

Abstract
Background and Purpose
Multidrug resistance (MDR), usually mediated by overexpression of efflux transporters such as P-gp, ABCG2 and/or MRP1, remains a major obstacle hindering successful cancer chemotherapy. There has been great interest in the development of inhibitors towards these transporters to circumvent resistance. However, since the inhibition of transporter is not specific to cancer cells, a decrease in the cytotoxic drug dosing may be needed to prevent excess toxicity, thus undermining the potential benefit brought about by a drug efflux inhibitor. The design of potent MDR modulators specific towards resistant cancer cells and devoid of drug-drug interactions will be needed to effect MDR reversal.
Experimental Approach
Recent evidence suggests that the PTEN/PI3K/Akt pathway may be exploited to alter ABCG2 subcellular localization, thereby circumventing MDR. Three PPARγ agonists (telmisartan, pioglitazone and rosiglitazone) that have been used in the clinics were tested for their effect on the PTEN/PI3K/Akt pathway and possible reversal of ABCG2-mediated drug resistance.
Key Results
The PPARγ agonists were found to be weak ABCG2 inhibitors by drug efflux assay. They were also shown to elevate the reduced PTEN expression in a resistant and ABCG2-overexpressing cell model, which inhibit the PI3K-Akt pathway and lead to the relocalization of ABCG2 from the plasma membrane to the cytoplasma, thus apparently circumventing the ABCG2-mediated MDR.
Conclusions and Implications
Since this PPARγ/PTEN/PI3K/Akt pathway regulating ABCG2 is only functional in drug-resistant cancer cells with PTEN loss, the PPARγ agonists identified may represent promising agents targeting resistant cells for MDR reversal.
Keywords: ABCG2, multidrug resistance (MDR), PPARγ, phosphatase and tensin homologue deleted on chromosome 10 (PTEN), telmisartan, pioglitazone, rosiglitazone
Introduction
Despite the advances in the development of novel chemotherapeutic drugs, the occurrence of multidrug resistance (MDR) remains a major unresolved clinical problem hindering successful cancer chemotherapy. The most common mechanism of resistance is the active efflux of a variety of structurally unrelated anticancer drugs by ATP-binding cassette (ABC) transporters including P-glycoprotein (P-gp), ABCG2 and MRP1 (Gottesman et al., 2002). An obvious strategy to restore drug sensitivity in MDR cancer cells is therefore to block drug efflux mediated by the ABC transporters. Over the past decade, tremendous efforts have been made to discover and develop such inhibitors or modulators in the clinic against P-gp, the most extensively studied MDR transporter (Sandor et al., 1998; Tamaki et al., 2011; Amiri-Kordestani et al., 2012). However, the lack of specific and potent inhibitors against the MDR transporters and unpredictable pharmacokinetic drug-drug interactions has significantly hindered the development of transporter inhibitors in the clinic. Moreover, since the inhibition of transporters is not specific to cancer cells, it could also lead to impairment of the natural protective mechanism in the gastrointestinal tract and the drug excretion process at the hepatobiliary canaliculi. As a result, a decrease in the cytotoxic drug dosing may be needed to prevent excess toxicity, thus undermining the potential benefit obtained from having a drug efflux inhibitor. New transporter modulators with novel mechanisms targeting the resistance cancer cells are needed to realize the MDR reversal hypothesis.
ABCG2 is a relatively newer ABC transporter known to confer MDR, which has attracted wide attention recently because of its role in stem cell biology (Zhou et al., 2001; Bunting, 2002) and the association of its genetic polymorphism with gout (Matsuo et al., 2011). Growing evidence suggests that ABCG2 underlies the MDR of clinical samples from different cancer types (Ross et al., 2000). Besides mediating MDR, ABCG2 is also known to play critical roles in absorption, drug elimination and tissue protection against toxins and xenobiotics in various organs (Kerr et al., 2011). Due to the complex role played by ABCG2 in the body, it will be especially challenging to devise an ABCG2 inhibitor for MDR reversal.
The PI3K-Akt signalling pathway is a key regulator of cell cycle proliferation, growth, survival, protein synthesis and glucose metabolism, which therefore is implicated in many human diseases including cancer (Vivanco and Sawyers, 2002). PTEN (phosphatase and tensin homologue deleted on chromosome 10) is an important tumour suppressor gene that negatively regulates the PI3K/Akt pathway by dephosphorylating the lipid signalling intermediate PIP3 (Maehama and Dixon, 1998). Importantly, somatic inactivation of PTEN is prevalent in various cancer types and is associated with cancer susceptibility and tumour progression (Eng, 2003). Overactivation of the PI3K/Akt pathway as a result of PTEN loss is known to facilitate cancer growth and may predict resistance to anticancer drugs (Hafsi et al., 2012). To this end, novel strategies to elevate PTEN expression and/or inhibit the PI3K/Akt pathway have been proposed for cancer prevention and chemotherapy (Patel et al., 2001; Yap et al., 2008; Cheng et al., 2012; Garcia-Cao et al., 2012).
Recently, inhibition of the PTEN/PI3K/Akt signalling pathway has been shown to modulate ABCG2-mediated drug transport via the translocation of ABCG2 from the plasma membrane to intracellular compartments in different cell model systems including side population cells in the bone marrow (Mogi et al., 2003), gallbladder epithelial cells (Aust et al., 2004), polarized ABCG2-overexpressing porcine kidney LLCPK1 cells (Takada et al., 2005), hepatocellular carcinoma-derived cells (Hu et al., 2008), glioma-derived stem-like cells (Bleau et al., 2009) and ABCG2-overexpressing extracellular vesicles derived from MCF-7/MR breast cancer cells (Goler-Baron et al., 2012). Importantly, Patel et al. have provided compelling data demonstrating that a synthetic PPARγ agonist can up-regulate PTEN in human colorectal carcinoma Caco-2 cells and breast cancer MCF-7 cells, which correlated well with the suppression of proliferation in the treated cells. This activation of PTEN by a PPARγ agonist was later shown to cause inhibition of the PI3K/Akt pathway, as evidenced by Akt dephosphorylation, thereby inhibiting cell proliferation (Teresi et al., 2006). We therefore proposed to exploit PPARγ agonist activity of existing drugs in clinical use to interact with the PTEN/PI3K/Akt pathway and probably also interfere with ABCG2 cellular localization in an attempt to circumvent MDR.
Angiotensin II receptor blockers (ARBs) represent a well-tolerated and increasingly prescribed class of anti-hypertensive agents used widely in combination drug regimens (Benndorf and Boger, 2008). Among the clinically available ARBs, telmisartan is well-known for its unique PPARγ activating activities at therapeutically relevant concentrations (Benson et al., 2004; Schupp et al., 2004). Moreover, telmisartan and some other ARBs have been found to interact with ABC transporters including P-gp and ABCG2 (Kamiyama et al., 2010; Weiss et al., 2010), although the potential use of ARBs to reverse MDR has not been explored. It is also noteworthy that ARBs are generally considered to have low potential for drug-drug interaction because of their lack of significant interactions with cytochrome P450 monoxygenase (Schmidt and Schieffer, 2003).
Thiazolidinediones (TZDs), such as rosiglitazone and pioglitazone, are synthetic PPARγ ligands and were used as insulin-sensitizing antidiabetic drugs (Staels and Fruchart, 2005). They work by enhancing the insulin sensitivity in liver, muscle and adipose cells and by modulating lipid metabolism. Apart from their antidiabetic activity, some TZDs have in recent years caught attention for their potential anti-cancer activity. In particular, triglitazone and rosiglitazone were found to induce growth arrest and apoptosis in a broad spectrum of cancer cells, at least partially through the PPARγ pathway (Elstner et al., 1998; Koeffler, 2003).
The aim of this study was to evaluate the potential circumvention of ABCG2-mediated MDR by these commonly prescribed PPARγ agonists. Intriguingly, remarkable down-regulation of PTEN was observed in our resistant and ABCG2-overexpressing cancer cell line model. Our data indicates that PPARγ agonists up-regulate PTEN in the resistant cells to apparently circumvent drug resistance, probably via a dual mechanism by (i) inhibition of the PI3K/Akt pathway and (ii) driving the internalization of ABCG2 to the cytoplasm.
Methods
Chemicals
Mitoxantrone was purchased from Sigma Chemical (St Louis, MO, USA). Pheophorbide A (PhA) and fumitremorgin C (FTC) were kind gifts obtained from Dr Susan Bates (National Cancer Institute, NIH, Bethesda, MD, USA). Telmisartan was purchased from Changzhou Yabang Pharmaceutical Co., Ltd (Jiangsu Province, China). Rosiglitazone was obtained from GlaxoSmithKline. Pioglitazone was purchased from Amylin Pharmaceuticals, Inc (San Diego, CA, USA, USA). Irbesartan was from Zhejiang Apeloa Jiayuan Pharmaceutical Co. Ltd. (Zhejiang Province, China). Losartan and valsartan were purchased from Cayman Chemical (Ann Arbor, MI, USA). GW9662 was obtained from Sigma-Aldrich (St Louis, MO, USA). LY294002 was purchased from Selleck Chemicals (Houston, TX, USA).
Cell culture
MCF-7 and its drug-selected resistant subline MCF-7 FLV1000, and pcDNA3- or ABCG2-stably transfected HEK293 cells were obtained from Dr Susan Bates (National Cancer Institute). MCF-7 FLV1000 is overexpressing ABCG2, contributing to drug resistance, and was maintained in 1000 nM flavopiridol. No P-glycoprotein or MRP1 was detected in this resistant cell line. MCF-7 and MCF-7 FLV1000 cells, and the transfected HEK293 cells were maintained in DMEM and MEM medium, respectively, supplemented with 10% FBS, 100 units mL−1 streptomycin sulfate and 100 units mL−1 penicillin G sulfate, and incubated at 37°C in 5% CO2. The resistant cells were grown in drug-free culture medium for more than 2 weeks before analysis. The ABCG2-transfected HEK293 cells expressed the wild type ABCG2 (i.e. R482) and were maintained at 1 mg mL−1 G418.
