

SUMMARY
An RNA enzyme has been developed that catalyzes the joining of oligonucleotide substrates to form additional copies of itself, undergoing self-replication with exponential growth. The enzyme also can cross-replicate with a partner enzyme, resulting in their mutual exponential growth and enabling self-sustained Darwinian evolution. The opportunity for inventive evolution within this synthetic genetic system depends on the diversity of the evolving population, which is limited by the catalytic efficiency of the enzyme. Directed evolution was used to improve the efficiency of the enzyme and increase its exponential growth rate to 0.14 min−1, corresponding to a doubling time of 5 min. This is close to the limit of 0.21 min−1 imposed by the rate of product release, but sufficient to enable more than 80 logs of growth per day.
INTRODUCTION
A key distinguishing feature of living systems is their capacity to undergo Darwinian evolution in response to natural selection. Darwinian evolution requires the propagation of genetic information, from parent to progeny, through processes of molecular self-replication. In contemporary biology the genetic material is copied by a complex protein machinery, but at the time of life’s origins the replicative process must have been more rudimentary. One attractive hypothesis is that the original genetic material was composed of RNA rather than DNA, and its replication was catalyzed by simple enzymes that themselves were composed of RNA (Woese 1967; Crick 1968; Orgel, 1968). In support of this hypothesis, RNA enzymes have been developed that catalyze the RNA-templated polymerization of RNA utilizing NTPs substrates (Ekland and Bartel, 1996; Johnston et al., 2001). These enzymes accurately copy certain RNA sequences up to 95 nucleotides in length (Zaher and Unrau, 2007; Wochner et al., 2011), and when made to operate at near-freezing temperatures for 7 days can generate products up to 206 nucleotides in length (Attwater et al., 2013). Thus far, however, these enzymes are not sufficiently robust to enable the replication of RNA, let alone replication of the RNA enzyme that catalyzes the polymerization reaction.
A somewhat different approach relies on RNA enzymes with RNA-templated RNA ligase activity to join oligonucleotide substrates to form complementary RNA products. It has been proposed that the first replicating, evolving systems on Earth operated by this mechanism and only later came to depend upon residue-by-residue polymerization (James and Ellington, 1999; Levy and Ellington, 2001). The set of substrates needed to support RNA replication through RNA-templated ligation might include the 16 possible dinucleotides, the 64 possible trinucleotides, or perhaps a subset of the much larger number of longer oligonucleotides. As a demonstration of this mode of replication, an RNA ligase enzyme has been developed that catalyzes the production of additional copies of itself through the joining of two component oligonucleotide substrates (Paul and Joyce, 2002). The parent and progeny enzymes dissociate in a non-rate-limiting manner, resulting in exponential amplification.
To enable the propagation of genetic information, the self-replicating RNA ligase has been converted to a cross-replication format whereby two RNA enzymes catalyze each other’s synthesis from a total of four component substrates (Kim and Joyce, 2004). Information is transmitted between the parent and progeny enzymes through two regions of Watson-Crick pairing, each of which may contain many possible sequences. Recombination can occur between these two regions, resulting in novel variants that compete for utilization of the oligonucleotide substrates. Those variants that have faster exponential growth rates enjoy a selective advantage, resulting in the self-sustained Darwinian evolution of the fittest replicators (Lincoln and Joyce, 2009).
The self- and cross-replicating RNA enzymes are the only known informational macromolecules that bring about their own exponential amplification. They can do so indefinitely, so long as an ongoing supply of substrates is made available. At a constant temperature of 44 °C the exponential growth rate of the replicating enzymes is 0.03 min−1, corresponding to a doubling time of 20 min (Ferretti and Joyce, 2013). Amplification can be made dependent on recognition of a target ligand by appending a ligand-binding domain (aptamer) to the catalytic domain of the enzyme (Lam and Joyce, 2009; Lam and Joyce, 2011). The rate of amplification then depends on the concentration of the ligand relative to the Kd of the ligand-binding domain. In this format, the replicating RNA enzymes have been used to measure the concentration of various drugs and metabolites. The entire replication system can be constructed from non-natural L-ribonucleotides (Olea et al., 2012). The L-RNA replicators behave the same as their D-RNA counterparts, but are not susceptible to degradation by biological ribonucleases.
The cross-replicating RNA enzymes have not yet demonstrated the capacity for inventive Darwinian evolution. Existing function can be optimized within the system, but the invention of novel function requires more genetic information than currently can be supported (Sczepanski and Joyce, 2012). The chief reason for this limitation is that the Km of the enzymes for their oligonucleotide substrates is in the range of 1–8 μM, but supplying complex mixtures of substrates at this concentration becomes problematic when there are thousands of variants. In addition, the substrates tend to form non-productive complexes that sequester these materials and reduce their effective concentration (Ferretti and Joyce, 2013). As a result, the enzymes do not operate close to saturation, which causes their observed rate of reaction to be substantially slower than their inherent catalytic rate.
A previous study demonstrated that if it were possible to increase the catalytic efficiency of the replicating enzymes without causing product dissociation to become rate limiting, the exponential growth rate could be increased by several-fold, resulting in a doubling time of less than 5 min (Ferretti and Joyce, 2013). Increased catalytic efficiency also would allow the enzymes to operate at lower substrate concentrations, thus enabling the utilization of more complex mixtures of substrates. The present study describes the directed evolution of replicating RNA enzymes that operate with an exponential growth rate of 0.14 min−1, corresponding to a doubling time of 5 min. Each parental enzyme can give rise to thousands of copies per hour, and each of these copies in turn can do the same, all the while transmitting molecular information across the generations.
