypanosoma brucei. Mol. Biochem. Parasitol. 2009;163:67–76. doi: 10.1016/j.molbiopara.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petriv O.I., Tang L., Titorenko V.I., Rachubinski R.A. A new definition for the consensus sequence of the peroxisome targeting signal type 2. J. Mol. Biol. 2004;341:119–134. doi: 10.1016/j.jmb.2004.05.064. [DOI] [PubMed] [Google Scholar]
- Räz B., Iten M., Grether-Bühler Y., Kaminsky R., Brun R. The Alamar Blue assay to determine drug sensitivity of African trypanosomes in vitro. Acta Trop. 1997;68:139–147. doi: 10.1016/s0001-706x(97)00079-x. [DOI] [PubMed] [Google Scholar]
- Rodenko B., van der Burg A.M., Wanner M.J., Kaiser M., Brun R., Gould M., de Koning H.P., Koomen G.J. 2, N6-disubstituted adenosine analogs with antitrypanosomal and antimalarial activities. Antimicrob. Agents Chemother. 2007;51:3796–3802. doi: 10.1128/AAC.00425-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roditi I., Schwarz H., Pearson T.W., Beecroft R.P., Liu M.K., Richardson J.E., Biihring H., Pleiss J., Billow R., Williams R.O., Overath P. Procyclin gene expression and loss of the variant surface glycoprotein during differentiation of Trypanosoma brucei. J. Cell Biol. 1989;108:737–746. doi: 10.1083/jcb.108.2.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro T.A., Were J.B., Danso K., Nelson D.J., Desjardins R.E., Pamplin C.L., 3rd Pharmacokinetics and metabolism of allopurinol riboside. Clin. Pharmacol. Ther. 1991;49:506–514. doi: 10.1038/clpt.1991.61. [DOI] [PubMed] [Google Scholar]
- Shih S., Hwang H.Y., Carter D., Stenberg P., Ullman B. Localization and targeting of the Leishmania donovani hypoxanthine-guanine phosphoribosyltransferase to the glycosome. J. Biol. Chem. 1998;273:1534–1541. doi: 10.1074/jbc.273.3.1534. [DOI] [PubMed] [Google Scholar]
- Shih S., Stenberg P., Ullman B. Immunolocalization of Trypanosoma brucei hypoxanthine-guanine phosphoribosyltransferase to the glycosome. Mol. Biochem. Parasitol. 1998;92:367–371. doi: 10.1016/s0166-6851(98)00007-3. [DOI] [PubMed] [Google Scholar]
- Spector T., Jones T.E., LaFon S.W., Nelson D.J., Berens R.L., Marr J.J. Monophosphates of formycin B and allopurinol riboside. Interactions with leishmanial and mammalian succino-AMP synthetase and GMP reductase. Biochem. Pharmacol. 1984;33:1611–1617. doi: 10.1016/0006-2952(84)90282-x. [DOI] [PubMed] [Google Scholar]
- Taliaferro W.H., D’Alesandro P.A. Trypanosoma lewisi infection in the rat: effect of adenine. Proc. Natl. Acad. Sci. USA. 1971;68:1–5. doi: 10.1073/pnas.68.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertommen D., Van Roy J., Szikora J.P., Rider M.H., Michels P.A., Opperdoes F.R. Differential expression of glycosomal and mitochondrial proteins in the two major life-cycle stages of Trypanosoma brucei. Mol. Biochem. Parasitol. 2008;158:189–201. doi: 10.1016/j.molbiopara.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Vodnala M., Fijolek A., Rofougaran R., Mosimann M., Mäser P., Hofer A. Adenosine kinase mediates high affinity adenosine salvage in Trypanosoma brucei. J. Biol. Chem. 2008;283:5380–5388. doi: 10.1074/jbc.M705603200. [DOI] [PubMed] [Google Scholar]
- Wallace L.J.M., Candlish D., de Koning H.P. Different substrate recognition motifs of human and trypanose nucleobase transporters: selective uptake of purine antimetabolites. J. Biol. Chem. 2002;277:26149–26156. doi: 10.1074/jbc.M202835200. [DOI] [PubMed] [Google Scholar]
- Wallace L.J., Candlish D., Hagos A., Seley K.L., de Koning H.P. Selective transport of a new class of purine antimetabolites by the protozoan parasite Trypanosoma brucei. Nucleosides, Nucleotides Nucleic Acids. 2004;23:1441–1444. doi: 10.1081/NCN-200027660. [DOI] [PubMed] [Google Scholar]
- Wenzler T., Steinhuber A., Wittlin S., Scheurer C., Brun R., Trampuz A. Isothermal microcalorimetry, a new tool to monitor drug action against Trypanosoma brucei and Plasmodium falciparum. PLoS Negl. Trop. Dis. 2012;6:e1668. doi: 10.1371/journal.pntd.0001668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarella-Boitz J.M., Rager N., Jardim A., Ullman B. Subcellular localization of adenine and xanthine phosphoribosyltransferases in Leishmania donovani. Mol. Biochem. Parasitol. 2004;134:43–51. doi: 10.1016/j.molbiopara.2003.08.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phylogenetic tree of all the hits in the predicted proteomes of Trypanosoma brucei (red), Trypanosoma cruzi (blue), Leishmania major (green) and Homo sapiens (black) with an E-value below 10−10 against Pfam profile PF00156 (‘Phosphoribosyl transferase domain’) as calulated by HMMer. Predicted glyocosomal location is indicated with one (PTS1), respectively two asterisks (PTS2). Bootstrap values are in percent positives of 1000 rounds. The scale bar indicates number of amino acid substitutions per site. APRT, adenine phosphoribosyltransferases (EC 2.4.2.7); OPRT, orotate phosphoribosyltransferases (EC 2.4.2.10); PRS, phosphoribosyl pyrophosphate synthetases (ribose-phosphate pyrophosphokinases, EC 2.7.6.1); HGXPRT, hypoxanthine/guanine/xanthine phosphoribosyltransferases (EC 2.4.2.8, 2.4.2.22).