Growth inhibition assay
Growth inhibitory effect of mitoxantrone with or without the concomitant treatment of the tested PPARγ agonists was evaluated by the sulforhodamine B assay (Skehan et al., 1990). Cells were seeded into 96-well microtitre plates in 100 μL at a plating density of 5000 cells per well and allowed to incubate overnight. The cells were then treated with mitoxantrone at a range of concentrations in the presence or absence of a fixed concentration of tested PPARγ agonist and allowed to incubate at 37°C in 5% CO2 for 72 h. Each drug concentration was tested in quadruplicate and controls were tested in replicates of eight. The fold reversal of resistance was calculated by dividing the IC50 for mitoxantrone or cisplatin (non-ABCG2 substrate as negative control) in the absence of PPARγ agonist by that obtained in the presence of PPARγ agonist. Each experiment was carried out independently at least three times. To determine whether differences between IC50 values were significant, a Student's t-test was performed with P < 0.05 being considered significant.
Reverse transcription and real-time PCR
Total RNA was isolated using the Trizol reagent (Invitrogen, Carlsbad, CA, USA). RNA (1 μg) was reverse transcribed using the Transcriptor High Fidelity cDNA Synthesis Kit (Roche Applied Science, Indianapolis, IN, USA). Quantitative real-time PCR was performed to measure ABCG2 transcript level using the KAPA SYBR FAST qPCR Kit (KapaBiosystems, Woburn, MA, USA) in a LightCycler 480 Instrument I (Roche Applied Science). The human GAPDH RNA was amplified in parallel as the internal control for normalization purpose. The specific primers used are as follows: ABCG2 (forward) 5′-TTTCCAAGCGTTCATTCAAAAA-3′ (reverse) 5′-TACGACTGTGACAATGATCTGAGC-3′ (To et al., 2008): GAPDH (forward) 5′-AGCCACATCGCTCAGACAC-3′ (reverse) 5′-GTTCAAACTTCTGCTCCTGA-3′; MDR1 (forward) 5′-CCCATCATTGCAATAGCAGG-3′ (reverse) 5′-GTTCAAACTTCTGCTCCTGA-3′. PCRs were performed at 95°C for 5 min, followed by 50 cycles of 95°C for 10 s and 60°C for 10 s. Fluorescence signal was acquired at the end of the elongation step of every PCR cycle (72°C for 10 s) to monitor the increasing amount of amplified DNA. ΔCt was calculated by subtracting the Ct of GAPDH from that of ABCG2. ΔΔCt was then calculated by subtracting the ΔCt of the untreated cells from the ΔCt of the treated cells. Fold change of gene expression was calculated by the equation 2-ΔΔCt.
Western blot analysis
Whole cell lysates prepared from MCF-7 or MCF-7 FLV1000 were separated by SDS-PAGE and subjected to immunoblot analysis with the respective antibodies (ABCG2, Kamiya Biomedical, Seattle, WA, USA; PPARγ, Akt and pAkt, Cell Signaling Technology, Danvers, MA, USA; PTEN and GAPDH, Abcam, Cambridge, MA, USA respectively). The blots were developed with ECL Plus Western Blotting Detection System (GE Healthcare, Piscataway, NJ, USA) and analysed with the FluroChem Q imaging system (Alpha Innotech, San Jose, CA, USA).
RNA interference
To construct a small interference hairpin-loop (sh) silencing vector against the human PPARγ, two complementary oligonucleotides were synthesized, annealed and ligated into the Bgl II and EcoR I site of the pU6 vector (a kind gift from Dr Chen Yang Chao, School of Biomedical Sciences, CUHK, Hong Kong). The sequences were 5′-TGAATTATCTGATTGAGGCTTAttcaagagaTAAGCTTCAATCGGATGGTTCTTTTTTC-3′ (forward) and 5′-TCGAGAAAAAAGAACCATCCGATTGAAGCTTAtctcttgaaTAAGCCTCAATCAGATAATTCA-3′ (reverse), which contained the oligonucleotide encoding a 20-mer hairpin sequence specific to the human PPARγ mRNA [designed by using the siRNA Selection Program at the Whitehead Institute for Biomedical Research (http://sirna.wi.mit.edu)] (Yuan et al., 2004), a TTCAAGAGA loop sequence separating the two complements, and a TTTTT terminator at the 3′ end. A negative control shRNA vector targeting firefly luciferase (pU6-Luc) was constructed similarly with a target sequence of 5′-TGCGCTGCTGGTGCCAACCCTATTCT-3′ (To et al., 2012). MCF-7 and MCF-7 FLV1000 cells were transfected with pU6-Luc or pU6-PPARγ using Lipofectamine 2000 (Invitrogen). Silencing efficiency was assessed by reverse transcription (RT)-PCR and immunoblot analysis.
Flow cytometry
The cell surface ABCG2 expression and ABCG2-mediated efflux activity (i.e. transport activity) in MCF-7 FLV1000 cells with or without the PPARγ agonist pretreatment (24 h) were determined by flow cytometric assays as described previously (Robey et al., 2004). To measure the cell surface ABCG2 expression, trypsinized cells were incubated in 2% BSA/Dulbecco's PBS with either phycoerythrin-conjugated anti-ABCG2 antibody 5D3 (eBioscience, San Diego, CA, USA) or phycoerythrin-conjugated mouse IgG2b negative control antibody (eBioscience) according to manufacturer's instructions for 30 min at room temperature. The anti-ABCG2 5D3 antibody recognizes an extracellular epitope of the protein (Ozvegy-Laczka et al., 2005; Polgar et al., 2010), therefore its signal represents cell surface ABCG2 expression. The cells were then washed with Dulbecco's PBS and subsequently analysed. Surface expression of ABCG2 was calculated as the difference in mean channel numbers between the 5D3 antibody and the negative control antibody histograms. ABCG2 transport activity was also examined in the PPARγ agonists-pretreated cells. MCF-7 and MCF-7 FLV1000 after 24-h pretreatment of PPARγ agonists (telmisartan 10 μM, pioglitazone 5 μM, rosiglitazone 25 μM) were trypsinized and were incubated in 1 μM PhA with or without 10 μM ABCG2-specific inhibitor FTC in complete medium (phenol red-free RPMI 1640 with 10% FBS) at 37°C in 5% CO2 for 30 min. Subsequently, the cells were washed with cold complete medium and then incubated for 1 h at 37°C in PhA-free medium continuing with FTC to generate the FTC/efflux histogram, or without FTC to generate the efflux histogram. The FTC-inhibitable PhA efflux was determined as the difference in mean fluorescence intensity (ΔMFI) between the FTC/efflux and efflux histograms, which indicates the ABCG2-mediated transport activity. The evaluation of ABCG2 transport activity was also repeated by replacing PhA with a fluorescent anticancer drug (mitoxantrone 10 μM). Cells were finally washed with cold Dulbecco's PBS and placed on ice in the dark until analysis by flow cytometry.
Telmisartan, pioglitazone and rosiglitazone were also evaluated for direct competition with PhA for ABCG2-mediated efflux. In these experiments, an ABCG2-stable transfected HEK293 and an ABCG2-overexpressing drug resistant MCF-7 FLV1000 cell line were used. The cells, without drug pretreatment, were incubated directly with the three individual PPARγ agonists in complete medium for 30 min at 37°C. Subsequently, the cells were washed with cold complete medium and then incubated for 1 h at 37°C in PhA-free medium continuing with the same PPARγ agonist to obtain the efflux histogram. The ABCG2-specific inhibitor FTC was used as a control for comparison. Inhibitors of the transporter will shift the inhibitor/efflux histogram to the right, indicating retention of the fluorescent substrate in the cells.
Samples were analysed on an LSRFortessa Cell Analyzer (BD Biosciences, San Jose, CA, USA). Phycoerythrin fluorescence was detected with a 488-nm argon laser and a 585-nm bandpass-filter. PhA fluorescence was detected with a 488-nm argon laser and a 670-nm bandpass filter. Mitoxantrone fluorescence was detected with a 635-red diode laser and a 670-nm bandpass filter. At least 10 000 events were collected for all flow cytometry studies. Cell debris was eliminated by gating on forward versus side scatter and dead cells were excluded based on propidium iodide staining. All assays were performed in three independent experiments.
Analysis of transporter inhibition kinetics
The inhibition kinetic of the ABCG2-mediated efflux of PhA (an ABCG2 specific substrate) by the tested PPARγ agonists was followed as described previously with minor modification (Zhang et al., 2010). Briefly, ABCG2-stably transfected HEK293/ABCG2 cells were incubated with various concentration of PhA (1, 2, 5 and 10 μM) in the presence of four designated concentrations of the PPARγ agonists for 3 h at 37°C. The cells were then collected, centrifuged and washed once with cold PBS, and resuspended in the medium without PhA in the absence or presence of the PPARγ agonists. Subsequently, cells were incubated for 10 min at 37°C to allow for efflux, centrifuged and washed three times with cold PBS. In the control samples, the incubations were kept at 0°C. Finally, the intracellular concentration of PhA was determined by flow cytometric analysis as above. The quantity of PhA efflux by ABCG2 was calculated by subtracting values obtained at 37°C from that at 0°C. The inhibitory effect of the three PPARγ agonists on ABCG2 was then analysed by Dixon plot. In the Dixon plot, the Ki was estimated from the linear regression of reciprocal plot of PhA efflux rate versus the PPARγ agonist concentration.
Immunofluorescence
Immunofluorescence studies were performed as previously described (Polgar et al., 2004). Briefly, cells were seeded on sterile glass coverslips in 6-well culture dishes and grown overnight. Drug treatments with the various PPARγ agonists (24 h) were carried out in these cells growing on glass coverslips. Afterwards, the cells were fixed with 4% formaldehyde and permeabilized with ice-cold methanol, and followed by blocking in a buffer containing 2 mg mL−1 BSA, 0.1% Triton-X100 and 5% goat serum. Immunostaining was performed by incubation with a 1:100 dilution of the mouse monoclonal anti-ABCG2 antibody, BXP-21 (Kamiya Biomedical) and a 1:1000 dilution of the rabbit polyclonal anti-calnexin antibody (Santa Cruz Biotechnology, Dallas, TX, USA) for 2 h at room temperature. Non-specific antibody binding was then removed by washing twice in PBS. This was followed by incubation with secondary Rhodamine-conjugated donkey anti-mouse antibody and FITC-conjugated donkey anti-rabbit antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Nuclei were counterstained with the DNA dye DAPI at 0.5 μg mL−1. Confocal images were generated on an Olympus FV1000-ZCD laser scanning confocal system fitted with an IX81 inverted microscope.
