

Abstract
Susceptibility to aggregation is general to proteins because of the potential for intermolecular interactions between hydrophobic stretches in their amino acid sequences. Protein aggregation has been implicated in several catastrophic diseases, yet we still lack in-depth understanding about how proteins are channeled to this state. Using a predominantly β-sheet protein whose folding has been explored in detail: cellular retinoic acid-binding protein 1 (CRABP1), as a model, we have tackled the challenge of understanding the links between a protein’s natural tendency to fold, ‘breathe’, and function with its propensity to misfold and aggregate. We identified near-native dynamic species that lead to aggregation and found that inherent structural fluctuations in the native protein, resulting in opening of the ligand entry portal, expose hydrophobic residues on the most vulnerable aggregation-prone sequences in CRABP1. CRABP1 and related intracellullar lipid-binding proteins have not been reported to aggregate inside cells, and we speculate that the cellular concentration of their open, aggregation-prone conformations is sufficient for ligand binding but below the critical concentration for aggregation. Our finding provides an example of how nature fine-tunes a delicate balance between protein function, conformational variability, and aggregation vulnerability, and implies that with the evolutionary requirement for proteins to fold and function, aggregation becomes an unavoidable but controllable risk.
Keywords: functional dynamics, near-native intermediate, conformational landscape, ligand binding cavity, folding, misfolding, aggregation propensity, intracellular lipid-binding protein, nuclear magnetic resonance
Protein sequences are under multiple evolutionary pressures based on Darwinian competition for the fitness of their host organism. Proteins must be synthesized in the right amount and at the right time, fold successfully to their native states in physiologically appropriate times, avoid aggregation and its accompanying deleterious effects, and perform their functions. Inevitably, these pressures compete, creating tension and consequent inherent suboptimal individual properties. It has recently been recognized that the inherent properties of polypeptides cause all proteins to have intrinsic tendencies to aggregate. (1) What then protects them sufficiently to enable their host organism to thrive? It might seem that encoding an ever more stable native state could provide protection against aggregation. But in order to function proteins must sample multiple conformations and retain significant dynamics, (2, 3) and it is a conundrum how the ability to ‘breath’ and populate multiple functional states has been preserved without creating unacceptable aggregation propensity. (4) Surprisingly, only recently has the tension between function and aggregation begun to be explored. (5, 6) It is thus of great interest to investigate in detail the balance of tensions on a given protein sequence between folding, aggregation and function.
Extensive work over the last several years on mechanisms of folding and linkages to aggregation mechanisms has led to the view that the folding and aggregation energy landscapes overlap and share common precursors. (1, 7, 8) The states that are aggregation-prone are in some cases quite unfolded, (4, 9, 10) while in others, near-native states have emerged as the precursors to aggregation. (11, 12) Since aggregation is generally mediated by association of hydrophobic sequences in extended conformations, local fluctuations in the native state may give rise to near-native, aggregation-prone states, which in turn may lead to partial solvent exposure of hydrophobic stretches. (4, 13, 14) While mechanisms of aggregation have been studied in detail for a few relatively small globular proteins, (15–19) there is not yet a clear picture of the links between folding, function and aggregation.
The intracellular lipid-binding protein (iLBP) family exemplifies the folding-aggregation-function tension. The native structure of iLBPs is comprised of an up-down anti-parallel β-barrel made up of two orthogonal β-sheets with an open angle between (Figure 1A); most strands are linked by tight turns, except strands 7 and 8, which are linked by an omega loop, and strands 1 and 2, which are linked by two α-helices. (20, 21) The hydrophobic ligands of iLBPs bind inside the barrel and interact with helix 2, turn II (between strands 3 and 4), and turn IV (between strands 5 and 6) (Figure 1C, red). (21) We have extensively studied a representative iLBP, cellular retinoic acid-binding protein 1 (CRABP1), and are well positioned to relate its folding and aggregation landscapes. (22–25) While wild type (WT) CRABP1 is soluble when overexpressed in E. coli, either single residue substitutions or mild perturbations in solution conditions lead to the formation of amorphous aggregates containing β-lamellar structures. (22, 26) Previously, we found that two distinct regions of CRABP1–one linking strand 3, turn II, and strand 4, and the other linking strands 9 and 10 (Figure 1B)–are sequestered in the resulting aggregates, and that these regions correspond closely with predicted aggregation-prone sequences. (25) The fact that the aggregation-prone regions are strongly conserved in the iLBP family suggests a compromise between requirements for function and risk of aggregation. Excitingly, early folding events (docking of strands 1 and 10 during barrel closure) appear to provide protection for aggregation-prone regions in CRABP1. (25) Particularly, early barrel closure also prevents exposure of intrinsically labile strand 9. These findings suggest that the folding and aggregation energy landscapes of iLBPs may overlap and offer clues as to how a protective strategy may have been evolutionarily encoded in the folding mechanism of this protein.
Figure 1.

Structural features of folded CRABP1 and its amorphous aggregates. A. Overlay of the X-ray structures of holo-CRABP1 (PDB ID 1CBR, chain A, white) and apo-CRABP1 (PDB ID 1CBI, chain A, red and B, pink). The RA molecule is shown in green. B. Two aggregation cores of CRABP1 (strand 3-turn II-strand 4 and strand 9 and 10, shown in orange) mapped onto holo-CRABP1 (PDB ID 1CBR, chain A) (as reported by Budyak et al .(25)). C. Conserved ligand-binding residues (red spheres) in the iLBP family (as defined by Marcelino et al. (49) mapped on the X-ray structures of holo-CRABP1 (PDB ID 1CBR, chain A). The RA molecule is shown in green.
In the present study, we have delved more deeply into the nature of the CRABP1 conformational flexibility that is required for ligand binding and how it is linked to aggregation-prone sequences. Ligand entry and egress necessarily require conformational changes to allow access of the ligand to its binding site in the cavity, (20, 27, 28) and inspection shows that these conformational changes increase exposure of the predicted aggregation-prone regions (Figure 1A). Coping with the consequent increased risk of aggregation places tension on the sequence and conformational landscape of CRABP1. We find that mild perturbations (such as low urea concentrations or single-point mutations) affect near-native conformational ensemble of CRABP1 by enhancing the population of a near-native, aggregation-prone conformation, thus leading to increased aggregation propensity. Aggregates formed upon introduction of point mutations sequester the same aggregation-prone sequences whether formed in vitro or in vivo, arguing that the conformational landscapes for the native and near-native states are very similar in the cellular environment. Intriguingly, we demonstrate that under native conditions, apo-CRABP1 partially populates the same vulnerable conformation, where highly aggregation-prone sequences are exposed to solution. Thus, we conclude that the functional energy landscape of CRABP1 and other iLBPs includes this aggregation-prone state. Taken together, our findings suggest an important evolutionary constraint on the iLBP family: To avoid the risk of aggregation, iLBPs must either minimize the population of this aggregation-prone species or if significant populations of these species are required for function, limit the aggregation propensity of exposed, vulnerable sequences. Hence, the representative iLBP, CRABP1, serves as a paradigm for the complex and intimate connections between folding, functional dynamics, and aggregation of proteins.
MATERIALS AND METHODS
Expression and Purification of CRABP1
Expression and purification of a variant of murine CRABP1 constructs with an N-terminal (His)10-tag and a stabilizing R131Q mutation (referred to as WT*; in all cases the CRABP1 constructs in this study harbors this stabilizing mutation) were performed according to published methods. (25) Briefly, the CRABP1 WT* in pET16b expression vector was transformed into E. coli strain BL21(DE3) (Novagen) and grown in M9 minimal medium to OD600 of 0.8. To produce uniformly 15N-labeled samples for NMR analysis, media were supplemented by 1 g/L 15NH4Cl as the sole nitrogen source. Protein expression was induced using 0.4 mM IPTG, and cells were grown for an additional 4 hours at 30 °C. Cells were re-suspended in 50 mM sodium phosphate buffer (pH 8.0) containing 300 mM NaCl and disrupted using a Microfluidizer M-110L processor (Microfluidics). CRABP1 WT* protein was purified from the soluble fraction of the lysate using Ni-NTA affinity chromatography (Qiagen). Protein concentration was determined using a molar extinction coefficient of ε280=20,970 M−1cm−1. Fractions containing pure protein were pooled and dialyzed against 10 mM ammonium bicarbonate buffer and lyophilized.