RESULTS
Directed Evolution
A directed in vitro evolution strategy was used to optimize the catalytic efficiency of the replicating RNA enzymes. This strategy allowed exploration of a much larger population of variants than would have been possible through self-sustained evolution of the replicating enzymes. However, a directed evolution strategy required selecting for the ability to catalyze a simple ligation reaction, rather than replication itself. Thus three measures were taken to ensure that the fruits of directed evolution would be applicable to self-sustained evolution. First, the ligation reaction was carried out in two different formats, with either the 3′-hydroxyl-bearing substrate (substrate A) or the 5′-triphosphate-bearing substrate (substrate B) being provided as a separate biotinylated molecule (Figure 1). In either case, the product of the reaction was a single biotinylated molecule with the substrate joined to the enzyme via a newly-formed 3′,5′-phosphodiester linkage. The second measure was to provide recognition regions between enzyme and substrate that mimicked those required for replication, but using substrates that lacked nucleotides needed to form another catalytic center. The third measure was to limit randomization of the starting enzyme to the catalytic center, while maintaining fixed sequences throughout the regions involved in base pairing with the substrates. This was done to avoid selecting enzyme variants with altered modes of substrate binding that might slow the rate of product dissociation.
Figure 1.
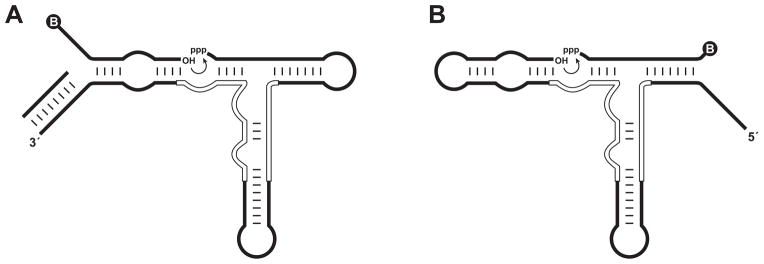
Formats for in vitro evolution of RNA ligation activity. (A) Substrate-A format, employing a separate 5′-biotinylated molecule bearing a 3′-hydroxyl. The primer for reverse transcription is pre-hybridized to the 3′ end of the enzyme to prevent interaction of this region with the substrate. (B) Substrate-B format, employing a separate 3′-biotinylated molecule bearing a 5′-triphosphate. Curved arrow indicates the site of ligation. Circled “B” indicates the biotin moiety. Open lines within the enzyme indicate nucleotides that were mutagenized at 21% degeneracy in the starting population.
A population of 1014 variants of the starting enzyme (E1) was constructed, introducing random mutations at a frequency of 21% per nucleotide position over 25 positions that encompassed the catalytic center (Figure 1). This population encompassed all possible variants containing up to 10 mutations relative to the wild type. Members of the population were challenged to perform ligation in either the substrate-A or substrate-B format, with interconversion between the two formats achieved by PCR amplification using appropriate primers (see Experimental Procedures). Ten rounds of directed evolution were carried out, selecting the reacted, biotinylated products by capturing them on a streptavidin-agarose resin. Following thorough washing of the resin, the captured materials were further selected by RT-PCR using primers specific for the covalently joined product. The stringency of selection was increased progressively by decreasing both the concentration of substrate and the reaction time over the course of the 10 rounds of evolution (Table 1).
Table 1.
Parameters for Successive Rounds of In Vitro Evolution
| Round | Formata | Enzyme (pmol)b | [Substrate] (nM) | Time (min) |
|---|---|---|---|---|
| 1 | A | 830 | 250 | 60 |
| 2 | A | 200 | 100 | 5 |
| 3 | A | 100 | 50 | 2.5 |
| 4 | B | 10 | 2 | 0.5 |
| 5 | B | 10 | 2 | 0.5 |
| 6 | A | 25 | 50 | 1 |
| 7 | A | 25 | 50 | 1 |
| 8 | A | 25 | 25 | 0.5 |
| 9 | A | 5 | 5 | 0.08 |
| 10 | A | 5 | 5 | 0.08 |
Reaction with either A or B as the separate substrate.
Concentration of enzyme was half that of substrate in rounds 1–8 and equal to that of substrate in rounds 9 and 10.
After each round, the ligation activity of the population was assayed in both the substrate-A and substrate-B formats, using 250, 100, or 50 nM substrate (Figure 2). These data were used to guide the choice of reaction conditions for the following round. The observed rate of reaction of the starting population was about 8-fold lower than that of E1 when measured in the presence of 250 nM substrate, and undetectable when measured in the presence of either 100 or 50 nM substrate. Activity improved about 10-fold per round over the first three rounds of evolution, then continued to improve at a more modest pace through the eighth round. After the eighth round, the observed rate of reaction for the evolved population was 90-fold faster in the presence of 50 nM substrate compared to that of E1 in the presence of 250 nM substrate. Two final rounds were carried out to cull the most reactive variants from the population, employing only 5 nM substrate and a reaction time of only 5 s.
Figure 2.