Results
PPARγ agonists sensitized ABCG2-overexpressing resistant cancer cells
The cytotoxicity of mitoxantrone (ABCG2 substrate) or cisplatin (non-ABCG2 substrate) was evaluated in the absence or presence of telmisartan, pioglitazone or rosiglitazone in MCF-7 and its ABCG2-overexpressing resistant MCF-7 FLV1000 cells. This MCF-7 FLV1000 cell line has been routinely used as a reliable model for studying ABCG2-mediated multidrug resistance because only ABCG2 is up-regulated among all other major transporters (Robey et al., 2001). The concentration of the PPARγ agonists tested alone in the assays did not affect cell growth. Of note, pioglitazone was only used at concentrations up to 5 μM, which is limited by its aqueous solubility. As summarized in Table 2012 (upper panel), all three PPARγ agonists tested were able to sensitize MCF-7 FLV1000 cells to mitoxantrone but not a non-ABCG2 substrate cisplatin. Similar potentiation of mitoxantrone cytotoxicity was also obtained in an ABCG2-stably transfected HEK293 human kidney embryonic cell line (Table 2012, lower panel), albeit a lower degree of resistance reversal was attained.
Table 1.
Cell proliferation was determined by SRB assay as described in Materials and Methods. Data are presented as means ± SDs from at least three independent experiments performed in quadruplicate. The fold reversal of resistance was calculated by dividing the IC50 for mitoxantrone (Top Panel) or cisplatin (Bottom Panel) in the absence of PPARγ agonist by that obtained in the presence of PPARγ agonist. The specific ABCG2 inhibitor, FTC, was used as the positive control. The three PPARγ agonists when tested alone, telmisartan, pioglitazone and rosiglitazone, did not affect cell proliferation at concentrations below 20 μM, 5 μM and 60 μM respectively.
| IC50 ± SD (fold reversal) | ||||
|---|---|---|---|---|
| MCF-7 | MCF-7 FLV1000 (ABCG2-overexpression) | HEK293/pcDNA3 (stably transfected) | HEK293/ABCG2 (stably transfected) | |
| Mitoxantrone alone | 0.0097 ± 0.0035 μM | 2.116 ± 0.261 μM | 0.83 ± 0.42 nM | 46.2 ± 3.5 nM |
| +FTC 5 μM | 0.0100 ± 0.0021 μM (1.0) | 0.023 ± 0.010 μM (92) | 0.79 + 0.37 nM (1.1) | 1.08 ± 0.36 nM (42.8) |
| +Telmisartan 1 μM | 0.0152 ± 0.0251 μM (0.6) | 0.378 ± 0.101* μM (5.6) | 1.42 ± 0.69 nM (0.6) | 28.8 ± 6.9 nM (1.6) |
| +Telmisartan 2 μM | 0.0087 ± 0.0022 μM (1.1) | 0.168 ± 0.038* μM (12.6) | 1.80 ± 0.72 nM (0.5) | 9.6 ± 2.1* nM (4.8) |
| +Telmisartan 10 μM | 0.0110 ± 0.0260 μM (0.9) | 0.048 ± 0.035* μM (44.1) | 1.08 ± 0.63 nM (0.8) | 1.6 ± 0.5* nM (28.9) |
| +Pioglitazone 1 μM | 0.0109 ± 0.0312 μM (0.9) | 1.519 ± 0.253 μM (1.4) | 0.94 ± 0.52 nM (0.9) | 35.2 ± 5.4 nM (1.3) |
| +Pioglitazone 2 μM | 0.0081 ± 0.0045 μM (1.2) | 0.369 ± 0.062* μM (5.7) | 0.88 ± 0.49 nM (0.9) | 24.1 ± 3.6 nM (1.9) |
| +Pioglitazone 5 μM | 0.0042 ± 0.0044 μM (2.3) | 0.162 ± 0.031* μM (13.1) | 1.12 ± 0.46 nM (0.7) | 10.5 ± 2.8* nM (4.4) |
| +Rosiglitazone 1 μM | 0.0126 ± 0.0035 μM (0.8) | 0.175 ± 0.032* μM (12.1) | 0.86 ± 0.26 nM (1.0) | 29.5 ± 3.2 nM (1.6) |
| +Rosiglitazone 10 μM | 0.0082 ± 0.0041 μM (1.2) | 0.119 ± 0.031* μM (17.8) | 1.19 ± 0.35 nM (0.7) | 9.2 ± 2.4* nM (5.0) |
| +Rosiglitazone 50 μM | 0.0039 ± 0.0042 μM (2.5) | 0.095 ± 0.021* μM (22.3) | 1.18 ± 0.48 nM (0.7) | 4.8 ± 1.0* nM (9.6) |
| Cisplatin alone | 11.23 ± 2.11 μM | 12.45 ± 1.43 μM | 2.02 ± 0.34 μM | 2.15 ± 0.86 μM |
| +FTC 5 μM | 12.56 ± 1.87 μM (1.0) | 11.98 ± 1.37 μM (1.0) | 1.99 ± 0.36 μM (1.0) | 2.35 ± 0.76 μM (0.9) |
| +Telmisartan 2 μM | 10.98 ± 1.87 μM (1.0) | 13.15 ± 2.11 μM (0.9) | 2.26 ± 0.65 μM (0.9) | 1.98 ± 0.74 μM (1.1) |
| +Telmisartan 10 μM | 11.25 ± 2.09 μM (1.0) | 12.78 ± 1.88 μM (1.0) | 2.19 ± 0.49 μM (0.9) | 2.05 ± 0.68 μM (1.0) |
| +Pioglitazone 2 μM | 10.76 ± 2.02 μM (1.0) | 11.67 ± 1.54 μM (1.1) | 1.86 ± 0.59 μM (1.1) | 2.11 ± 0.48 μM (1.0) |
| +Pioglitazone 5 μM | 12.98 ± 1.72 μM (0.9) | 12.19 ± 1.76 μM (1.0) | 2.01 ± 0.43 μM (1.0) | 1.98 ± 0.56 μM (1.1) |
| +Rosiglitazone 10 μM | 11.76 ± 2.14 μM (1.0) | 12.64 ± 1.76 μM (1.0) | 1.59 ± 0.86 μM (1.3) | 2.08 ± 0.59 μM (1.0) |
| +Rosiglitazone 50 μM | 10.88 ± 1.65 μM (1.0) | 11.54 ± 1.28 μM (1.1) | 1.89 ± 0.46 μM (1.1) | 2.16 ± 0.47 μM (1.0) |
P < 0.05, versus that obtained in the absence of inhibitor
PPARγ agonists inhibited ABCG2-mediated drug efflux
To understand the underlying mechanism(s) of resistance reversal, the competition between the PPARγ agonists and a fluorescent ABCG2 substrate (PhA) for efflux was studied in the ABCG2-overexpressing MCF-7 FLV1000 and HEK293/ABCG2 cells by flow cytometric analysis. The read-out of the assay is the accumulation of the fluorescent ABCG2 substrate (PhA) after a 1-h drug-free efflux. Inhibition of ABCG2-mediated efflux is indicated by a shift to higher intracellular fluorescence signal. As illustrated in Figure 1, all three PPARγ agonists were found to inhibit the efflux of the fluorescent ABCG2 substrate in a concentration-dependent manner, with telmisartan being the most potent one followed by rosiglitazone and pioglitazone. Using the specific and potent ABCG2 inhibitor (FTC) as a reference for comparison, the inhibition of ABCG2 by the three PPARγ agonists was found to be only mild to moderate.
Figure 1.
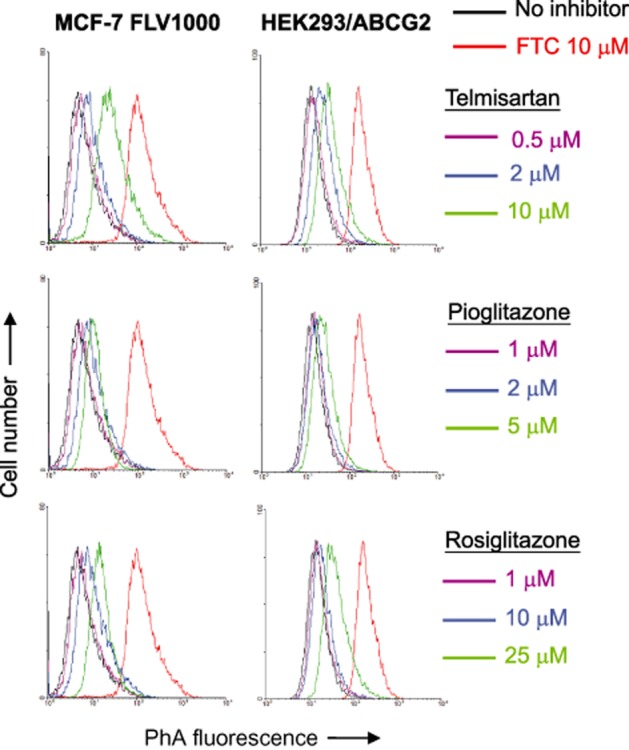
Direct inhibition of ABCG2-mediated pheophorbide A (PhA) efflux in ABCG2-overexpressing MCF-7 FLV1000 and ABCG2-stably transfected HEK293/ABCG2 cells. Cells (no pretreatment) were incubated with 1 μM PhA alone (black), 1 μM PhA with the various PPARγ agonists at the indicated concentrations (various colours) or 1 μM PhA with 10 μM FTC (red) at 37°C for 30 min. PhA fluorescence retention in the cells after a 1-h PhA-free efflux was measured by flow cytometry. Representative histograms from three independent experiments are shown.
Inhibition kinetics of ABCG2-mediated PhA efflux by PPARγ agonists
To further elucidate the mode of interaction between the PPARγ agonists with ABCG2, the efflux of the fluorescent ABCG2 substrate (PhA) at four different concentrations was studied in the absence and presence of the PPARγ agonists. The resulting inhibition curves were transformed to a Dixon plot and analysed by linear regression. As demonstrated in the Dixon plots (Figure 2), all curves intersect at the x-axis, suggesting non-competitive inhibition. The Ki values for telmisartan, pioglitazone and rosiglitazone were found to be 14.3, 28.5 and 25.9 μM, respectively, indicating a relatively weak interaction with ABCG2. For comparison, we have identified axitinib (a molecular targeting tyrosine kinase inhibitor) as a potent ABCG2 inhibitor from an independent project with a Ki of 0.85 μM (detailed graph is not shown).