Protein Partitioning Experiments
Single residue substitutions were introduced into CRABP1 WT* in a pET16b plasmid by site-directed mutagenesis using a QuikChange protocol (Stratagene). The E. coli BL21(DE3) (Novagen) strain transformed with plasmids containing sequences for CRABP1 WT* variants was grown in Luria Bertani medium to an OD600 of 0.8. Protein expression was induced with 0.4 mM IPTG for 3 hours at 37 °C. Cells were lysed using BPER II bacterial protein extraction reagent (Thermo Scientific). Lysates were spun down at 20,000 × g for 5 minutes to separate pellet and soluble fractions. The pellets were dissolved in equal volumes of 8 M urea. Soluble and insoluble fractions were run on 12% Tricine SDS-PAGE. Protein bands were stained with Coomassie blue. The partitioning of CRABP1 mutant proteins between soluble and insoluble fractions was determined by measuring the band intensities using a GelDoc system (BioRad).
Purification of Bacterial Inclusion Bodies
Uniformly 13C,15N-labeled inclusion body samples were obtained for I52A, F71A, and L118V CRABP1 WT* variants by carrying out protein expression in BL21(DE3) E. coli strain in M9 minimum medium supplemented by 1 g/L 15NH4Cl and 2 g/L 13C-glucose as the only nitrogen and carbon sources. Cells were grown to an OD600 of 0.8, and protein expression was induced with 0.4 mM IPTG for 5–6 hours at 37 °C. Inclusion bodies of aggregation-prone mutant proteins were purified using BPER II reagent (Thermo Scientific) following the manufacturer’s instructions with minor modifications. Briefly, purification of inclusion bodies was performed on 15 mL cell culture aliquots for efficient separation of contaminating cellular components. Cells containing inclusion bodies were collected by centrifugation and re-suspended in BPER II at 1:10 (BPER II: bacterial growth culture (OD>1.0) ratio). The pellet was collected and re-suspended in the same volume of BPER II and treated with lysozyme (0.4 mg/mL). The pellet was washed with twenty-fold diluted BPER II reagent and spun down at 20,000 × g for 5 minutes. Pellet wash steps were repeated twice. Final wash steps were performed using 10 mM Tris-HCl pH 8.0 to eliminate residual detergent. Purity of the inclusion bodies was checked by re-suspending samples in 8 M urea and analyzing them on a 12% tricine-SDS PAGE. Protein concentrations in the inclusion bodies were estimated using band intensities of known concentrations of purified CRABP1 WT* samples.
Hydrogen Exchange NMR of Aggregates
The aggregation core residues of inclusion bodies for CRABP1 WT* variants were identified using the DMSO-quenched hydrogen-deuterium (H/D) exchange approach, monitoring deuterium incorporation by solution NMR spectroscopy as described previously. (25, 29) Briefly, 15N,13C-labeled inclusion bodies of F71A, L118V and I52A CRABP1 WT* variants were incubated in D2O containing 0.025% (w/v) NaN3 for four weeks at 4 °C. Aggregates were collected, lyophilized, and then re-suspended in d6-DMSO containing 0.1% (v/v) TFA, 50 mM DTT and 5% (v/v) D2O (pD 3.0–3.5) to a final protein concentration of at least 200 μM. The solution was immediately transferred to an NMR tube, and an amide heteronuclear single quantum coherence (1H-15N HSQC) spectrum was recorded at 26 °C on a 600-MHz Bruker Avance spectrometer equipped with a TXI cryoprobe. For unexchanged samples, inclusion bodies were re-suspended in water containing 0.025% (w/v) NaN3 and incubated at 4 °C for four weeks. NMR data were processed using NMRPipe. (30) To identify the highly protected residues of CRABP1 (i.e., those for which the amide peaks retained significant intensities after 4 weeks of exchange), previously reported backbone assignments of DMSO-dissolved CRABP1 WT*(25) were used.
In Vitro Aggregation Assays
Purified CRABP1 WT* aliquots with known concentrations were lyophilized and re-suspended in buffer (10 mM sodium phosphate, 5 mM DTT pH 7.0) containing different urea concentrations (0 to 3.0 M) to a total reaction volume of 100 μL and a total protein concentration of 200 μM. Aggregation reactions were incubated and agitated at 1000 rpm at 37 °C for 12 hours. The fraction of aggregated protein was determined as: fAgg=(Co−Cn)/Co, where Co is an initial concentration of soluble CRABP1 WT* (total protein concentration), and Cn is the protein concentration in the soluble fraction after 12 hours of incubation.
To determine the effect of retinoic acid (RA) on CRABP1 WT* aggregation, protein samples containing 200–300 μM of CRABP1 WT*, equimolar amounts of RA and 1.5 M urea were incubated at 37 °C for two hours. The amount of aggregation was determined by measuring the concentration of soluble CRABP1 before and after incubation. Multiple independent aggregation reactions (76 reactions for the apo protein and 69 for RA-bound CRABP1) were used for final analysis. Because of the stochastic nature of aggregation reactions, for analysis of data a histogram with equal bin widths showing a fraction of values lying in each bin in the presence and in the absence of RA was plotted as a function of aggregation propensity (a fraction of insoluble protein). In addition, data were partitioned into clusters and mean values and standard deviations were calculated for each cluster using the Euclidean distance function. The data analysis was performed using Wolfram Mathematica 8 (Wolfram Research, Inc.).
Urea-mediated Unfolding of CRABP1 WT* Monitored by Fluorescence
Unfolding of protein samples (about 5 μM protein concentration) in 10 mM Tris-HCl pH 8.0 containing 1 mM dithiothreitol (DTT) and varying urea concentrations, and equilibrated overnight (16–18 h) at 25 °C were monitored by Trp fluorescence (excitation at 280 nm, emission at 350 nm). The unfolding transition data were analyzed by a two-state model using the linear extrapolation method. (31)
Urea-mediated Conformational Changes and Denaturation of CRABP1 WT* Monitored by NMR
Lyophilized 15N-labeled CRABP1 WT* was dissolved in NMR buffer (20 mM Tris, 5 mM DTT, 5% (v/v) D2O, pH=8.0) to a final protein concentration of about 500 μM. The urea concentration was increased in ~ 0.5 M steps in the range of from 0.5 to 8 M. For each urea concentration, a 2D 1H-15N HSQC spectrum of CRABP1 was recorded at 26 °C using a 600-MHz Bruker Avance spectrometer equipped with a TXI cryoprobe. Data were processed using NMRPipe. (30) The backbone chemical shift assignments for near-native CRABP1 WT* conformations were transferred from previous assignments (BMRB accession number 19271) and verified using triple-resonance HNCO and HNCACB experiments recorded for NMR samples containing 0 and 2.5 M urea.
Fluorescein-labeling of CRABP1 WT*
The solvent accessibility of cysteine residues in CRABP1 WT* and its variants (F71M, L118V, C129A and Cys95, the C81A/C129A mutant containing only a single cysteine) under native condition was assessed using fluorescein maleimide labeling. The N-(5-fluoresceinyl)maleimide (Sigma, 10 mM stock prepared in dimethyl formamide) was added in about five- or 15-fold molar excess to protein in 20 mM Tris, pH 8.0, and reaction was carried out at 37 °C for 2 minutes. The reaction was quenched using SDS-gel buffer containing a large excess of β-mercaptoethanol, and the samples were analyzed using 10% tricine-SDS PAGE. The fluorescein fluorescence was monitored on a UV-transilluminator (AlphaImager gel documentation system, Protein Simple), and band quantitation was carried out using ImageJ software. (32) A comparison of extent of fluorescein fluorescence associated with the protein was made by considering fluorescence normalized with respect to the amount of protein, as estimated from the band intensity of Coomassie brilliant blue staining of gels.