Assay of ligation activity for successive populations of enzymes over the course of in vitro evolution. Reactions were performed in the substrate-A format for the E1 enzyme (wt), starting population (0), and populations obtained after rounds 1–10, employing 250 (black), 100 (gray), or 50 (white) nM substrate, always in two-fold excess over enzyme. Reaction conditions: 25 mM MgCl2, pH 8.5, 42 °C.
Analysis of Individual Evolved Enzymes
Individuals were cloned from the population following rounds 6, 8, and 10, then sequenced. Alignment of 31 cloned sequences following the 10th round revealed that a majority conformed to a consensus, albeit with minor variation both within and outside the region that was originally mutagenized (Table S1). Only 4 of the 31 sequences did not conform to this consensus, although these 4 share strong sequence similarity among themselves. No clone contained fewer than 6 nor more than 10 mutations compared to E1. Nine of the 25 positions that were originally mutagenized remained invariant among all the clones, and one position (A38→G) was mutated in all cases. Mutations can arise outside the region that was originally mutagenized due to polymerase errors during amplification. These mutations occurred sporadically and appear to have little functional consequence.
Because the ultimate objective was to improve the replicative efficiency of the enzyme, individual clones isolated following rounds 6, 8, and 10 were screened for self-replication activity rather than simple ligation activity. The standard substrate concentrations for self- and cross-replication are 10 and 5 μM, respectively, but a more stringent condition employing only 1 μM substrates was used to assay the various clones (Figure S1). All but one clone exhibited self-replication activity exceeding that of E1, and activity was generally greater for clones isolated from the later rounds. The fastest clone tested (round 10, clone 1) had a 31-fold faster exponential growth rate compared to that of E1 under the conditions tested. Interestingly, this clone was one of the few that did not conform to the consensus sequence. It subsequently was determined that it and other clones that contain a deletion at position A48 perform poorly in the cross-replication format. Reverting this deletion restored cross-replication activity, but not to the level seen with the most active clones.
Clone 37 from round 10 performed especially well in both the self- and cross-replication assays. Its sequence conforms closely to the consensus sequence of all clones isolated after round 10 (Table S1), and its self-replication activity is 23-fold greater than that of E1 when measured in the presence of 1 μM substrates at 42 °C. This clone, hereafter referred to as the “F1” enzyme, was chosen for more detailed study. It contains six mutations relative to E1, all within the catalytic center (Figure 3). Two other clones isolated after round 10 had the exact same sequence as F1 and four other clones differed by only one nucleotide.
Figure 3.

Sequence and secondary structure of the evolved enzyme, bound to the substrates used in self-replication. Mutations in F1 relative to the E1 enzyme are highlighted by black circles. Nucleotide positions 10, 20, 30, 40, and 50 are numbered. Boxed regions indicate nucleotides that differ in F1′, the partner for F1 in cross-replication.
The catalytic rate of the F1 enzyme is too fast to measure using manual pipetting methods, necessitating the use of a rapid-quench device to provide reaction times as short as 80 msec (see Experimental Procedures). Ligation reactions were carried out in the substrate-A format (Figure 1A), employing trace concentrations of radiolabeled substrate A, saturating concentrations of substrate B, and varying concentrations of enzyme that spanned the Km. The data fit well to the Michaelis-Menten equation, with a kcat of 16.6 ± 0.4 min−1 and Km of 7.1 ± 0.5 μM (Figure S4). This compares with values of 1.7 ± 0.1 min−1 and 7.4 ± 1.1 μM, respectively, for the E1 enzyme operating in the same reaction format (Ferretti and Joyce, 2013). Thus the 10-fold improvement in catalytic efficiency of F1 compared to E1 is almost entirely attributable to an improvement in kcat. Similar behavior was observed for the cross-catalytic partner of the F1 enzyme (Figure S4).
The exponential growth rate of both the E1 and F1 enzymes varies as a function of temperature. Temperature has a complex influence on the overall rate of replication, affecting the stability and catalytic rate of the productive enzyme-substrate complex, the rate of product dissociation, and the stability of competing non-productive substrate complexes (Ferretti and Joyce, 2013). The temperature optimum for E1 is 42–46 °C, whereas the optimum for F1 is 46–50 °C (Figure S2). At its temperature optimum, F1 has an exponential growth rate of 0.14 min−1, measured in the presence of 10 μM substrates at 48 °C (Figure 4A). This corresponds to a doubling time of only 5 min, whereas E1 has a doubling time of 20 min at its optimal temperature of 44 °C. The exponential growth rate of both enzymes decreases with decreasing substrate concentration (Figure S3). In the presence of 2 μM substrates and at their respective temperature optima, the F1 and E1 enzymes have doubling times of 36 and 99 min, respectively (Figure 4B).
Figure 4.

Self-replication with exponential growth, comparing the starting and evolved enzymes. Reactions were performed using either E1 at 44 °C (gray) or F1 at 48 °C (black), in the presence of either (A) 10 μM or (B) 2 μM substrates. Inset in (A) shows the behavior of F1 over the first 10 min of the reaction. The data were fit to the logistic growth equation, which gave an exponential growth rate of 0.035 and 0.14 min−1 for E1 and F1, respectively, in the presence of 10 μM substrates, and 0.019 and 0.0070 min−1 for E1 and F1, respectively, in the presence of 2 μM substrates. Reaction conditions: 25 mM MgCl2, pH 8.5.