Figure 2.
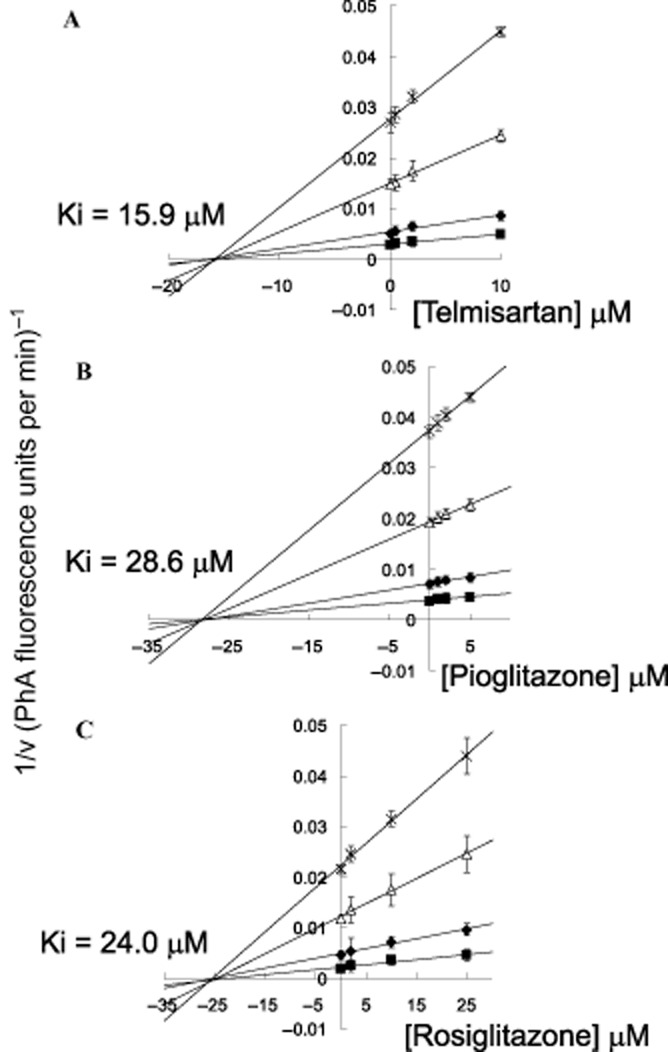
Inhibition kinetic of ABCG2-mediated efflux of PhA by PPARγ agonists. HEK293/ABCG2 cells were incubated with different concentrations of PhA (1, 2, 5, and 10 μM) in the presence of four concentrations of telmisartan (0, 0.5, 2, or 10 μM), pioglitazone (0, 1, 2, or 5 μM) or rosiglitazone (0, 2, 10, or 25 μM) for 3 h. After a brief wash, the cells were allowed to incubate in PhA-free medium continuing with or without the PPARγ agonist incubations to allow for efflux. The quantity of PhA efflux was measured for 10 min by flow cytometry, which was calculated by subtracting the fluorescence signal obtained at 37°C from that at 0°C. The reciprocal of PhA efflux rate is plotted against the concentration of the PPARγ agonist using the Dixon plot. The intersection point represents the Ki value. Since the lines converge on the x axis, the PPARγ agonists tested are likely non-competitive inhibitors of ABCG2. Each data point is presented as the mean ± SEM from three independent experiments.
ABCG2 mRNA and total cellular protein levels were not affected by PPARγ agonists
Besides the direct inhibition of ABCG2 transport function, the reversal of ABCG2-mediated MDR could also be mediated by decreased transporter expression. The effect of the three PPARγ agonists on ABCG2 mRNA and protein expression was examined in MCF-7 and MCF-7 FL1000 cells by RT-real time PCR and immunoblot analysis. Our results indicated that 24-h treatment with the three PPARγ agonists (at concentrations up to 10, 5, and 25 μM for telmisartan, pioglitazone and rosiglitazone, respectively) did not affect ABCG2 expression at both mRNA and protein levels (Figure 3).
Figure 3.
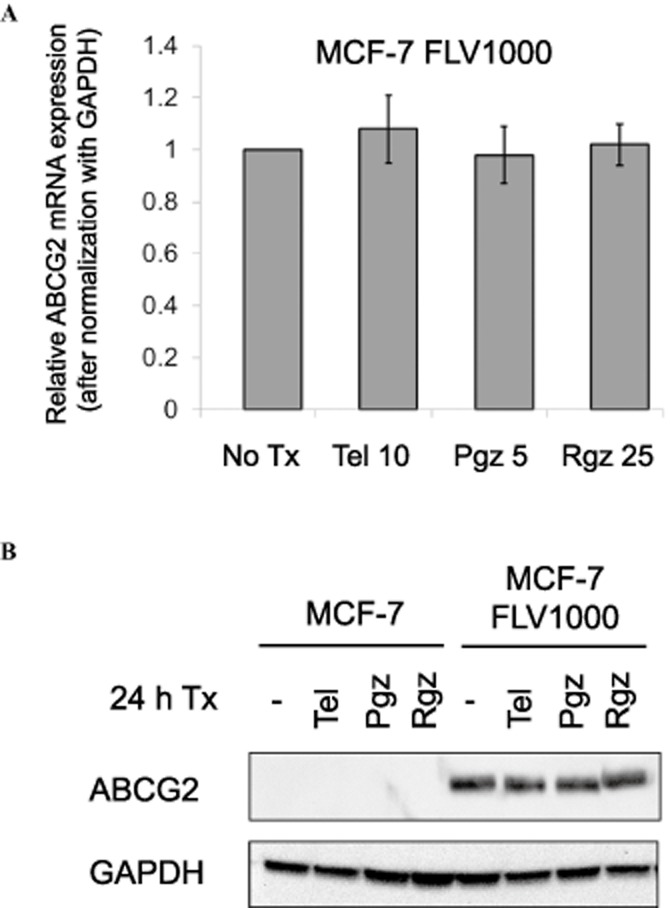
Quantitative PCR and immunoblot analysis showing that treatment with PPARγ agonists did not affect ABCG2 expression at both mRNA and total cellular protein levels. (A) PCR analysis in MCF-7 FLV1000 cells treated with the three PPARγ agonists for 24 h [at concentrations found to inhibit ABCG2-mediated transport; telmisartan (Tel): 10 μM; pioglitazone (Pgz): 5 μM; rosiglitazone (Rgz): 25 μM]. mRNA expression were normalized with GAPDH. Relative ABCG2 mRNA levels were expressed relative to that in the untreated resistant MCF-7 FLV1000 cells. ABCG2 expression in the parental MCF-7 cells is only about 1/1500 times that in the resistant MCF-7 FLV1000 cells, which was also not affected by the PPARγ agonists treatment (not shown in the figure). Mean ± SD from three independent experiments is shown. (B) Immunoblot analysis under the same experimental conditions as in (A) above.
PPARγ agonists reduce surface expression of ABCG2 protein and transport activity
Since the direct inhibition of ABCG2 transport activity by the three PPARγ agonists was only fairly moderate (Figure 1), it is unlikely to contribute to the remarkable enhancement of mitoxantrone cytotoxic effect especially in the ABCG2-overexpressing MCF-7 FLV1000 cells (Table 2012). As shown in Figure 3, total cellular ABCG2 expression was not affected; therefore, we sought to investigate the possible change in localization of the transporter upon PPARγ agonists treatment.
By flow cytometric analysis, ABCG2 surface expression was found to be substantially reduced in the PPARγ agonists 24-h pretreated MCF-7 FLV1000 cells (Figure 4). The effect on the parental MCF-7 cells was very minimal because of the low ABCG2 expression level in these cells. Consistent with the reduction in ABCG2 surface expression, FTC-inhibitable efflux of two ABCG2 fluorescent substrates [PhA and mitoxantrone (mito)] in MCF-7 FLV1000 (i.e. ΔMFI), representing the ABCG2-mediated transport activity, was also significantly decreased in the PPARγ agonists 24-h treated MCF-7 FLV1000 cells (Figure 5; left panel – PhA and right panel – mito).
Figure 4.
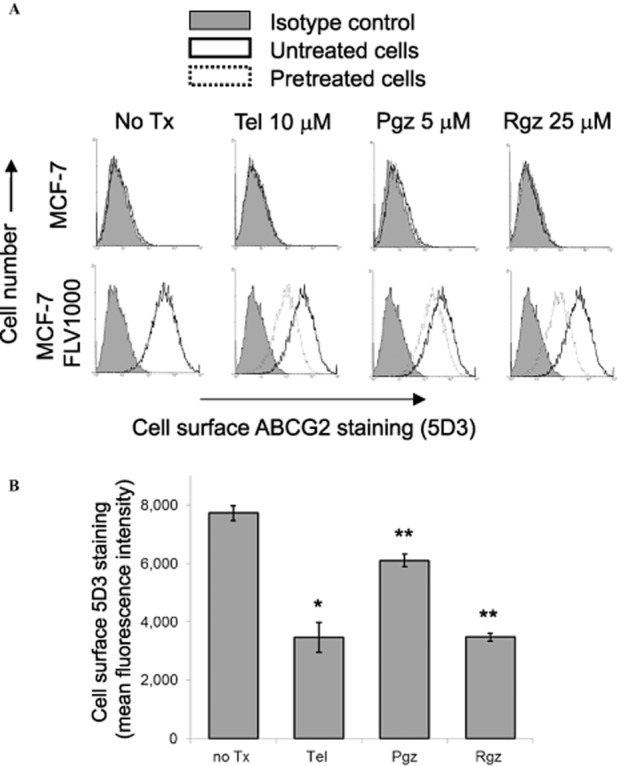
ABCG2 surface expression of PPARγ agonists-treated MCF-7 and its ABCG2-overexpressing MCF-7 FLV1000 cells (24-h treatment). (A) Representative histograms showing the cell surface staining of ABCG2 by the 5D3 monoclonal antibody, which recognizes an extracellular epitope of ABCG2, of the untreated and PPARγ agonists-treated cells. Cells were trypsinized and incubated for 30 min in phycoerythrin (PE)-labelled negative control antibody (shaded histogram) or 5D3 antibody (solid line: untreated cells; dashed line: PPARγ agonists-treated cells) and analysed in a FACSsort flow cytometry. The distance between the 5D3 histogram (solid or dashed lines representing untreated and pretreated cells, respectively) and the shaded negative control antibody histogram provide an indication of the amount of ABCG2 protein expressed on the cell surface. The assays were repeated in three independent experiments. (B) Cell surface 5D3 staining of the resistant MCF-7 FLV1000 cells before and after a 24-h treatment with the indicated PPARγ agonists (telmisartan 10 μM, pioglitazone 5 μM, or rosiglitazone 25 μM) was quantified by subtracting the fluorescence signal from 5D3 labelling by that from the control IgG isotype labelling. Mean ± SD from three independent experiments is shown. *, P < 0.05; **, P < 0.01; compared with the untreated MCF-7 FLV1000 cells.