In the case of fluorescein labeling of holo-CRABP1, 15-fold molar excess of RA (10 mM stock in ethanol) was added to the protein in 20 mM Tris, pH 8.0. The complex formation was carried out at 37 °C for 10 minutes before proceeding with fluorescein labeling as described above.
RESULTS
CRABP1 Aggregation Does Not Require Significant Protein Destabilization
Interestingly, CRABP1 has not been reported to aggregate in mammalian cells under physiological conditions. Moreover, in our hands, a stabilized variant of CRABP1 containing R131Q mutation (referred to as WT*) remains soluble at millimolar concentrations in vitro and upon over-expression in E. coli. Nonetheless, we find that any of several point mutations introduced into CRABP1 WT* increases its aggregation propensity (Table S1). Intriguingly, in vitro, even very minor perturbations, e.g., the presence of low concentration of denaturant (2 M urea, well below the denaturation midpoint) causes tetra-Cys CRAPB1 WT*, a tetra-Cys harboring omega loop variant of CRABP1 (which was designed for in-cell stability measurements), to form amorphous β-lamellar aggregates. (22, 26)
To delve more deeply into the correlation between stability and aggregation in CRABP1 variants, we measured the aggregation propensity of several previously characterized well-folded CRABP1 variants by determining the fraction of protein expressed in E. coli cells as inclusion bodies. This assay has been found to report aggregation propensities semi-quantitatively and to yield the same trends as those observed in vitro under mildly destabilizing conditions. (33) The results (Figure 2A, Table S1) show a rough correlation between the extent of destabilization of the native conformation, ΔΔG0, (as a result of mutations) and the aggregation propensity for the corresponding mutant, suggesting that increased population of the unfolded state, U, presages increased risk of aggregation. However, closer inspection reveals that a strikingly different change in global stability may result in similar aggregation propensities (e.g., red in Figure 2A). Moreover, more stable CRABP1 variants may result in the larger amount of the protein in the inclusion body fraction. For example, the introduction of an R79A mutation destabilizes CRABP1 WT* by 3.1 ± 0.2 kcal/mol, and leads to 30% of the protein in the inclusion body fraction, while a F50M mutation destabilizes the protein by 2.0 ± 0.2 kcal/mol but causes 50% of the protein to partition into inclusion bodies. These data suggest that the origin of aggregation propensity in CRABP1 is more complex than simple increased fraction of the U state.
Figure 2.
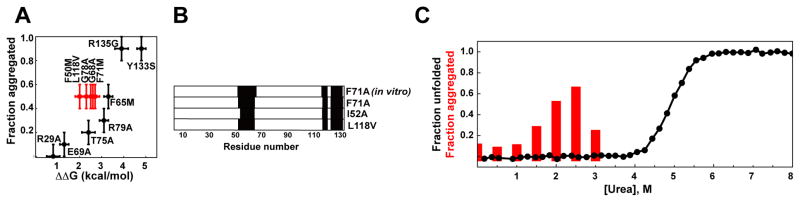
Origin of aggregation propensity in CRABP1 is more complex than simple increased fraction of the unfolded protein. A. Aggregation propensity (a fraction of insoluble protein) of different CRABP1 WT* mutants as a function of protein destabilization upon single point mutations (see also Table S1). B. Aggregation cores of different CRABP1 WT* aggregation-prone variants (F71A, I52A and L118V) obtained from E. coli inclusion bodies using DMSO-quenched H/D-exchange experiments and previously reported aggregation cores obtained in vitro using the F71A variant of CRABP1 WT*. (25) C. Aggregation propensity of CRABP1 WT* (red, monitored as a fraction of insoluble protein) and fraction of unfolded CRABP1 WT* (black, monitored by intrinsic tryptophan fluorescence spectroscopy) as a function of urea concentrations.
It is formally possible that different mutations are giving rise to aggregates with different structure, implying a different aggregation pathway and confounding these comparisons. To check this possibility, we tested whether CRABP1 aggregates formed in vitro and in vivo (as inclusion bodies) share the same structural features. We performed DMSO-quenched hydrogen-deuterium (H/D) exchange NMR experiments on inclusion bodies obtained from E. coli cells overexpressing (15N,13C)-labeled F71A CRABP1 WT*. Remarkably, the same regions (i.e., strand 3-turn II-strand 4 and strands 9 and 10) were highly protected after 4 weeks of exchange (Figure 1B), as reported previously for in vitro aggregates of the aggregation-prone variant of CRABP1 WT*, F71A, (25) providing strong evidence that in vivo aggregation arises from the same aggregation-prone sequences and arguing that the aggregation pathway in vivo and in vitro is the same. This important result validates the comparative use of either in vitro aggregation reactions or inclusion body formation to derive in vivo aggregation propensities.
To check that the aggregation processes triggered by different point mutations can justifiably be compared to one another, we performed similar H/D exchange NMR experiments on inclusion bodies obtained from E. coli cells overexpressing two another highly aggregation-prone mutants of CRABP1 WT*, I52A and L118V. Strikingly, in all cases, despite the very different sites of the mutation in the protein’s structure, identical sequences were found to constitute the protected cores of the aggregates (Figure 2B).
The finding that several point mutations and a variety of different conditions led to aggregates with the same core sequences, together with the fact that the same aggregation-prone sequences were predicted for CRABP1 WT* by several sequence-based aggregation prediction algorithms, (25) suggests that these aggregation-prone stretches are intrinsically encoded in the CRABP1 sequence. One might wonder why regions such as these are retained in a protein sequence, despite the exacerbated risk of aggregation from their presence. Two factors seem to be acting here to offset any selective pressure against retention of these aggregation-prone sequences: First, CRABP1, like the other members of the iLBP family, binds hydrophobic ligands that are sparingly soluble in water. Thus, ligand binding requires an array of hydrophobic side chains. Many of the ligand-binding residues occur in the aggregation-prone regions of CRABP1 (Figure 1C). Second, a number of residues in the aggregation-prone regions have been identified to be involved in a network of conserved hydrophobic-hydrophobic interactions, (34), (25) and thus the risk of aggregation is counterbalanced by requirements for stability and folding.
Aggregation Propensity of CRABP1 WT* is Increased by Low Concentrations of Denaturant, Well Below Global Melting
As noted above, the lack of correlation of aggregation propensity of CRABP1 mutants with destabilization of the native conformation argues that aggregation is not simply arising from increased population of U. We thus titrated CRABP1 WT* with denaturant and monitored both the fraction of aggregated CRABP1 (Figure 2C, red) and the amount of unfolded protein at different urea concentrations (as monitored by tryptophan fluorescence, Figure 2C, black). We found that the CRABP1 aggregation propensity increases sharply at low urea concentrations (from 0 to 2.5 M). Importantly, in this urea range, CRABP1 is well folded, and the population of the unfolded state is almost negligible (as monitored by tryptophan fluorescence, Fig. 2C), showing directly that CRABP1 unfolding is not required for CRABP1 aggregation. Moreover, at urea concentrations higher than 3 M, the fraction of aggregated protein dropped dramatically, presumably because CRABP1 aggregates are not stable at high urea concentration – the same behavior (urea instability) has been previously observed for other amorphous aggregates. (35) These results suggest that CRABP1 aggregation is triggered by mild perturbations that cause shifts in the population of states on the CRABP1 energy landscape in the region of the native protein, rather than by global unfolding.