Self-replication with Exponential Growth
The exponential growth behavior of the self-replicating F1 enzyme is seen by the sigmoidal shape of the reaction profile (Figure 4A). This profile fits well to the logistic growth equation:
where [E]t is the concentration of enzyme at time t, a is the maximum extent of growth, b is the degree of sigmoidicity, and c is the exponential growth rate.
Another measure of exponential growth is obtained by determining the initial velocity of the reaction as a function of the starting concentration of enzyme, fitting the data to the equation:
where the initial velocity, (d[E]/dt)0, is proportional to the starting enzyme concentration, [E]0, raised to the reaction order p. A plot of (d[E]/dt)0 vs. [E]0 p has a slope equal to the autocatalytic rate constant, kauto, and a y-intercept equal to the rate of reaction in the absence of enzyme, kspont. Exponential growth occurs when the reaction order is 1.0.
Self-replication of F1 was carried out in the presence of either 5 or 10 μM substrates at 48 °C, initiating the reaction with various starting concentrations of enzyme over the range 0–2 μM. The initial velocity was determined over the first 10% of the reaction by fitting the data to a simple exponential equation and calculating the value for (d[E]/dt)0. The calculated values for initial velocity then were plotted as a function of [E]0 (Figure 5). A nonlinear least-squares fit of the data gave values for p of 0.92 and 0.91 for experiments employing 5 μM and 10 μM substrates, respectively. The quality of the fits were not statistically different from those obtained when the reaction order was fixed as 1.0 (r = 0.996 and 0.991, respectively). For the reaction with 10 μM substrates, the calculated autocatalytic rate constant is 0.083 min−1, which is slightly lower than that measured by fitting the logistic growth equation to the entire amplification profile (Figure 4A). The efficiency of replication of the F1 enzyme, kauto/kspont, is 1 × 107 M−1, which is the same as that measured previously for E1 (Ferretti and Joyce, 2013). Thus, the enhanced catalytic efficiency of F1 compared to E1 in the simple ligation reaction is captured as a corresponding increase in both the autocatalytic and spontaneous rates in the replication reaction.
Figure 5.

Exponential growth deduced from the initial rate of reaction as a function of the starting concentration of enzyme. Reactions were carried out using either 5 (gray) or 10 (black) μM substrates. Initial velocity was measured over the first 10% of the reaction, and the data were fit to an equation with reaction order 1.0. Reaction conditions: 25 mM MgCl2, pH 8.5, 48 °C.
Cross-replication and Sustained Exponential Growth
Cross-replication employs two different RNA enzymes that catalyze each other’s synthesis and enable the transfer of genetic information from parent to progeny molecules (Kim and Joyce, 2004; Lincoln and Joyce, 2009). The catalytic center is typically (but not necessarily) the same for the two enzymes, with the symmetry broken in the central and flanking base-paired regions (Figure 3). Optionally, the distal portion of the central stem-loop can be altered to provide a size difference between the two enzymes or to incorporate an aptamer or some other functional domain.
The F1 enzyme was converted to a pair of cross-replicating enzymes (F1 and F1′), which were supplied with 5 μM each of the four substrates needed to support exponential growth. The cross-replicating enzymes have a slightly lower temperature optimum compared to the self-replicating enzyme (Figure S2). At their temperature optimum of 47 °C, the exponential growth rate of F1 and F1′ is 0.11 min−1, corresponding to a doubling time of 6 min (Figure 6A). This is slightly slower than the rate of self-replication of F1 at 48 °C, and substantially faster than the rate of cross-replication of the wild-type enzymes (E1 and E1′) at 44 °C.
Figure 6.

Cross-replication and sustained exponential growth. (A) One round of cross-replication, beginning with 0.02 μM each of F1 (black) and F1′ (gray). The data were fit to the logistic growth equation, which gave an exponential growth rate of 0.11 min−1 for both enzymes. Dashed vertical line indicates the yield at 45 min, which was the time of serial transfer. (B) Fifty successive rounds of cross- replication, with transfer of 1% of reacted materials (100-fold dilution) after each round. The yield of newly-synthesized F1 and F1′ was measured after each round and the compounded yield was plotted as a function of time, giving a growth rate of 2.67 logs/h−1. Reaction conditions: 5 μM each substrate, 25 mM MgCl2, pH 8.5, 47 °C.
Cross-replication employing 5 μM substrates reaches a maximum extent of about 4 μM (75%). Similar behavior is seen with the E1 and E1′ enzymes, which has been attributed to sequence heterogeneity at the 5′ end of the 5′-triphosphorylated substrates (Olea et al., 2012). When the substrates were prepared synthetically, a maximum extent of >90% was achieved. In addition, there is some degradation of the RNA during the course of the reaction, occurring at a constant rate of ~0.1% min−1 for both the enzymes and substrates.
One way to overcome the limitation of maximum extent is to provide what is effectively an infinite supply of substrates. This can be achieved either in a continuous flow reactor or through a serial transfer procedure. The latter involves periodically transferring a portion of a completed reaction mixture to a new reaction mixture that contains a fresh supply of substrates. No enzymes are added to the new reaction other than those that are carried over in the transfer. A serial transfer procedure was used previously to carry out the self-sustained evolution of a population of cross-replicating RNA enzymes based on the E1 motif (Lincoln and Joyce, 2009; Sczepanski and Joyce, 2012). The F1 enzyme, with its faster doubling time, allows such experiments to be carried out at a much faster tempo.