Figure 5.
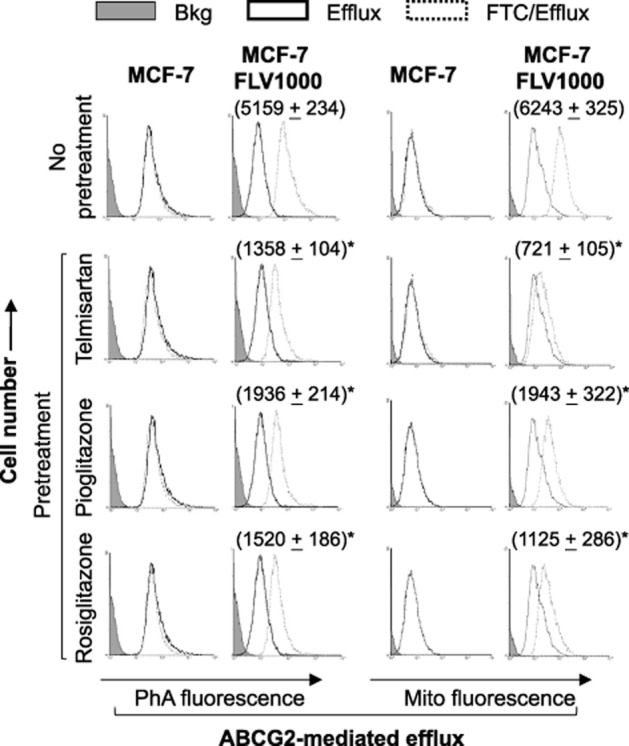
Flow cytometric analysis of ABCG2 transport activity in cells pretreated with PPARγ agonists (telmisartan 10 μM; pioglitazone 5 μM; rosiglitazone 25 μM; 24-h treatment). Left panel: Cells were incubated for 30 min in complete media containing 1 μM pheophorbide A (PhA) with or without the ABCG2-specific inhibitor FTC (solid line – efflux/without FTC; dashed line – efflux/with FTC). The fluorescence signal between the efflux/with and efflux/without FTC histograms (i.e. ΔMFI) represents the ABCG2-mediated transport activity in the cells. Right panel: The same analysis as the left panel but PhA was replaced with a fluorescent anticancer drug mitoxantrone 10 μM (mito). Negligible ΔMFI was obtained for the parental MCF-7 cells. ΔMFI values for the resistant MCF-7 FLV1000 under the different PPARγ agonist treatments are reported in parenthesis (* P < 0.05, compared with untreated MCF-7 FLV1000 cells).
PPARγ agonists led to the internalization of ABCG2 into cell cytoplasm
The decrease in the cell surface expression of ABCG2 (Figure 4) without a significant change in the total protein level (Figure 3) suggest a possible alteration in the cellular localization of the transporter after treatment with the three PPARγ agonists. To further investigate the subcellular localization of ABCG2 after PPARγ agonist treatment, cells were examined by immunofluorescence confocal microscopy. The untreated MCF-7 FLV1000 cells showed primarily plasma membrane staining with little intracellular signal (Figure 6). However, both intracellular and cell surface staining could be observed in the PPARγ agonists-treated cells (Figure 6). The overlapping staining of ABCG2 (red) with the ER marker (green) in MCF-7 FLV1000 cells treated with the three PPARγ agonists provides strong evidence for the subcellular location of ABCG2. The indispensable role of PPARγ activation in the internalization of ABCG2 was further demonstrated by the recovery of cell surface expression of ABCG2 via genetic knockdown (by siRNA; Figure 6) or chemical antagonism of PPARγ (by GW9662 200 nM; Supporting information Figure S1).
Figure 6.
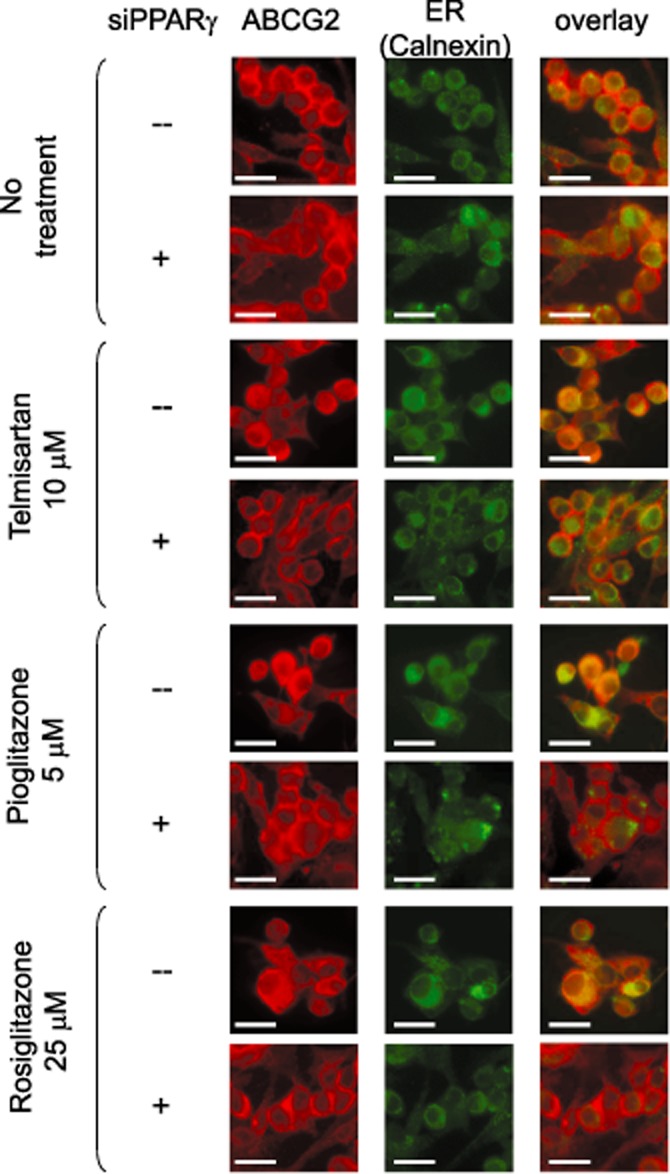
Immunofluorescence analyses of the translocation of ABCG2 in MCF-7 FLV1000 cells before and after treatment with PPARγ agonists. Confocal microscopy of untreated MCF-7 FLV1000 cells or cells after a 24-h treatment with telmisartan (10 μM), pioglitazone (5 μM), or rosiglitazone (25 μM) was performed following fixation with paraformaldehyde and permeabilization with ice-cold methanol. Immunostaining was conducted for 2 h at room temperature with the anti-ABCG2 BXP-21 antibody and anti-calnexin (ER marker) antibody. Nuclei were counter-stained with DAPI (blue), which is not shown for clarity. Control untreated MCF-7 FLV1000 cells shows predominantly cell surface expression of ABCG2 (red). In contrast, MCF-7 FLV1000 cells treated with the various PPARγ agonists exhibited both cell surface and intracellular compartment localization of ABCG2. This is illustrated by the colocalization of ABCG2 with the ER marker calnexin (green) after drug treatment. When PPARγ was silenced by siRNA, the predominant cell surface expression of ABCG2 was restored in PPARγ agonists-treated MCF-7 FLV1000 cells. Scale bar, 50 μm.
PPARγ agonists reverse the PTEN loss in MCF-7 FLV1000 cells and inhibit Akt activation, presumably leading to internalization of ABCG2
Since the PTEN/PI3K/Akt signalling pathway has been proposed to regulate plasma membrane localization of ABCG2 (Bleau et al., 2009), PTEN status and inhibition of Akt were examined in the PPARγ agonists treated MCF-7 and MCF-7 FLV1000 cells. A remarkably lower PTEN protein expression was noted in the resistant MCF-7 FLV1000 than in the parental MCF-7 cells (Figure 7). Interestingly, the down-regulated PTEN level in the MCF-7 FLV1000 cells was found to be elevated by treatment with all three PPARγ agonists tested. This up-regulation of PTEN was dependent on PPARγ because it was abolished by the chemical PPARγ antagonist (GW9662, 200 nM) or genetic knockdown by PPARγ siRNA. Active PTEN is known to inhibit the activation of its downstream effector Akt (i.e. phosphorylation) via its lipid phosphatase activity to inhibit cell proliferation. According to our immunoblot analysis (Figure 7), phosphorylated Akt (p-Akt) was only detected at a basal level in MCF-7 cells because of normal cellular proliferation whereas elevated p-Akt was observed in the resistant MCF-7 FLV1000 cells probably because of PTEN down-regulation in the latter cells. Intriguingly, treatment of MCF-7 FLV1000 cells with all three PPARγ agonists resulted in a significant decrease in p-Akt, concomitant with the increase in PTEN expression described above. The decrease in p-Akt by the PPARγ agonists is PPARγ-dependent because it was not observed in cells concomitantly exposing to the PPARγ antagonist GW9662 or PPARγ specific siRNA. The involvement of PTEN/PI3K/Akt pathway in the internalization of ABCG2 by PPARγ agonists was further examined by the use of PI3K inhibitor (LY294002). As illustrated in Figure 8, treatment of MCF-7 FLV1000 by LY294002 for 16 h was found to cause concentration-dependent translocation of ABCG2 from plasma membrane to the subcellular compartment (Figure 8A and B), mirroring the effect of the three PPARγ agonists (Figure 6).
Figure 7.