CRABP1 Aggregation Occurs from a Near-native Conformation
To characterize the nature of the aggregation-prone state of CRABP1, we took advantage of the observation that the CRABP1 aggregation-prone state was maximally populated at 2.5 M urea and recorded NMR spectra at different urea concentrations. Importantly, CRABP1 aggregation is slow enough that we could carry out NMR characterization of the CRABP1 conformation at different urea concentrations before aggregation occurred. NMR spectra of CRABP1 WT* in the absence of urea have all the features of a well-folded protein (Figure 3A, left). In agreement with tryptophan fluorescence experiments (Figure 2C, black), at urea concentrations higher than 4 M, NMR spectra of CRABP1 are indicative of global unfolding as a slow-exchange process on the NMR time scale: In addition to peaks corresponding to the residual fraction of native CRABP1, a new set of peaks appears when the urea concentration exceeds 4 M (Figure 3A, middle and right panels). The limited proton chemical shift dispersion for this new set of peaks (Figure 3A, light blue) argues that these peaks correspond to unfolded CRABP1. From 4 to 6 M urea, peaks corresponding to the native conformation gradually disappear with almost no urea-dependent changes in peak positions, and at urea concentrations higher than 6 M, the set of peaks corresponding to the unfolded state are the only ones observed (Figure 3A, right). The presence of two sets of peaks in the spectra in the intermediate urea concentration range (4 to 6 M), corresponding to native and unfolded CRABP1 conformational ensembles (Figure 3A, middle), indicates that the interconversion rate between these two conformations, unfolded and native, is slow on the NMR time scale, as expected from a large energetic barrier between the unfolded and native CRABP1 states (ΔG0U-N ~ 10 kcal/mol (25)).
Figure 3.

Urea results in two transitions in CRABP1 WT*: near-native transition at low urea concentrations and protein unfolding at high urea concentrations. A. 1H,15N HSQC NMR spectra of CRABP1 WT* in the absence (left) and presence of 5 M (middle) and 8 M (right) of urea. B. 1H,15N HSQC NMR spectra of CRABP1 WT* in the absence (black) and presence of 0.5–3.1 M urea (colored from yellow to red as the urea concentration increased). C. Amide proton chemical shift (CS) perturbations upon the presence of 0.5–3.1 M urea, mod[Δδ(urea) − Δδ (no urea)]; colored as in B. D. CS dependences of amide 1H (red) and 15N (blue) as a function of urea concentration for a representative residue (V33); see also Fig. S1. E. Aggregation propensity of CRABP1 WT* (gray bars, same as red bars in Figure 2) and fractions of the aggregation-prone near-native conformation (calculated as normalized CS dependences of amide 1H (red) and 15N (blue) [Δδ(urea) − Δδ(no urea)]/[Δδ(3.5 M urea) − Δδ(no urea)]; individual data points correspond to particular highly affected residues), as a function of urea concentration; see also Fig. S1. A green region indicates the higher urea concentrations that result in aggregate instability.
But the most exciting and informative observation in the NMR urea titration is that, in addition to its global unfolding transition, at low urea concentrations (below 4 M) CRABP1 undergoes a previously uncharacterized conformational transition that precedes global unfolding. In the urea range from 0 to 3 M, several CRABP1 residues in 1H-15N HSQC spectra experience small but distinct chemical shift changes (Figure 3B and C). Importantly, throughout the urea range from 0 to 4 M, only one set of peaks is present in all NMR spectra, consistent with a low barrier, fast exchange interconversion between native-like states. Both 1H and 15N amide chemical shift changes have a sigmoidal dependence on urea concentration up to 3 M (Figure 3D, Fig. S1), after which there are essentially no further chemical shift changes were observed. Moreover, with only one exception (G104), which experience very small shifts that are near to experimental error, the amide chemical shift dependence for the rest of affected residues is very similar (Fig. S1). Thus, the low urea NMR titration reports on a cooperative conformational transition, which we hypothesize represents the population of an aggregation-prone, near-native state. The fact that the observed chemical shift perturbations are relatively small (less than 0.25 ppm for proton chemical shifts, Figure 3C) and that only low amounts of urea are required to increase its population, argues that the aggregation-prone state of CRABP1 populated at low urea is structurally and energetically very close to the native CRABP1 state.
To assess whether population of this near-native conformation correlates with enhanced CRABP1 aggregation, we compared the fraction of the near-native conformation for each urea concentration from the sigmoidal dependence of 1H and 15N chemical shifts (CS) (obtained by normalizing different residue CS values) (Figure 3E, red for the 1H CSs and blue for 15N CSs) with the fractions of aggregated CRABP1 at each urea concentration (as previously shown in Figure 2C in red and now included in Figure 3E as black bars). Increased aggregation propensity at low urea concentrations closely correlates with the fraction of the new near-native conformation up to 2.5 M urea (note that at higher urea concentrations CRABP1 amorphous aggregates became unstable, as discussed above), arguing compellingly that the near-native conformation populated in the presence of low urea is indeed the state responsible for CRABP1 aggregation.
Tryptophan fluorescence data (Figure 2C, black) and relatively small chemical shift perturbations observed in the presence of low urea (Figure 3C) argue that the aggregation-prone conformation of CRABP1 (favored at low urea concentration) is not structurally very distinct from native CRABP1 (favored at zero urea). To characterize the structural features of the aggregation-prone state, we used 1–2 M urea and relatively low CRABP1 WT* concentrations (about 150 uM) to maintain a population of the aggregation-prone CRABP1 conformation that is below 50% and, by doing so, prevent the protein from aggregation during relatively long NMR experiments. First, we probed hydrogen bonding by measuring amide temperature coefficients using HSQC spectra of CRABP1 WT* recorded at several temperatures, and found them to be indistinguishable in the presence and in the absence of 1.5 M urea (Figure 4A), suggesting that the hydrogen-bonding patterns of residues (Figure 4C) are unperturbed in the aggregation-prone conformation relative to the CRABP1 native state. 1H-15N HSQC hydrogen-deuterium exchange experiments were completely consistent with the amide temperature coefficients: Residues that are highly protected from H/D exchange (Figure 4D) are the same in the absence and in the presence of low urea (Figure 4B for 1M urea, similar results were obtained in the presence of 2 M urea, data not shown), with the only difference that in the presence of urea H/D exchange rates uniformly increase for all residues as expected from decreased thermodynamic stability in the presence of low urea. The absence of residue-specific differences in the H/D exchange rates indicates that the aggregation-prone state has the same patterns of residues protected from the solvent as the native CRABP1 conformation (Figure 4D). Finally, only minor if any changes were observed for Cα chemical shifts of CRABP1 WT* in urea concentrations ranging from 0 to 3 M (data not shown), indicating that secondary structure elements also remain largely unperturbed in the aggregation-prone state. We also compared amide NMR spectra for several of the aggregation-prone variants, F50M, F71M and F65M (24) and G78A (data not shown) and found only local chemical shift perturbations upon mutations, indicating that the corresponding mutants adopt the same or very similar structure as CRABP1 WT*, even though they are aggregation-prone in the absence of denaturant.
Figure 4.

The native and aggregation-prone CRABP1 conformations share the same structural features. A. Amide temperature coefficients (ΔδHN/ΔT) of CRABP1 WT* in the absence (black) and presence (red) of 1.5 M urea. B. Deuterium incorporation after 2 and 24 h of H/D exchange in the presence (red) and absence (black) of 1 M urea, I(time)/I(1h), where I(time) is a peak intensity after 2 or 24 h of exchange and I(1h) is a intensity for the corresponding peak after 1 h exchange.C) CRABP1 WT* residues involved in hydrogen bonding as determined from the amide temperature coefficients from A (i.e., ΔδHN/ΔT > −4.5 ppb/K) mapped on the X-ray structure of CRABP1 (PDB ID 1CBR, chain A). The same residues were identified in the absence and presence of 1.5 M urea (see A). D. Mapping of CRABP1 WT1* residues that are highly (red, I(24)/I(1) > 0.5) and moderately (yellow, I(24)/I(1) < 0.5 and I(24)/I(1) > 0.2) protected from the solvent as determined from H/D exchange measurements. The same groups of residues were identified in the absence and presence of 1 M urea (see B).
All told, these findings suggest that low urea conditions shift the CRABP1 population to a new, near-native, aggregation-prone state that retains the overall protein fold of native CRABP1, but must be altered in some way so as to increase aggregation propensity.