As a demonstration of the remarkable ability of the optimized cross-replicating enzymes to withstand serial dilution, successive reaction mixtures were prepared, each containing 5 μM substrates. A starting concentration of 0.02 μM each of F1 and F1′ was added to the first mixture and allowed to undergo exponential amplification for 45 min, resulting in 2–3 μM products. Then a 100-fold dilution was carried out, transferring 0.02–0.03 μM of both enzymes to the second reaction mixture. This process was repeated for 50 successive transfers as the replicating molecules withstood an overall dilution of 10100-fold (Figure 6B). Amplification was highly consistent over the total reaction period of 37.5 h, occurring at a pace of 464-fold h−1.
DISCUSSION
The replicating RNA enzyme is the only known molecule that can undergo self-sustained Darwinian evolution, but it has limited genetic complexity, and therefore limited capacity for the invention of novel function. Recent kinetic studies have pointed out the key shortcomings of the original form of this enzyme (Ferretti and Joyce, 2013), which motivated the present study to develop an improved version that could replicate faster and/or in the presence of lower concentrations of substrates. It at first seemed surprising that the original enzyme (E1) has a catalytic rate for ligation of 2–5 min−1, but an observed rate in the complete replication system of only 0.03 min−1, the latter coinciding with the exponential growth rate. It was determined that the 100-fold slower rate of replication compared to ligation is not due to limiting product release, which occurs at a rate of 0.21 min−1, but rather to the formation of non-productive substrate complexes that reduce the effective substrate concentrations to well below the Km of the corresponding enzyme-substrate interactions (Ferretti and Joyce, 2013). In self-replication, non-productive complexes form between A and B, whereas in cross-replication they form between both A and B′ and A′ and B, due to the inherent complementarity (or cross-complementarity) of the system.
If the catalytic efficiency of the original replicating enzyme could be improved, by either increasing kcat or decreasing Km, that would be expected to increase the rate of exponential growth. However, it likely would not be possible to achieve an exponential growth rate of >0.21 min−1, which would exceed the rate of product release. Thus the aim of the present study was to develop an optimized form of the enzyme with an exponential growth rate approaching 0.2 min−1. Such an improvement would be reflected exponentially, and therefore would have a dramatic effect on the tempo of self-sustained evolution.
Directed in vitro evolution was carried out, applying strong selection pressure to drive the improvement of ligation efficiency, which was parlayed to an improvement in the rate of exponential amplification. The resulting optimized enzyme (F1) undergoes self-replication with an exponential growth rate of 0.14 min−1 (employing 10 μM substrates at 48 °C; Figure 4A), and undergoes cross-replication with an exponential growth rate of 0.11 min−1 (employing 5 μM substrates at 47 °C; Figure 6A). There is the potential for slight further improvement of these rates, but substantial improvement could not be achieved without also increasing the rate of product release.
For both the E1 and F1 enzymes, the exponential growth rate increases linearly with increasing substrate concentrations (Figure S3). Because of its greater catalytic efficiency, F1 has an exponential growth rate in the presence of 1 μM substrates that is comparable to that of E1 in the presence of 10 μM substrates. This makes it possible for F1 to operate at lower substrate concentrations, which is beneficial for carrying out self-sustained evolution using complex mixtures of substrates. Prior self-sustained evolution experiments with the E1 enzyme employed either 12 variants of each substrate at a total concentration of 60 μM (Lincoln and Joyce, 2009), or 64 variants of each substrate at a total concentration of 50 μM (Sczepanski and Joyce, 2012). The number of potential enzyme variants in these experiments was either 144 (12 × 12) or 4,096 (64 × 64), respectively, reflecting the combinatorial complexity of the ligated products. It was not possible to use 256 variants of each substrate at a total concentration of 50 μM because that gave poor saturation of the enzyme-substrate complexes, nor was it possible to increase the total concentration of substrates above 100 μM because that increased the formation of non-productive substrate complexes, including complexes of partially mismatched substrates.
Two innovations were needed to increase the complexity of the system of self-evolving RNA enzymes. The first was to devise a means for implementing genetic codes that allow greater information content and are less susceptible to the formation of mismatched complexes. This was achieved by a split-and-pool synthetic method for preparing the substrates, enabling the implementation of codes of arbitrary design, including those that enforce more distinct genotypes. (Sczepanski and Joyce, 2012). The second innovation, reported here, was to improve the enzyme itself so that it can operate at lower concentrations of substrates, or with a similar total concentration of substrates shared among a larger number of variants.
The self-replicating RNA enzyme has applications based on its ability to undergo ligand-dependent exponential amplification at constant temperature (Lam and Joyce, 2009). For these applications the rate of exponential amplification is important. Measurement of ligand concentration is carried out in a manner similar to quantitative PCR, but without thermal cycling. The time required to reach a defined threshold of amplified products is determined and then related to a standard plot of time-to-threshold versus ligand concentration (Lam and Joyce, 2011). With the E1 enzyme, the typical time-to-threshold for sensitive detection is a few hours, whereas for the F1 enzyme it is less than one hour.
The improved replicator has an exponential growth rate approaching that needed to generate traveling waves of replicating RNAs that could be observed directly. The wave front velocity is given by 2(κD)½, where κ is the exponential amplification rate and D is the diffusion constant (Bauer et al., 1989). Assuming the diffusion constant of the RNA enzyme is similar to that of tRNA (Rhee et al., 1981), the calculated wave front velocity would be ~0.2 cm/h. For those with less patience, RNA replication in homogeneous solution is recommended.