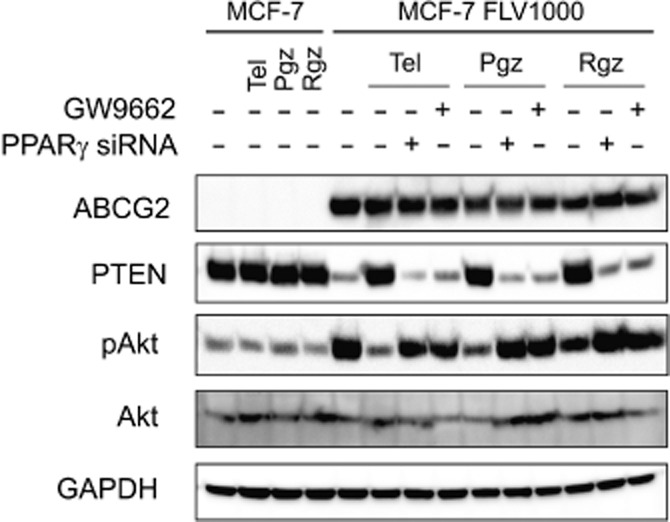
Immunoblot analysis showing that ABCG2 overexpression in the resistant MCF-7 FLV1000 cells is associated with PTEN loss and activation of Akt. The three PPARγ agonists tested (Tel: telmisartan 10 μM; Pgz: pioglitazone 5 μM; Rgz: rosiglitazone 25 μM; 24-h treatment) were found to correct this PTEN loss and decrease Akt phosphorylation. The specific effect of the PPARγ agonists on PTEN and Akt was confirmed by PPARγ knockdown with the chemical inhibitor GW9662 (200 nM) and a PPARγ specific siRNA. Representative results from three independent and reproducible experiments were shown.
Figure 8.
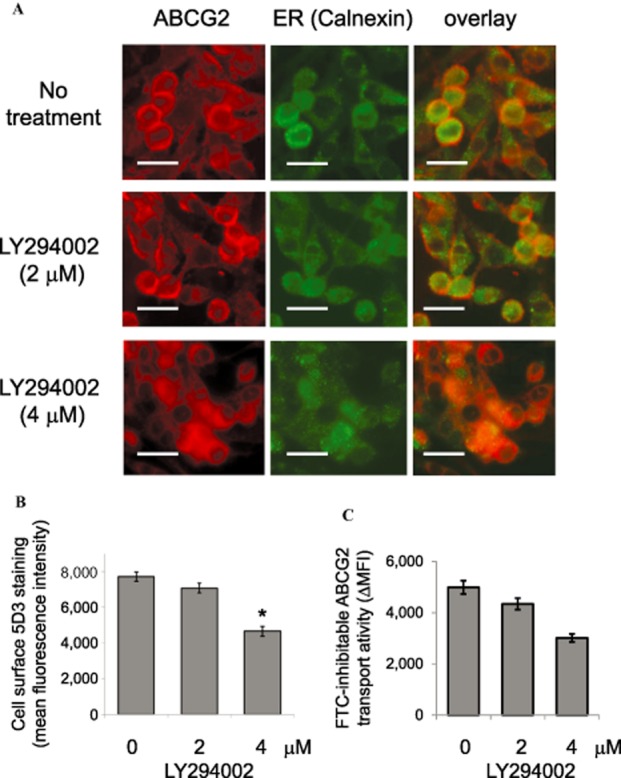
Immunofluorescence and flow cytometric analyses of the translocation of ABCG2 in MCF-7 FLV1000 cells before and after treatment with the PI3K inhibitor LY294002. (A) Confocal microscopy of MCF-7 FLV1000 cells after a 16-h treatment with 2 μM or 4 μM of LY294002 was performed as described in Figure 6. Representative images taken from three independent experiments are shown. Scale bar, 50 μm. (B) Cell surface 5D3 staining of MCF-7 FLV1000 cells after treatment with LY294002 was performed as described in Figure 4. (C) ABCG2 transport activity (FTC-inhibitable PhA efflux) in MCF-7 FLV1000 cells treated with LY294002 (16 h; 2 or 4 μM) was examined as in Figure 5. * P < 0.05, compared with untreated MCF-7 FLV1000 cells.
Discussion and conclusions
Various ABC transporters, including ABCG2, are often overexpressed in multidrug resistance cancer cells. There has been great interest in the development of novel inhibitors towards these ABC transporters as a strategy to circumvent multidrug resistance. Post-translational regulation of ABCG2 has been extensively studied in relation to the PI3K/PTEN/Akt signalling pathway in a variety of cancer cells (Mogi et al., 2003; Aust et al., 2004; Takada et al., 2005; Hu et al., 2008; Bleau et al., 2009; Goler-Baron et al., 2012). Inhibition of this pathway appears to facilitate the translocation of ABCG2 from plasma membrane to intracellular compartment, thus leading to apparent circumvention of the transporter-mediated drug resistance. Since the PI3K/Akt pathway is also regulating a number of other oncogenic effectors driving tumour growth and survival (Kitamura et al., 2001; Chang et al., 2003), inhibition of this pathway may be especially beneficial and may represent a druggable target for sensitization of cancer cells to chemotherapy.
PTEN is an important tumour suppressor gene that inhibits the PI3K/Akt pathway to mediate cell cycle arrest and apoptosis (Stambolic et al., 1998; Salmena et al., 2008). While PTEN down-regulation is frequently observed in cancer, we have observed a remarkable decrease in PTEN expression in our resistant and ABCG2-overexpressing cancer cell line model relative to the respective parental cells (Figure 7). It follows that strategies to up-regulate PTEN level may be used to specifically target these ABCG2-overexpressing resistant cancer cells. Since PPARγ is known to up-regulate PTEN transcriptionally (Patel et al., 2001), three PPARγ agonists (telmisartan, rosiglitazone and pioglitazone) which have been in clinical use were investigated for their modulation on the PTEN/PI3K/Akt pathway and possible circumvention of ABCG2-mediated drug resistance. Telmisartan is the only member of the ARB class of antihypertensive agents exhibiting PPARγ activity whereas the TZD-class antidiabetic drugs (i.e. rosiglitazone and pioglitazone) represent the first few synthetic compounds investigated as PPARγ ligands (Zhang et al., 1996). Our goal is to generalize some insights for further clinical development. To this end, the MDR reversing concentration of the three PPARγ agonists revealed from the present study (Table 2012) is in fact achievable in vivo. According to a few recent pharmacokinetic studies, Cmax of telmisartan 80 mg, pioglitazone 40 mg and rosiglitazone 8 mg was found to be around 1.3 (Smith et al., 2000), 2.4 (Jaakkola et al., 2005) and 1.7 μM (Cox et al., 2000) respectively.
Our working model is presented in Figure 9. Briefly, the PTEN/PI3K/Akt signalling pathway is believed to regulate the subcellular localization of ABCG2 in cancer cells. In our resistant cell line model, significant PTEN down-regulation was noted, which is believed to allow activation of Akt for predominant ABCG2 expression on cell surface. PPARγ agonists, by transcriptionally up-regulating PTEN, inhibit Akt phosphorylation and lead to translocation of ABCG2 from the plasma membrane to the intracellular compartment.
Figure 9.
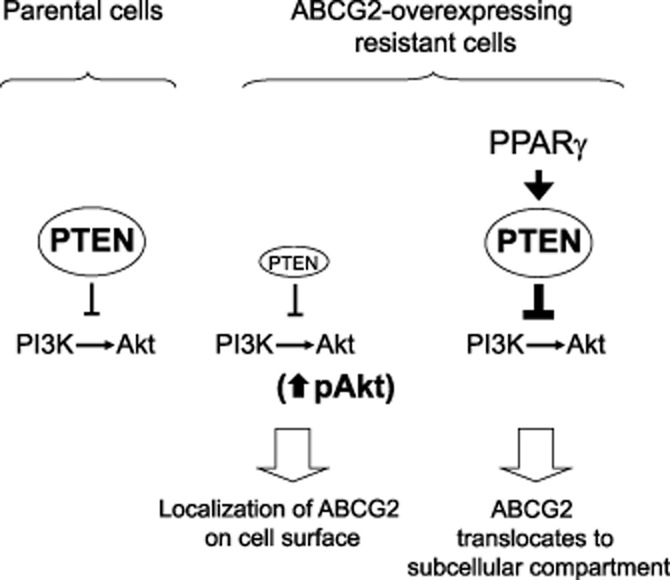
A working model illustrating the involvement of PTEN loss and the activation of Akt in the localization of ABCG2 to the cell surface in the resistant MCF-FLV1000 cells. While the aetiology for the dramatic decrease in PTEN expression in the resistant cells is still not clear, PPARγ agonist treatments were found to reverse the PTEN loss in the resistant cells, subsequently inhibiting Akt, and leading to the re-localization of ABCG2 from the cell surface to the cytoplasm.
However, there have been some controversies about the involvement of the PI3K/Akt axis in regulating plasma membrane localization of ABCG2 (Hegedus et al., 2012; Imai et al., 2012). In these studies, ABCG2 translocation was not observed upon treatment with inhibitors of the downstream effectors of the PI3K/Akt/mTOR pathway (i.e. PI3K inhibitors: LY294002 and wortmannin; mTOR inhibitor: rapamycin). In our ABCG2-overepressing resistant MCF-7 FLV1000 cell line model, translocation of ABCG2 from cell surface to subcellular compartment (Figure 8A and B) and decreased ABCG2 transport activity (Figure 8C) was observed when cells were treated with LY294002 (a PI3K inhibitor) for 16 h, which mirrors the effect of the three PPARγ agonists tested (Figure 5 and 6). The underlying reason for the discrepancy is not clear. Nonetheless, our observed circumvention of ABCG2-mediated MDR is likely associated with this PPARγ/PTEN/PI3K/Akt pathway because the concomitant administration of the chemical PPARγ antagonist GW9662 or genetic knockdown of PPARγ were found to abolish the observed elevation of PTEN (Figure 7), inhibition of Akt (Figure 7), internalization of ABCG2 (Figures 6 and Supporting information Figure S1) and apparent reversal of ABCG2-mediated resistance (Supporting information Table S1) by the PPARγ agonists.