The Aggregation-Prone Near-native CRABP1 Conformation Is Closely Related to an ‘Open’ State that Is Required for Ligand-binding Function
To better understand the nature of the aggregation-prone state, we mapped chemical shift changes observed in the presence of low urea (Figure 5A) into the X-ray structure of CRABP1 (Figure 5B). A majority of the residues experiencing relatively large chemical shift perturbations in the presence of low urea are located either around the ligand-entry portal – a dynamic gateway for CRABP1 ligands – or around the N-terminal region, including three residues F3, Q49 and A114 (Figure 5B, red). While flexibility near the N-terminus has not been shown to play any physiological role, conformational changes in the portal region are absolutely essential for CRABP1 function (27, 28, 36–40). Restriction of this conformational flexibility (for example, by crosslinking of flexible elements of the portal with each other) results in inability of CRABP1 to bind and release ligand. (41) It has been shown that apo-CRABP1 in the absence of its hydrophobic ligands, CRABP1 is significantly more dynamic than RA-bound CRABP1. (28) In crystal structures, CRABP1 populates two distinct conformations, closed for the RA-bound protein and open for the apo-protein, which have overall identical structures elsewhere but around the ligand-entry portal (Figure 1A, see introduction for more details). This ligand-entry portal comprises helix 2, turn II (between strands 3 and 4), and turn IV (between strands 5 and 6), and portal opening is required for ligand entry into the β-barrel. The ability of CRABP1 to interconvert between these two near-native conformations, open and closed, provides a plausible explanation for our results: Low denaturant conditions are shifting the near-native state CRABP1 ensemble towards an open, aggregation-prone state. The fact that the same regions in the protein were perturbed in the presence of low urea suggests that the urea-induced aggregation-prone conformation is closely related to the functionally important opening of the ligand-entry portal of CRABP1.
Figure 5.

The aggregation-prone conformation of CRABP1 affects the ligand-entry portal. A. Chemical shift perturbations (CSPs) as a function of residual number. CSPs were determined as √[ΔδH2+(0.154ΔδN)2], where ΔδH and ΔδN are amide proton and nitrogen differences in chemical shifts between CRABP1 WT* in the absence and presence of 3.5 M urea. Residues with large CSPs (> 0.14 ppm) and significant CSPs (> 0.035 ppm) are colored red and yellow, respectively; the rest are shown as gray. B. Mapping of CSPs from A into the X-ray structure of CRABP1 (PDB ID 1CBR, chain A). Residues with significant and large CSPs are colored as in A.
The fact that the interconversion in the near-native CRABP1 ensemble is in the fast (μs-ms) regime (as concluded from the presence of only one set of peaks at low urea concentrations, see above), while CRABP1 aggregation occurs on the slow (hours) time-scale suggests that the population of the open, near-native CRABP1 conformation triggers a complex cascade of CRABP1 transformations that result in its aggregation. In other words, the population of this near-native conformation is essential but is not the only step preceding CRABP1 aggregation.
Solvent Accessibility of Residues Facing the Ligand-binding Cavity Is Essential for CRABP1 Function but also Responsible for an Increased Risk of Aggregation
To understand how opening of the ligand portal promotes aggregation and investigate changes in exposure of aggregation-prone regions between open and closed CRABP1 conformations, we employed fluorescence labeling of cysteinyl thiols in CRABP1 with fluorescein maleimide. CRABP1 contains three intrinsic cysteine residues at positions 81, 95 and 129. In crystal structures of both apo- and holo-CRABP1, all three cysteines have their sulfhydryls facing the ligand-binding cavity in the native state of the protein and are completely inaccessible to solvent (Fig. S2B). However, under native conditions fluorescent labeling of WT* was still observed, indicating the presence of a small population of protein in a barrel-open state (Figure 6A). Further similar fluorescein-labeling of cysteine variants, C129A CRABP1 WT* and C81A, C129A CRABP1 WT* (a mutant CRABP1 containing only a single cysteine, Cys95), confirm that C129 and C95 are the two cysteine residues that are solvent-accessible and contribute to the fluorescent labeling of the WT* protein under native conditions (Figure 6A). It should be mentioned here that only a negligibly small fraction of the unfolded state is populated under our experimental conditions (see above, Figure 2C). These results suggest that observed thiol accessibility is a feature of the near-native CRABP1 ensemble rather than a result of unfolded fraction under these conditions: We conclude that portal opening of apo-CRABP1 required for ligand entry into the β-barrel allows fluorescein maleimide molecules to enter the cavity and label the barrel-facing cysteines.
Figure 6.
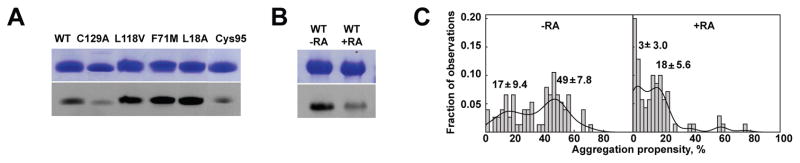
Transition to the aggregation-prone state of CRABP1 accompanies increased accessibility of the ligand-binding cavity. A. Fluorescein maleimide labeling of apo-CRABP1 WT* and its aggregation-prone mutants, L118V, F71M, and L18A and C129A and Cys95 (C81A, C129A) variants. B. Fluorescein maleimide labeling of CRABP1 WT* in the absence and presence of retinoic acid (RA). In A. and B., the lower panels represent the UV-exposed fluorescence image of the Coomassie stained gels shown in the upper panels (see also Fig. S2 and S3). C. Histograms showing aggregation propensity (the amount of insoluble protein) of CRABP1 WT*+1.5 M urea in the presence and absence of retinoic. Values of aggregation propensity obtained from multiple independent aggregation reactions (76 reaction for apo-CRABP1 and 69 reactions for the RA-bound protein) were used for the histograms. The histograms were plotted with equal bin widths showing the fraction of values lying in each bin. Experimental data obtained from multiple independent aggregation reactions were partitioned into clusters – the mean and standard deviations of clusters are shown on the plot. Two clusters corresponding to 17±9.4% and 49±7.8% of aggregation propensity were obtained in the absence of RA. Two overlapping clusters corresponding to 3±3% and 18±5.6% of aggregation propensity were identified in the presence of RA. The solid lines represent the normalized smooth kernel distributions.
Next, we explored whether aggregation-prone mutations in CRABP1 change thiol accessibility of cysteines facing the ligand-binding cavity. The fluorescein maleimide labeling experiment was performed for three aggregation-prone CRABP1 mutants, L18A, F71M and L118V. These three mutants are well-folded (data not shown) and bear no significant changes in an environment of cysteine residues (Fig. S2C). Moreover, despite very different substitution sites, at least two of these substitutions (in positions F71 and L118, Figure 2B) lead to aggregates with the same core sequences, suggesting a similar aggregation mechanism for these CRABP1 variants. In addition, we included here the L18A mutation in helix 1 of CRABP1, which presumably increases flexibility in the portal region and thus, leads to an increase in the population of the near-native conformation under native conditions. The L18A, F71M and L118V substitutions only slightly destabilize the protein and hence cause only negligible changes in the population of the unfolded fractions under near-native conditions. Therefore, fluorescein maleimide labeling of these mutants relies on barrel opening, and thus their labeling will report on the population of the open (near-native) conformation. Intriguingly, we found a significantly higher labeling for these aggregation-prone mutants than observed for CRABP1 WT* (Figure 6A) suggesting that these aggregation-inducing mutations indeed enhance barrel opening, resulting in labeling of thiol groups along with exposure of the aggregation-prone regions. In other words, these results argue that higher aggregation in these CRABP1 variants is a result of increased population of the open, near-native state.