SIGNIFICANCE
The fundamental measure of fitness of a replicating, evolving system is its net rate of production, measured as an exponential growth rate. A synthetic genetic system, based on populations of replicating RNA enzymes, was developed previously and shown to have limited capacity to undergo self-sustained Darwinian evolution. In order to increase that capacity, an improved version of the enzyme has now been developed using directed evolution. Strong selection pressure was applied to drive improvement of the catalytic efficiency of the enzyme, which resulted in an increased exponential growth rate. The improved catalytic efficiency also makes it possible for the enzyme to operate at lower substrate concentrations, enabling more complex populations to be maintained. These results, coupled with the recent development of a method for implementing user-specified genetic codes of arbitrary design, provides a synthetic genetic system with greater capacity for inventive evolution. The replicating enzyme also has been used to measure the concentration of target ligands by appending an aptamer domain to the catalytic domain of the enzyme, configured so that replication depends upon ligand binding to the aptamer domain. Those measurements are sensitive, quantitative, and made at a constant temperature, but previously had required several hours to perform. With the improved replicator, exponential amplification is accelerated and the time required to reach the threshold for detection is reduced to less than one hour. For both molecular evolution and molecular diagnostics, speed is of the essence.
EXPERIMENTAL PROCEDURES
Preparation of Starting Population of Enzymes
A library of DNA templates was prepared using standard phosphoramidite chemistry on an Applied Biosystems Expedite 8909 RNA/DNA synthesizer (see Table S2 for all sequences). Reagents for the 25 mutagenized positions were drawn from four vials, each containing the wild-type and other three nucleotides in amounts needed to achieve coupling ratios of 79:7:7:7, respectively (normalized for differences in phosphoramidite coupling efficiency). Following standard deprotection and purification by denaturing polyacrylamide gel electrophoresis (PAGE), 1 nmol of the DNA library was mixed with 2 nmol of the primer for second-strand synthesis, which were heated at 70 °C for 3 min, then cooled to room temperature. The primer was extended using 10 U/μL MMLV reverse transcriptase (Life Technologies, Carlsbad, CA) in a mixture containing 0.5 mM each dNTP, 3 mM MgCl2, 75 mM KCl, 10 mM DTT, and 50 mM Tris (pH 8.3), which was incubated at 42 °C for 1 h. A portion of the extended products (166 pmol = 1014 molecules) were transcribed in a mixture containing 15 U/μL T7 RNA polymerase, 0.001 U/μL inorganic pyrophosphatase (Sigma-Aldrich, St. Louis, MO), 5 mM each NTP, 25 mM MgCl2, 2 mM spermidine, 10 mM DTT, and 40 mM Tris (pH 7.9), which was incubated at 37 °C for 2 h. Then 0.5 U/μL DNase I (Roche Applied Science, Indianapolis, IN) was added and the mixture was incubated at 37 °C for an additional 1 h. The transcribed RNA was purified by PAGE and again treated with DNase, using 0.03 U/μL TURBO DNase (Life Technologies) in a mixture containing 10 mM MgCl2, 0.5 mM CaCl2, and 20 mM Tris (pH 7.5), which was incubated at 37 °C for 1 h. Following phenol/chloroform extraction and ethanol precipitation, the resulting RNA was used to initiate in vitro evolution in the substrate-A format.
RNA for the substrate-B format reactions was transcribed with 14 additional nucleotides at the 3′ end, which were removed using E. coli M1 RNA and an external guide RNA to generate a homogeneous terminus bearing a 2′- and 3′-hydroxyl (Forster and Altman, 1990). The cleavage reaction employed 20 μM RNA transcript and 20 μM guide RNA (Table S2), which were annealed by heating at 70 °C for 3 min and cooling to room temperature, then 2.5 μM M1 RNA was added, the mixture was incubated at 37 °C for 5 min, and cleavage was carried out in a mixture containing 100 mM MgCl2, 100 mM NH4Cl, and 50 mM Tris (pH 7.5), which was incubated at 37 °C for 2.5 h. The cleaved products were purified by PAGE.
Preparation of Substrates
The 3′-hydroxyl-bearing substrate (used in substrate-A format reactions) was prepared as a chimeric synthetic oligonucleotide containing a 5′-terminal biotin, 14 deoxynucleotides, and 11 ribonucleotides (Table S2). The 5′-triphosphate-bearing substrate (used in substrate-B format reactions) was prepared by transcription of a DNA template that had been assembled from two overlapping synthetic oligodeoxynucleotides (Table S2), which were cross-extended using reverse transcriptase, then transcribed as described above. The transcribed RNA contained an additional stem-loop at the 3′ end, mimicking the acceptor stem of precursor tRNA, which was removed using M1 RNA (without an external guide RNA), as described above. The cleaved products were purified by PAGE, then extended by a single 3′-azidoadenosine residue in a mixture containing 10 μM RNA, 60 U/μL yeast poly(A) polymerase (Affymetrix, Santa Clara, CA), 0.5 mM 2′,3′-dideoxy,3′-azidoadenosine 5′-triphosphate (Trilink, San Diego, CA), 100 μg/ml BSA, 0.6 mM MnCl2, 50 mM KCl, 20 μM EDTA, 1 mM spermidine, 0.2 mM DTT, 20 mM Tris (pH 7.0), and 10% glycerol, which was incubated at 37 °C for 8 h. The reaction products were thoroughly desalted using a 3-kDa-cutoff Amicon centrifugal filter (EMD Millipore, Billerica, MA), then coupled to biotin alkyne in a copper-catalyzed click reaction, employing 25 μM 3′-azido-RNA, 140 μM PEG4 carboxamide-propargyl biotin (Invitrogen), 0.4 μM CuSO4, 2 μM tris(3-hydroxypropyltriazolylmethyl)amine (a generous gift of M.G. Finn), 10 mM sodium ascorbate, 50 mM Na2HPO4 (pH 7.4), and 10% dimethyl sulfoxide, which were incubated under argon at 37 °C for 40 min. The resulting materials were purified by PAGE.