PPARγ is a ligand-mediated nuclear transcription factor that regulates gene expression by binding to PPARγ response elements (PPRE) within the promoter of its target gene. To this end, three functional PPRE have been identified within a 150-bp long enhancer region located at about 4 kb upstream of the human ABCG2 promoter and were shown to mediate the upregulation of ABCG2 in human myeloid dendritic cells (Szatmari et al., 2006). In our study, ABCG2 expression was not changed in the breast cancer cell lines used (Figure 3) and a few other colon cancer cell lines (data not shown). Various splice variants of the ABCG2 5′-untranslated leader exons have been reported and were shown to be related to alternative promoter utilization (Zong et al., 2006; Campbell et al., 2011; Natarajan et al., 2011). An alternative leader exon E1U located at approximately 73 kb upstream from the reported ABCG2 transcriptional start site, has been identified and studied in bone marrow samples (Campbell et al., 2011). To our knowledge, E1U is the only alternative leader exon identified that spans the PPRE. Coincidently, PPARγ activation has only been shown to increase the expression of the functional ABCG2 transporter in dendritic cells for protection against endogenous substances and xenobiotics (Szatmari et al., 2006). This tissue-specific alternative promoter usage may explain why ABCG2 is not changed in the breast cancer cell line tested in our study.
Telmisartan tested in our study was known to be unique among other ARB class of antihypertensive agents in that it is the only one exhibiting PPARγ agonist activity. Indeed, this PPARγ-mediated pleiotropic effect of telmisartan was shown to cause induction of insulin sensitivity and a reduction of low-density lipoprotein cholesterol (Benndorf and Boger, 2008), which will be beneficial in patients in need for diabetes and/or lipid control. For comparison, three other ARBs including the classical losartan and two newer ones valsartan and irbesartan, which have no or very minimal PPARγ agonist effect (Benson et al., 2004; Schupp et al., 2004), were also investigated for possible circumvention of ABCG2-mediated MDR. As presented in Supporting information Figure S2, all of these ARBs did not affect PTEN expression and consequently no inhibition of Akt was observed in MCF-7 FLV1000. They were also not able to alter the predominant cell surface localization of ABCG2 in the resistant MCF-7 FLV1000 (Supporting information Figure S3). Interestingly, these ARBs were also not found to be direct inhibitors for ABCG2 as demonstrated by flow cytometry-based drug efflux assay (Supporting information Figure S4). The possible pleiotropic effects of the tested PPARγ agonists on other ABC transporters associated with multidrug resistance (MDR-1/P-gp and MRP-1) was also evaluated by quantitative real-time PCR. While basal expression levels of MDR-1/P-gp and MRP-1 in our MCF-7 FLV1000 cell model were both lower than those in the parental MCF-7 cells (by about 50%), they were not altered by the treatment with the PPARγ agonists tested (Supporting information Figure S5). It is noteworthy that pharmacokinetic interaction between telmisartan and other ABCG2 substrate drugs is expected to be fairly minimal because of its weak inhibition on ABCG2-mediated transport activity (Figure 1). The final proof will await clinical pharmacokinetic study in patients concomitantly on telmisartan and other ABCG2 substrate drugs. To this end, although telmisartan has been shown to inhibit significantly P-gp transport (Weiss et al., 2010), it was not found to adversely affect the safety profile of digoxin (a well-known P-gp substrate drug) (Stangier et al., 2000).
In summary, the three PPARγ agonists tested were shown to reverse ABCG2-mediated MDR by (i) exerting moderate inhibitory effect on ABCG2 transport activity; and (ii) facilitating the translocation of ABCG2 from cell surface to intracellular compartments via a PTEN/PI3K/Akt pathway. Since PPARγ agonists are also known to exert tumour suppressor and apoptotic effects by mechanisms other than the PI3K/Akt signalling axis (Bonofiglio et al., 2011), the potential dual mechanism of resistance reversal advocates more in-depth investigation for their optimal clinical use for MDR circumvention.
Acknowledgments
We would like to thank Dr Susan Bates (NCI, NIH, USA) for the ABCG2-overexpressing cell line (MCF-7 FLV1000). This work was partly supported by a direct grant from the Faculty of Medicine, The Chinese University of Hong Kong (2041633).
Glossary
- ARBs
angiotensin II receptor blockers
- MDR
multidrug resistance
- PTEN
phosphatase and tensin homologue deleted on chromosome 10
- TZDs
thiazolidinediones
Conflict of interest
There are no potential conflicts of interest to be disclosed.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12367
Table S1 Cell proliferation data showing that GW9662 alone did not affect cell viability but it abolished the circumvention of ABCG2-mediated resistance in MCF-7 FLV1000 by the PPARγ agonists tested. Data are presented as means ± SDs from at least three independent experiments performed in quadruplicate. The fold reversal of resistance was calculated by dividing the IC50 for mitoxantrone in the absence of PPARγ agonist by that obtained in the presence of PPARγ agonist. *P < 0.05, versus that obtained in mitoxantrone alone in the same cell line.
Figure S1 Immunofluorescence analyses of the translocation of ABCG2 in MCF-7 FLV1000 cells before and after treatment with PPARγ agonists. Confocal microscopy was performed as described in Figure 8. In the presence of the chemical PPARγ antagonist (GW9662, 200 nM), the predominant cell surface expression of ABCG2 was restored in PPARγ agonists-treated MCF-7 FLV1000 cells. Scale bar, 50 μm.
Figure S2 Immunoblot analysis showing that other angiotensin II receptor blockers [ARBs; including losartan (Lo), valsartan (Val) and irbesartan (Irbe)], which have minimal PPARγ agonist effect, did not correct PTEN loss and affect Akt phosphorylation in MCF-7 FLV1000 cells. MCF-7 FLV1000 cells were treated for 24 h with these three ARBs at 50 μM before harvested for immunoblot analysis. Representative results from three independent and reproducible experiments are shown.
Figure S3 Immunofluorescence analyses of the plasma membrane localization of ABCG2 in MCF-7 FLV1000 cells before and after treatment with a few ARBs (losartan, valsartan, and irbesartan; at 50 μM for 24 h) with minimal PPARγ agonist effect. Confocal microscopy was performed as described in Figure 6. (A) Representative images taken from three independent experiments are shown. Predominant cell surface expression of ABCG2 was still observed after treatment with these ARBs. Scale bar, 50 μm. (B) Cell surface 5D3 staining of MCF-7 FLV1000 cells after the indicated treatments was performed as in Figure 4. Mean ± SD from three independent experiments is shown. There is no remarkable change in ABCG2 cell surface expression.
Figure S4 Flow cytometric PhA efflux analysis showing that losartan, valsartan and irbesartan did not directly compete for ABCG2-mediated PhA efflux. The assay was performed as described in Figure 1 in MCF-7 FLV1000 or HEK293/ABCG2 cells incubated for 30 min with the indicated concentrations of the three ARBs tested. PhA fluorescence retention in the cells after a 1-h PhA-free efflux was measured by flow cytometry. Representative histograms from three independent experiments are shown.
Figure S5 Quantitative PCR analysis showing that the tested PPARγ agonists did not affect expression levels of other ABC transporters [(A) MDR-1/P-gp and (B) MRP-1] commonly associated with multidrug resistance. Parental MCF-7 and resistant MCF-7 FLV1000 cells were treated with the three tested PPARγ agonists for 24 h [at concentrations found to inhibit ABCG2-mediated transport; telmisartan (Tel): 10 μM; pioglitazone (Pgz): 5 μM; rosiglitazone (Rgz): 25 μM]. Total RNA was then harvested for RT-PCR analysis. Relative MDR- 1/P-gp and MRP-1 mRNA levels were expressed relative to that in the untreated parental MCF-7 cells, after normalization with GAPDH. It is noted that the basal expression of both MDR-1/P-gp and MRP-1 are lower in the resistant MCF-7 FLV1000 than in the parental MCF-7 cells. The PPARγ agonists tested did not significantly affect MDR-1/P-gp and MRP-1 expression in MCF-7 FLV1000 cells. Mean ± SD from three independent experiments is shown.