Next, to further test the hypothesis that population of a near-native state is responsible for aggregation of CRABP1, we employed the fact that RA binding substantially reduces the dynamics of the ligand-portal region and stabilizes the closed state of CRABP1. (28) Not surprisingly, the presence of the retinoic acid of CRABP1 WT* significantly suppresses fluorescein labeling of CRABP1 (Figure 6B). If the open CRABP1 conformation were responsible for aggregation, one would expect that RA binding should significantly decrease the aggregation propensity of CRABP1, even when the open, aggregation-prone conformation is more populated in the absence of RA (i.e., in the presence of low urea or upon mutation), by shifting the conformational equilibrium to the closed state. To test this, we examined how RA binding affects CRABP1 aggregation propensity in the presence of 1.5 M urea (Figure 6C). In full agreement with our expectations, we found that the presence of RA significantly decreased the aggregation propensity of CRABP1 in the presence of low urea, providing strong evidence that the functionally essential population of the open conformation indeed is responsible for aggregation. Taken together, our results strongly suggest that the open, near-native CRABP1 conformation that is required for ligand-binding function also places the protein at risk for aggregation.
DISCUSSION
The fact that the ligand-entry portal of CRABP1 juxtaposes the hydrophobic, aggregation-prone sequences located in the strand 3-turn II-strand 4 suggests a plausible mechanism for CRABP1 aggregation: As a result of cooperative motions of the helical region and widening of the gap between strands 3–4 and strand 5 to allow ligand entry to the β-barrel, the aggregation-prone sequences are transiently exposed to the solvent such that they may facilitate aggregation (Figure 7A). The fact that CRABP1 aggregation has not been observed in mammalian cells under physiological conditions raises an intriguing question: Why do the functionally important conformational dynamics not result in aggregation in vivo? In line with a previously observed correlation between protein aggregation propensity and expression level, (42) lower cellular concentrations of CRABP1 (about 1–10 μM) may be an important factor that allows CRABP1 to function with optimal efficiency and avoid the risk to aggregate. Indeed, cellular concentrations of CRABP1 are significantly lower than the concentrations required to promote CRABP1 aggregation in vitro and under overexpressed conditions in E. coli cells. (43) In addition to low cellular concentrations of CRABP1, our results also suggest another mechanism to control CRABP1 aggregation through fine-tuning of the near-native conformational ensemble in order to optimize population of a vulnerable, aggregation-prone conformation without compromising protein function (Figure 7B, black). Indeed, the open state of CRABP1 is the most ‘at-risk’ conformation with exposed aggregation-prone regions. We found that only a small fraction of CRABP1 populates this functionally important, but vulnerable conformation of CRABP1, enabling efficient ligand binding, but such a low population is not sufficient for protein aggregation. Indeed, sigmoidal dependencies of chemical shifts upon increasing urea concentrations from 0 to 3 M (Figure 3E) revealed that the population of the open, aggregation-prone conformation in the absence of urea does not exceed 10%. In line with these NMR results, only a small fraction of cavity-exposed cysteine residues are labeled by fluorescein maleimide (Figure 6A), suggesting that indeed the majority of the protein adopts the closed conformation. These finding fully agrees with our previous NMR study that revealed only very limited (however significant and functionally important) flexibility around the ligand-entry region of CRABP1. (28)
Figure 7.
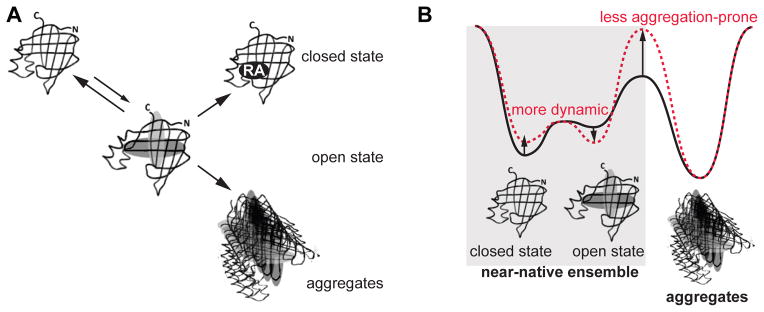
Interplay between conformational flexibility and aggregation in the iLBP family. A. A schematic representation of transitions from the near-native CRABP1 ensemble: The population of the open, aggregation-prone conformation is required for ligand binding but also results in aggregation. B. The energy landscape of iLBPs under native conditions comprises a delicate balance between protein conformational flexibility and aggregation. The iLBP near-native conformational ensemble comprises at least two conformations: close, ‘secure’ conformation and open, ‘at-risk’ conformation that is essential for ligand binding but may promote aggregation. The fine-tuning of thermodynamic and kinetic features of the landscape prevents different iLBP members from aggregation and allows them to perform their specific function. Rigid iLBPs such as CRABP1 (black) minimize the amount of time that proteins spend exposing the hydrophobic regions to be efficient enough for ligand binding but not sufficient for aggregation. The Lb-FABP/LFABP/ILBP subfamily members (red) spend significant amount of time in the open conformation allowing binding to larger ligands. To avoid aggregation, regions exposed in the open conformation are less hydrophobic.
In general, the fact that CRABP1 aggregation does not require significant protein unfolding and thus occurs from a near-native protein conformation is reminiscent of the aggregation mechanism suggested for another globular β-strand protein, the acylphosphatase AcPDro2 (44). Under some conditions (e.g., in the presence of very low concentrations of trifluoroethanol) AcPDro2 populates an aggregation-prone state that has native-like structure, folding stability and enzymatic activity. However, under normal physiological conditions the aggregation-prone state is energetically unfavorable, which prevents the AcPDro2 near-native ensemble from populating this dangerous aggregation-prone state. In the case of CRABP1, the population of its near-native aggregation-prone conformation (which exposures vulnerable hydrophobic regions) is essential for CRABP1 binding to retinoic acid and thus cannot be completely avoided. As a result, to prevent CRABP1 aggregation without compromising its function, flexibility of the ligand-entry portal is restricted to be sufficient for binding to retinoic acid but not to result in significant protein aggregation.
However, the balance between aggregation propensity of CRABP1 and its function is very sensitive and fragile: Even small enhanced conformational flexibility in the portal region of CRABP1 is directly associated with higher aggregation propensity that puts the protein in a higher aggregation risk. Indeed, as we discussed above, a very mild perturbation – low urea – results in enhanced dynamics around the ligand-entry portal and a higher population of the open, aggregation-prone CRABP1 conformation and thus, inevitably promotes formation of amorphous aggregates. Another illustration comes from aggregation-prone CRABP1 mutations that perturb the near-native CRABP1 conformational ensemble. For example, previously, we suggested that mutations in the positions F50, F65 and F71 perturb aromatic-aromatic contacts between the non-strands 3–4 and 5 and thus, stabilize the open, aggregation-prone conformation, exposing aggregation sequences located in the strand 3-turn II-strand 4 and facilitating aggregation. (24) Indeed, MD simulation for one of these mutants, F65M revealed that this aggregation-prone variant spends about 40% of simulation time (vs. less than 10% for CRABP1 WT) in the open conformation (Fig. S4) that explains the enhanced aggregation propensity for this mutant observed experimentally.