In Vitro Evolution
The RNA library (830 pmol in the first round, corresponding to an average of five copies of each template sequence) was mixed with a two-fold excess of the primer for reverse transcription (Table S2), incubated at 70 °C for 3 min, then cooled to room temperature. The hybridized materials were mixed with 25 mM MgCl2 and 50 mM EPPS (pH 8.5), incubated at 42 °C for 5 min, and the reaction was initiated by adding the appropriate substrate and incubating at 42 °C for the designated amount of time (Table 1). The reaction was quenched with EDTA and the biotinylated products were captured on a streptavidin-agarose resin (Sigma-Aldrich). The resin was washed successively with a 10-mL solution of 100 mM NaCl, 1 mM EDTA, and 50 mM Tris (pH 7.5); a 20-mL solution of 1 M NaCl, 1 mM EDTA, 0.1% Triton X-100, and 50 mM Tris (pH 7.5); a 10-mL solution of 100 mM NaCl, 1 mM EDTA, and 50 mM Tris (pH 7.5); and a 20-mL solution of 1 mM EDTA and 10 mM Tris (pH 7.5). The retained RNA was reverse transcribed on the resin using 40 U/μL MMLV and 5 μM primer (Table S2), as described above. The DNA, still within the resin slurry, was PCR amplified using appropriate primers for either the substrate-A or substrate-B format reaction (Table S2), then transcribed and purified as described above. Conversion from either the substrate-A or substrate-B format to the self-replication format was achieved by PCR using appropriate primers (Table S2).
Assay of Ligation Activity
The RNA enzyme was prepared by transcription in the presence of 1 μCi/μL [α-32P]ATP (Perkin-Elmer, Waltham, MA), as described above. Prior to use, the RNA enzyme was incubated at 70 °C for 3 min, then cooled to room temperature. For reactions in the substrate-A format, a two-fold excess of the reverse transcriptase primer was included. The materials were mixed with 25 mM MgCl2 and 50 mM EPPS (pH 8.5), incubated at 42 °C for 2 min, and the reaction was initiated by adding 50, 100, or 250 nM substrate (always in two-fold excess over enzyme) and incubating at 42 °C. Aliquots were taken from the mixture at various times and quenched by adding 71% formamide and 140 mM EDTA. The products were separated by PAGE and quantitated using a PharosFX molecular imager (Bio-Rad, Hercules, CA).
Formal determination of kcat and Km required use of a KinTek (Austin, TX) model RQF-3 rapid-quench device. Steady-state kinetic parameters for the F1 and F1′ enzymes were determined under enzyme-excess, single-turnover conditions, employing trace amounts (≤3 nM) of [5′-32P]-labeled A (or A′), saturating concentrations (100 μM) of B (or B′), and a range of enzyme concentrations (0.5–37.5 μM) that spanned the Km of the enzyme-substrate complex. Two mixtures, one containing F1 and A′ (or F1′ and A) and the other containing B′ (or B), both containing 25 mM MgCl2 and 50 mM EPPS (pH 8.5) at 42 °C, were combined to initiate the reaction. The reactions were sampled at various times over the first 12% of the reaction, then the products were purified by ethanol precipitation and analyzed by PAGE, as described above. Values for kobs were obtained by a linear fit of the fraction of labeled substrate converted to product as a function of time. Each fit included 4–6 data points and had a linear regression coefficient of >0.95. Values for kcat and Km were obtained from a plot of kobs versus [F1] (or [F1′]), fitting the data to the Michaelis-Menten equation.
Assay of Replication Activity
Unlabeled RNA enzyme was prepared as described above. Substrate A was radiolabeled by first dephosphorylating using Antarctic phosphatase (New England Biolabs, Ipswich, MA), then phosphorylating using T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP (Perkin-Elmer). Replication was performed using 1–10 μM each of substrates A and B, including a trace amount (<10 nM) of 5′-[32P]-labeled substrate A. Two mixtures, one containing substrate A and the other containing the enzyme and substrate B, and both containing 25 mM MgCl2 and 50 mM EPPS (pH 8.5), were incubated separately at 42–50 °C for 2 min, then combined to initiate the reaction. Aliquots were taken from the mixture at various times, quenched, and analyzed as described above.
Cross-replication was performed similarly, except those reactions also contained 5′-[32P]-labeled substrate A′ and unlabeled substrate B′ (Figure 3). For serial transfer experiments, cross-replication was performed in a 10-μL volume containing 5 μM each of substrates A, A′, B, and B′, 25 mM MgCl2 and 50 mM EPPS (pH 8.5), which was incubated at 47 °C for 45 min. The first reaction mixture was seeded with 0.02 μM each of F1 and F1′, and subsequent mixtures were seeded by carrying over 0.1 μL of material from the previous mixture. In each case, mixtures containing the A and A′ substrates and the B and B′ substrates were pre-incubated separately at 47 °C for 2 min, then combined to initiate the reaction. The products were analyzed by PAGE, as described above.