All together these observations argue that enhanced portal mobility indeed unavoidably increases the risk of CRABP1 aggregation. Amazingly, enhanced portal flexibility is functionally essential for most iLBP family members, particularly for the most primitive Lb-FABP/LFABP/ILBP subfamily, as its members are required to bind large, bulky ligands. (45) Consistent with their ligand size, the Lb-FABP/LFABP/ILBP family members experience substantially greater flexibility for the ligand-entry portal and larger opening of the gap between the strands 3–4 and 5. (46, 47) How, despite this enhanced flexibility, does the Lb-FABP/LFABP/ILBP subfamily cope with aggregation risk and remain functional at the same time? Since the exposure of strand 3-turn II-strand 4 sequences is unavoidable for the Lb-FABP/LFABP/ILBP subfamily, one plausible way for Nature to prevent aggregation is for this region to be less aggregation-prone (Figure 7B, red). To test this possibility, we used a sequence-based aggregation prediction algorithm, Zyggregator (48) and identified aggregation-prone sequences for the Lb-FABP/LFABP/ILBP subfamily members, ILBP and IFABP. Note that for CRABP1, the two aggregation cores (in strand 3-turn II-strand 4 and strands 9–10) identified experimentally correspond closely with sequences predicted by the Zyggregator, and these regions remain conserved through the entire iLBP family. (25) In full agreement with previous results, for ILBP and IFABP, Zyggregator predicted similar aggregation regions (Fig. S5). However, the aggregation core located around strand 3, turn II, and strand 4 is significantly less extensive in both ILBP and IFABP than in their more rigid homolog CRABP1 (Fig. S5). Intriguingly, while for many iLBPs family members, residues located in the strand 3-turn II-strand 4 are highly conserved, and many of them are directly involved in several hydrophobic ligand interactions for most iLBPs, (49) for the Lb-FABP/LFABP/ILBP subfamily, this region is significantly more variable and less hydrophobic, resulting in a lower aggregation-propensity for this region (Fig. S5), but compromising ligand specificity and binding affinities as compared with the other iLBPs. (45, 49)
In summary, the iLBP family provides an elegant example of how evolution fine-tunes a delicate balance between protein function, conformational variability and aggregation (Figure 7). There are several ways Nature can control aggregation propensity in this protein family and still retain functionally important conformational flexibility. The primitive members of the iLBP family (Lb-FABP/LFABP/ILBP) are very dynamic and spend a significant amount of time in the barrel-open conformation to allow ligand binding. However, regions exposed in this open conformation are less hydrophobic than in CRABP1, for example, which allows binding to bulky ligands without the risk of aggregation but with reduced ligand specificity and affinity. In order to increase specificity and affinity of binding to hydrophobic ligands, the other iLBP family members have evolved such that the ligand-binding region located in the strand 3, turn II and strand 4 became more hydrophobic. As a price for these features that required exposure of the hydrophobic, aggregation-prone sequences in the open conformation, these iLBPs became significantly more rigid to minimize the amount of time that proteins spend exposing the hydrophobic regions to be efficient enough for ligand binding but not sufficient for aggregation.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by an NIH Director’s Pioneer Award (OD000945).
ABBREVIATIONS
- CRABP1
cellular retinoic acid-binding protein
- iLBP
intracellular lipidbinding proteins
- Lb-FABP
liver basic fatty acid binding protein
- LFABP
liver-type fatty acid-binding protein
- ILBP
ileal lipid binding protein
- WT
wild type
- DTT
dithiothreitol
- NMR
nuclear magnetic resonance
- HSQC
heteronuclear single quantum coherence
- CS
chemical shift
- CSP
chemical shift perturbation
- MD
molecular dynamics
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. Aggregation propensities for different CRABP1 WT* mutants are tableted in Table S1. SI figures S1–S5 demonstrate urea-dependences of chemical shifts, cysteine accessibilities, open-closed transitions observed in MD simulations, and Zyggregator predictions of aggregation-prone regions for different iLBPs. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Jahn TR, Radford SE. Folding versus aggregation: polypeptide conformations on competing pathways. Arch Biochem Biophys. 2008;469:100–117. doi: 10.1016/j.abb.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tzeng SR, Kalodimos CG. Allosteric inhibition through suppression of transient conformational states. Nat Chem Biol. 2013;9:462–465. doi: 10.1038/nchembio.1250. [DOI] [PubMed] [Google Scholar]
- 3.Boehr DD, McElheny D, Dyson HJ, Wright PE. Millisecond timescale fluctuations in dihydrofolate reductase are exquisitely sensitive to the bound ligands. Proc Natl Acad Sci U S A. 2010;107:1373–1378. doi: 10.1073/pnas.0914163107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neudecker P, Robustelli P, Cavalli A, Walsh P, Lundstrom P, Zarrine-Afsar A, Sharpe S, Vendruscolo M, Kay LE. Structure of an intermediate state in protein folding and aggregation. Science. 2012;336:362–366. doi: 10.1126/science.1214203. [DOI] [PubMed] [Google Scholar]
- 5.Pastore A, Temussi PA. The two faces of Janus: functional interactions and protein aggregation. Current Opin Struct Biol. 2012;22:30–37. doi: 10.1016/j.sbi.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 6.Masino L, Nicastro G, Calder L, Vendruscolo M, Pastore A. Functional interactions as a survival strategy against abnormal aggregation. FASEB J. 2011;25:45–54. doi: 10.1096/fj.10-161208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brockwell DJ, Radford SE. Intermediates: ubiquitous species on folding energy landscapes? Current Opin Struct Biol. 2007;17:30–37. doi: 10.1016/j.sbi.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clark PL. Protein folding in the cell: reshaping the folding funnel. Trends Biochem Sci. 2004;29:527–534. doi: 10.1016/j.tibs.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 9.Calamai M, Chiti F, Dobson CM. Amyloid fibril formation can proceed from different conformations of a partially unfolded protein. Biophys J. 2005;89:4201–4210. doi: 10.1529/biophysj.105.068726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McParland VJ, Kad NM, Kalverda AP, Brown A, Kirwin-Jones P, Hunter MG, Sunde M, Radford SE. Partially unfolded states of beta(2)-microglobulin and amyloid formation in vitro. Biochemistry. 2000;39:8735–8746. doi: 10.1021/bi000276j. [DOI] [PubMed] [Google Scholar]
- 11.Chiti F, Dobson CM. Amyloid formation by globular proteins under native conditions. Nature Chem Biol. 2009;5:15–22. doi: 10.1038/nchembio.131. [DOI] [PubMed] [Google Scholar]
- 12.Lim KH, Dyson HJ, Kelly JW, Wright PE. Localized structural fluctuations promote amyloidogenic conformations in transthyretin. J Mol Biol. 2013;425:977–988. doi: 10.1016/j.jmb.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bemporad F, De Simone A, Chiti F, Dobson CM. Characterizing intermolecular interactions that initiate native-like protein aggregation. Biophys J. 2012;102:2595–2604. doi: 10.1016/j.bpj.2012.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sabate R, Espargaro A, Grana-Montes R, Reverter D, Ventura S. Native structure protects SUMO proteins from aggregation into amyloid fibrils. Biomacromolecules. 2012;13:1916–1926. doi: 10.1021/bm3004385. [DOI] [PubMed] [Google Scholar]
- 15.Hamada D, Tanaka T, Tartaglia GG, Pawar A, Vendruscolo M, Kawamura M, Tamura A, Tanaka N, Dobson CM. Competition between folding, native-state dimerisation and amyloid aggregation in β-lactoglobulin. J Mol Biol. 2009;386:878–890. doi: 10.1016/j.jmb.2008.12.038. [DOI] [PubMed] [Google Scholar]
- 16.Booth DR, Sunde M, Bellotti V, Robinson CV, Hutchinson WL, Fraser PE, Hawkins PN, Dobson CM, Radford SE, Blake CC, Pepys MB. Instability, unfolding and aggregation of human lysozyme variants underlying amyloid fibrillogenesis. Nature. 1997;385:787–793. doi: 10.1038/385787a0. [DOI] [PubMed] [Google Scholar]