Supplementary Material
Research Highlights.
An optimized self-replicating RNA enzyme was obtained by directed evolution.
The enzyme undergoes sustained exponential growth with a doubling time of 5 minutes.
Amplification of 10100-fold was achieved over a period of 37.5 hours.
The optimized enzyme will enable more complex synthetic genetic systems.
Acknowledgments
This work was supported by grant NNX07AJ23G from NASA, grant R01GM065130 from the NIH, and by The Skaggs Institute for Chemical Biology at The Scripps Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Attwater J, Wochner A, Holliger P. In-ice evolution of RNA polymerase ribozyme activity. Nat Chem. 2013;5:1011–1018. doi: 10.1038/nchem.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer GJ, McCaskill JS, Otten H. Traveling waves of in vitro evolving RNA. Proc Natl Acad Sci USA. 1989;86:7937–7941. doi: 10.1073/pnas.86.20.7937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick FHC. The origin of the genetic code. J Mol Biol. 1968;38:367–379. doi: 10.1016/0022-2836(68)90392-6. [DOI] [PubMed] [Google Scholar]
- Ekland EH, Bartel DP. RNA-catalysed RNA polymerization using nucleoside triphosphates. Nature. 1996;382:373–376. doi: 10.1038/382373a0. [DOI] [PubMed] [Google Scholar]
- Ferretti AC, Joyce GF. Kinetic properties of an RNA enzyme that undergoes self-sustained exponential amplification. Biochemistry. 2013;52:1227–1235. doi: 10.1021/bi301646n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster AC, Altman S. External guide sequence for an RNA enzyme. Science. 1990;249:783–786. doi: 10.1126/science.1697102. [DOI] [PubMed] [Google Scholar]
- James KD, Ellington AD. The fidelity of template-directed oligonucleotide ligation and the inevitability of polymerase function. Orig Life Evol Biosph. 1999;29:375–390. doi: 10.1023/a:1006544611320. [DOI] [PubMed] [Google Scholar]
- Johnston WK, Unrau PJ, Lawrence MS, Glasner ME, Bartel DP. RNA-catalyzed RNA polymerization: accurate and general RNA-templated primer extension. Science. 2001;292:1319–1325. doi: 10.1126/science.1060786. [DOI] [PubMed] [Google Scholar]
- Kim DE, Joyce GF. Cross-catalytic replication of an RNA ligase ribozyme. Chem Biol. 2004;11:1505–1512. doi: 10.1016/j.chembiol.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Lam BJ, Joyce GF. Autocatalytic aptazymes enable ligand-dependent exponential amplification of RNA. Nat Biotechnol. 2009;27:288–292. doi: 10.1038/nbt.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam BJ, Joyce GF. An isothermal system that couples ligand-dependent catalysis to ligand-independent exponential amplification. J Am Chem Soc. 2011;133:3191–3197. doi: 10.1021/ja111136d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy M, Ellington AD. The descent of polymerization. Nat Struct Biol. 2001;8:580–582. doi: 10.1038/89601. [DOI] [PubMed] [Google Scholar]
- Lincoln TA, Joyce GF. Self-sustained replication of an RNA enzyme. Science. 2009;323:1229–1232. doi: 10.1126/science.1167856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olea C, Jr, Horning DP, Joyce GF. Ligand-dependent exponential amplification of a self-replicating L-RNA enzyme. J Am Chem Soc. 2012;134:8050–8053. doi: 10.1021/ja302197x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orgel LE. Evolution of the genetic apparatus. J Mol Biol. 1968;38:381–393. doi: 10.1016/0022-2836(68)90393-8. [DOI] [PubMed] [Google Scholar]
- Paul N, Joyce GF. A self-replicating ligase ribozyme. Proc Natl Acad Sci USA. 2002;99:12733–12740. doi: 10.1073/pnas.202471099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee KW, Potts RO, Wang CC, Fournier MJ, Ford NC., Jr Effects of magnesium and ionic strength on the diffusion and charge properties of several single tRNA species. Nucleic Acids Res. 1981;9:2411–2420. doi: 10.1093/nar/9.10.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J, Joyce GF. The effect of cytidine on the structure and function of an RNA ligase ribozyme. RNA. 2001;7:395–404. doi: 10.1017/s135583820100228x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sczepanski JT, Joyce GF. Synthetic evolving systems that implement a user-specified genetic code of arbitrary design. Chem Biol. 2012;19:1324–1332. doi: 10.1016/j.chembiol.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wochner A, Attwater J, Coulson A, Holliger P. Ribozyme-catalyzed transcription of an active ribozyme. Science. 2011;332:209–212. doi: 10.1126/science.1200752. [DOI] [PubMed] [Google Scholar]
- Woese C. The Genetic Code. New York: Harper & Row; 1967. pp. 179–195. [Google Scholar]
- Zaher HS, Unrau PJ. Selection of an improved RNA polymerase ribozyme with superior extension and fidelity. RNA. 2007;13:1017–1026. doi: 10.1261/rna.548807. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.