- 17.Yamaguchi K, Katou H, Hoshino M, Hasegawa K, Naiki H, Goto Y. Core and heterogeneity of β2-microglobulin amyloid fibrils as revealed by H/D exchange. J Mol Biol. 2004;338:559–571. doi: 10.1016/j.jmb.2004.02.067. [DOI] [PubMed] [Google Scholar]
- 18.Villanueva J, Hoshino M, Katou H, Kardos J, Hasegawa K, Naiki H, Goto Y. Increase in the conformational flexibility of β2-microglobulin upon copper binding: a possible role for copper in dialysis-related amyloidosis. Protein Sci. 2004;13:797–809. doi: 10.1110/ps.03445704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pagano K, Bemporad F, Fogolari F, Esposito G, Viglino P, Chiti F, Corazza A. Structural and dynamics characteristics of acylphosphatase from Sulfolobus solfataricus in the monomeric state and in the initial native-like aggregates. J Biol Chem. 2010;285:14689–14700. doi: 10.1074/jbc.M109.082156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thompson JR, Bratt JM, Banaszak LJ. Crystal structure of cellular retinoic acid binding protein I shows increased access to the binding cavity due to formation of an intermolecular β-sheet. J Mol Biol. 1995;252:433–446. doi: 10.1006/jmbi.1995.0509. [DOI] [PubMed] [Google Scholar]
- 21.Kleywegt GJ, Bergfors T, Senn H, Le Motte P, Gsell B, Shudo K, Jones TA. Crystal structures of cellular retinoic acid binding proteins I and II in complex with all-trans-retinoic acid and a synthetic retinoid. Structure. 1994;2:1241–1258. doi: 10.1016/s0969-2126(94)00125-1. [DOI] [PubMed] [Google Scholar]
- 22.Ignatova Z, Krishnan B, Bombardier JP, Marcelino AM, Hong J, Gierasch LM. From the test tube to the cell: exploring the folding and aggregation of a bβ-clam protein. Biopolymers. 2007;88:157–163. doi: 10.1002/bip.20665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clark PL, Liu ZP, Zhang J, Gierasch LM. Intrinsic tryptophans of CRABPI as probes of structure and folding. Protein Sci. 1996;5:1108–1117. doi: 10.1002/pro.5560050613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Budyak IL, Zhuravleva A, Gierasch LM. The Role of Aromatic-Aromatic Interactions in Strand-Strand Stabilization of β-Sheets. J Mol Biol. 2013;425:3522–3535. doi: 10.1016/j.jmb.2013.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Budyak IL, Krishnan B, Marcelino-Cruz AM, Ferrolino MC, Zhuravleva A, Gierasch LM. Early folding events protect aggregation-prone regions of a β-rich protein. Structure. 2013;21:476–485. doi: 10.1016/j.str.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ignatova Z, Gierasch LM. Aggregation of a slow-folding mutant of a β-clam protein proceeds through a monomeric nucleus. Biochemistry. 2005;44:7266–7274. doi: 10.1021/bi047404e. [DOI] [PubMed] [Google Scholar]
- 27.Xiao H, Kaltashov IA. Transient structural disorder as a facilitator of protein-ligand binding: native H/D exchange-mass spectrometry study of cellular retinoic acid binding protein I. J Am Soc Mass Spectrom. 2005;16:869–879. doi: 10.1016/j.jasms.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 28.Krishnan VV, Sukumar M, Gierasch LM, Cosman M. Dynamics of cellular retinoic acid-binding protein I on multiple time scales with implications for ligand binding. Biochemistry. 2000;39:9119–9129. doi: 10.1021/bi000296l. [DOI] [PubMed] [Google Scholar]
- 29.Hoshino M, Katou H, Hagihara Y, Hasegawa K, Naiki H, Goto Y. Mapping the core of the β(2)-microglobulin amyloid fibril by H/D exchange. Nature Struct Biol. 2002;9:332–336. doi: 10.1038/nsb792. [DOI] [PubMed] [Google Scholar]
- 30.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 31.Pace CN. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 32.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferrolino MC. Program in Molecular and Cellular Biology. University of Massachusetts; Amherst, Amherst, MA: 2013. The Unavoidable Threat of Aggregation: Implicaiont for Folding and Function of a β-Rich Protein. [Google Scholar]
- 34.Gunasekaran K, Hagler AT, Gierasch LM. Sequence and structural analysis of cellular retinoic acid-binding proteins reveals a network of conserved hydrophobic interactions. Proteins. 2004;54:179–194. doi: 10.1002/prot.10520. [DOI] [PubMed] [Google Scholar]
- 35.Chow C, Kurt N, Murphy RM, Cavagnero S. Structural characterization of apomyoglobin self-associated species in aqueous buffer and urea solution. Biophys J. 2006;90:298–309. doi: 10.1529/biophysj.105.070227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu L, Kurian E, Prendergast FG, Kemple MD. Dynamics of palmitic acid complexed with rat intestinal fatty acid binding protein. Biochemistry. 1999;38:1554–1561. doi: 10.1021/bi982087v. [DOI] [PubMed] [Google Scholar]
- 37.Jamison RS, Newcomer ME, Ong DE. Detection of conformational changes in cellular retinoid-binding proteins by limited proteolysis. Methods Mol Biol. 1998;89:165–176. doi: 10.1385/0-89603-438-0:165. [DOI] [PubMed] [Google Scholar]
- 38.Donovan M, Olofsson B, Gustafson AL, Dencker L, Eriksson U. The cellular retinoic acid binding proteins. J Steroid Biochem Mol Biol. 1995;53:459–465. doi: 10.1016/0960-0760(95)00092-e. [DOI] [PubMed] [Google Scholar]
- 39.Jamison RS, Newcomer ME, Ong DE. Cellular retinoid-binding proteins: limited proteolysis reveals a conformational change upon ligand binding. Biochemistry. 1994;33:2873–2879. doi: 10.1021/bi00176a017. [DOI] [PubMed] [Google Scholar]
- 40.Hodsdon ME, Cistola DP. Discrete backbone disorder in the nuclear magnetic resonance structure of apo intestinal fatty acid-binding protein: implications for the mechanism of ligand entry. Biochemistry. 1997;36:1450–1460. doi: 10.1021/bi961890r. [DOI] [PubMed] [Google Scholar]
- 41.Sjoelund V, Kaltashov IA. Transporter-to-trap conversion: a disulfide bond formation in cellular retinoic acid binding protein I mutant triggered by retinoic acid binding irreversibly locks the ligand inside the protein. Biochemistry. 2007;46:13382–13390. doi: 10.1021/bi700867c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tartaglia GG, Pechmann S, Dobson CM, Vendruscolo M. Life on the edge: a link between gene expression levels and aggregation rates of human proteins. Trends Biochem Sci. 2007;32:204–206. doi: 10.1016/j.tibs.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 43.Donovan M, Olofsson B, Gustafson AL, Dencker L, Eriksson U. The cellular retinoic acid binding proteins. J Steroid Biochem Mol Biol. 1995;53:459–465. doi: 10.1016/0960-0760(95)00092-e. [DOI] [PubMed] [Google Scholar]
- 44.De Simone A, Dhulesia A, Soldi G, Vendruscolo M, Hsu ST, Chiti F, Dobson CM. Experimental free energy surfaces reveal the mechanisms of maintenance of protein solubility. Proc Natl Acad Sci U S A. 2011;108:21057–21062. doi: 10.1073/pnas.1112197108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schaap FG, van der Vusse GJ, Glatz JF. Evolution of the family of intracellular lipid binding proteins in vertebrates. Mol Cell Biochem. 2002;239:69–77. [PubMed] [Google Scholar]
- 46.Lucke C, Fushman D, Ludwig C, Hamilton JA, Sacchettini JC, Ruterjans H. A comparative study of the backbone dynamics of two closely related lipid binding proteins: bovine heart fatty acid binding protein and porcine ileal lipid binding protein. Mol Cell Biochem. 1999;192:109–121. [PubMed] [Google Scholar]
- 47.Eliseo T, Ragona L, Catalano M, Assfalg M, Paci M, Zetta L, Molinari H, Cicero DO. Structural and dynamic determinants of ligand binding in the ternary complex of chicken liver bile acid binding protein with two bile salts revealed by NMR. Biochemistry. 2007;46:12557–12567. doi: 10.1021/bi7013085. [DOI] [PubMed] [Google Scholar]
- 48.Tartaglia GG, Vendruscolo M. The Zyggregator method for predicting protein aggregation propensities. Chem Soc Rev. 2008;37:1395–1401. doi: 10.1039/b706784b. [DOI] [PubMed] [Google Scholar]
- 49.Marcelino AM, Smock RG, Gierasch LM. Evolutionary coupling of structural and functional sequence information in the intracellular lipid-binding protein family. Proteins. 2006;63:373–384. doi: 10.1002/prot.20860. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.