References
- Amiri-Kordestani L, Basseville A, Kurdziel K, Fojo AT, Bates SE. Targeting MDR in breast and lung cancer: discriminating its potential importance from the failure of drug resistance reversal studies. Drug Resist Updat. 2012;15:50–61. doi: 10.1016/j.drup.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aust S, Obrist P, Jaeger W, Klimpfinger M, Tucek G, Wrba F, et al. Subcellular localization of the ABCG2 transporter in normal and malignant human gallbladder epithelium. Lab Invest. 2004;84:1024–1036. doi: 10.1038/labinvest.3700127. [DOI] [PubMed] [Google Scholar]
- Benndorf RA, Boger RH. Pleiotropic effects of telmisartan: still more to come? J Hypertens. 2008;26:854–856. doi: 10.1097/HJH.0b013e3282f76481. [DOI] [PubMed] [Google Scholar]
- Benson SC, Pershadsingh H, Ho C, Chittiboyina A, Desai P, Pravenec M, et al. Identification of telmisartan as a unique angiotensin II receptor antagonist with selective PPARgamma-modulating activity. Hypertension. 2004;43:993–1002. doi: 10.1161/01.HYP.0000123072.34629.57. [DOI] [PubMed] [Google Scholar]
- Bleau AM, Hambardzumyan D, Ozawa T, Fomchenko EI, Huse JT, Brennan CW, et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4:226–235. doi: 10.1016/j.stem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonofiglio D, Cione E, Vizza D, Perri M, Pingitore A, Qi H, et al. Bid as a potential target of apoptotic effects exerted by low doses of PPARγ and RXR ligands in breast cancer cells. Cell Cycle. 2011;10:2344–2354. doi: 10.4161/cc.10.14.15917. [DOI] [PubMed] [Google Scholar]
- Bunting KD. ABC transporters as phenotypic markers and functional regulators of stem cells. Stem Cells. 2002;20:11–20. doi: 10.1002/stem.200011. [DOI] [PubMed] [Google Scholar]
- Campbell PK, Zong Y, Yang S, Zhou S, Rubnitz JE, Sorrentino BP. Identification of a novel, tissue-specific ABCG2 promoter expressed in pediatric acute megakaryoblastic leukemia. Leuk Res. 2011;35:1321–1329. doi: 10.1016/j.leukres.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, et al. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003;17:590–603. doi: 10.1038/sj.leu.2402824. [DOI] [PubMed] [Google Scholar]
- Cheng Z, Yang N, Liang W, Yan X, Li L, Pan L. Effect of phosphatase and tensin homology deleted on chromosome 10 (PTEN) gene transfection on reversal of multidrug resistance in K562/ADM cells. Leuk Lymphoma. 2012;53:1383–1389. doi: 10.3109/10428194.2011.650695. [DOI] [PubMed] [Google Scholar]
- Cox PJ, Ryan DA, Hollis FJ, Harris AM, Miller AK, Vousden M, et al. Absorption, disposition, and metabolism of rosiglitazone, a potent thiazolidinedione insulin sensitizer, in humans. Drug Metab Dispos. 2000;28:772–780. [PubMed] [Google Scholar]
- Elstner E, Muller C, Koshizuka K, Williamson EA, Park D, Asou H, et al. Ligands for peroxisome proliferator-activated receptor gamma and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proc Natl Acad Sci U S A. 1998;95:8806–8811. doi: 10.1073/pnas.95.15.8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22:183–198. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- Garcia-Cao I, Song MS, Hobbs RM, Laurent G, Giorgi C, Boer de VC, et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell. 2012;149:49–62. doi: 10.1016/j.cell.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goler-Baron V, Sladkevich I, Assaraf YG. Inhibition of the PI3K-Akt signaling pathway disrupts ABCG2-rich extracellular vesicles and overcomes multidrug resistance in breast cancer cells. Biochem Pharmacol. 2012;83:1340–1348. doi: 10.1016/j.bcp.2012.01.033. [DOI] [PubMed] [Google Scholar]
- Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- Hafsi S, Pezzino FM, Candido S, Ligresti G, Spandidos DA, Soua Z, et al. Gene alterations in the PI3K/PTEN/AKT pathway as a mechanism of drug-resistance (review) Int J Oncol. 2012;40:639–644. doi: 10.3892/ijo.2011.1312. [DOI] [PubMed] [Google Scholar]
- Hegedus C, Truta-Feles K, Antalffy G, Brozik A, Kasza I, Nemet K, et al. PI3-kinase and mTOR inhibitors differently modulate the function of the ABCG2 multidrug transporter. Biochem Biophys Res Commun. 2012;420:869–874. doi: 10.1016/j.bbrc.2012.03.090. [DOI] [PubMed] [Google Scholar]
- Hu H, Li H, Li J, Zhu Z, Yin S, Hao X, et al. Analysis of ABCG2 expression and side population identifies intrinsic drug efflux in the HCC cell line MHCC-97L and its modulation of Akt signaling. Carcinogenesis. 2008;29:2289–2297. doi: 10.1093/carcin/bgn223. [DOI] [PubMed] [Google Scholar]
- Imai Y, Yoshimori M, Fukuda K, Yamagishi H, Ueda Y. The PI3K/Akt inhibitor LY294002 reverses BCRP-mediated drug resistance without affecting BCRP translocation. Oncol Rep. 2012;27:1703–1709. doi: 10.3892/or.2012.1724. [DOI] [PubMed] [Google Scholar]
- Jaakkola T, Backman JT, Neuvonen M, Laitila J, Neuvonen PJ. Effect of rifampicin on the pharmacokinetics of pioglitazone. Br J Clin Pharmacol. 2005;61:70–78. doi: 10.1111/j.1365-2125.2005.02515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiyama E, Nakai D, Mikkaichi T, Okudaira N, Okazaki O. Interaction of angiotensin II type 1 receptor blockers with P-gp substrates in Caco-2 cells and hMDR1-expressing membranes. Life Sci. 2010;86:52–58. doi: 10.1016/j.lfs.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Kerr ID, Haider AJ, Gelissen IC. The ABCG family of membrane-associated transporters: you don't have to be big to be mighty. Br J Pharmacol. 2011;164:1767–1779. doi: 10.1111/j.1476-5381.2010.01177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura S, Miyazaki Y, Hiraoka S, Nagasawa Y, Toyota M, Takakura R, et al. PPARgamma agonists inhibit cell growth and suppress the expression of cyclin D1 and EGF-like growth factors in ras-transformed rat intestinal epithelial cells. Int J Cancer. 2001;94:335–342. doi: 10.1002/ijc.1470. [DOI] [PubMed] [Google Scholar]
- Koeffler HP. Peroxisome proliferator-activated receptor gamma and cancers. Clin Cancer Res. 2003;9:1–9. [PubMed] [Google Scholar]
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-triphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- Matsuo H, Takada T, Ichida K, Nakamura T, Nakayama A, Suzuki H, et al. ABCG2/BCRP dysfunction as a major cause of gout. Nucleosides Nucleotides Nucleic Acids. 2011;30:1117–1128. doi: 10.1080/15257770.2011.633954. [DOI] [PubMed] [Google Scholar]
- Mogi M, Yang J, Lambert JF, Colvin GA, Shiojima I, Skurk C, et al. Akt signaling regulates side population cell phenotype via Bcrp translocation. J Biol Chem. 2003;278:39068–39075. doi: 10.1074/jbc.M306362200. [DOI] [PubMed] [Google Scholar]
- Natarajan K, Xie Y, Nakanishi T, Beck WT, Bauer KS, Ross DD. Identification and characterization of the major alternative promoter regulating Bcrp1/Abcg2 expression in the mouse intestine. Biochim Biophys Acta. 2011;1809:295–305. doi: 10.1016/j.bbagrm.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozvegy-Laczka C, Varady G, Koblos G, Ujhelly O, Cervenak J, Schuetz JD, et al. Function-dependent conformational changes of the ABCG2 multidrug transport modify its interaction with a monoclonal antibody on the cell surface. J Biol Chem. 2005;280:4219–4227. doi: 10.1074/jbc.M411338200. [DOI] [PubMed] [Google Scholar]
- Patel L, Pass I, Coxon P, Downes CP, Smith SA, Macphee CH. Tumor suppressor and anti-inflammatory actions of PPARgamma agonists are mediated via upregulation of PTEN. Curr Biol. 2001;11:764–768. doi: 10.1016/s0960-9822(01)00225-1. [DOI] [PubMed] [Google Scholar]
- Polgar O, Robey RW, Morisaki K, Dean M, Michejda C, Sauna ZE, et al. Mutational analysis of ABCG2: role of the GXXXG motif. Biochemistry. 2004;43:9448–9456. doi: 10.1021/bi0497953. [DOI] [PubMed] [Google Scholar]
- Polgar O, Lerano C, Tamaki A, Stanley B, Ward Y, Xia D, et al. Mutational analysis of threonine 402 adjacent to the GXXXG dimerization motif in TM1 of ABCG2. Biochemistry. 2010;49:2235–2245. doi: 10.1021/bi902085q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey RW, Medina-Perez WY, Nishiyama K, Lahusen T, Miyake K, Litman T, et al. Overexpression of the ATP-binding cassette half-transporter, ABCG2 (Mxr/BCRP/ABCP1), in flavopiridol-resistant human breast cancer cells. Clin Cancer Res. 2001;7:145–152. [PubMed] [Google Scholar]
- Robey RW, Steadman K, Polgar O, Morisaki K, Blayney M, Mistry P, et al. Pheophorbide A is a specific probe for ABCG2 function and inhibition. Cancer Res. 2004;64:1242–1246. doi: 10.1158/0008-5472.can-03-3298. [DOI] [PubMed] [Google Scholar]
- Ross DD, Karp JE, Chen TT, Doyle LA. Expression of breast cancer resistance protein in blast cells from patients with acute leukemia. Blood. 2000;96:365–368. [PubMed] [Google Scholar]
- Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403–414. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- Sandor V, Fojo T, Bates SE. Future perspectives for the development of P-glycoprotein modulators. Drug Resist Updat. 1998;1:190–200. doi: 10.1016/s1368-7646(98)80039-3. [DOI] [PubMed] [Google Scholar]
- Schmidt B, Schieffer B. Angiotensin II AT1 receptor antagonists. Clinical implications of active metabolites. J Med Chem. 2003;46:2261–2270. doi: 10.1021/jm0204237. [DOI] [PubMed] [Google Scholar]
- Schupp M, Janke J, Clasen R, Unger T, Kintscher U. Angiotensin type I receptor blockers induce peroxisome proliferator-activated receptor-γ activity. Circulation. 2004;109:2054–2057. doi: 10.1161/01.CIR.0000127955.36250.65. [DOI] [PubMed] [Google Scholar]
- Skehan P, Stornet R, Scudiero D, Monks A, McMahon J, Vistica D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- Smith DHG, Matzek KM, Kempthorne-Rawson J. Dose response and safety of telmisartan in patients with mild to moderate hypertension. J Clin Pharmacol. 2000;40:1380–1390. [PubMed] [Google Scholar]
- Staels B, Fruchart JC. Therapeutic roles of peroxisome proliferator-activated receptor agonists. Diabetes. 2005;54:2460–2470. doi: 10.2337/diabetes.54.8.2460. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Suzuki A, Pompa de la JL, Brothers GM, Mirtsos C, Sasaki T, et al. Negative regulation of PKB/AKT-dependent cell survival by tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- Stangier J, Su CA, Hendriks MG, Lier van JJ, Sollie FA, Oosterhuis B, et al. The effect of telmisartan on the steady-state pharmacokinetics of digoxin in healthy male volunteers. J Clin Pharmacol. 2000;40:1373–1379. [PubMed] [Google Scholar]
- Szatmari I, Vamosi G, Brazda P, Balint BL, Benkp S, Szeles L, et al. Peroxisome proliferators-activated receptor γ-regulated ABCG2 expression confers cytoprotection to human dendritic cells. J Biol Chem. 2006;281:23812–23823. doi: 10.1074/jbc.M604890200. [DOI] [PubMed] [Google Scholar]
- Takada T, Suzuki H, Gotoh Y, Sugiuama Y. Regulation of the cell surface expression of human BCRP/ABCG2 by the phosphorylation state of Akt in polarized cells. Drug Metab Dispos. 2005;33:905–909. doi: 10.1124/dmd.104.003228. [DOI] [PubMed] [Google Scholar]
- Tamaki A, Ierano C, Szakacs G, Robey RW, Bates SE. The controversial role of ABC transporters in clinical oncology. Essays Biochem. 2011;50:209–232. doi: 10.1042/bse0500209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teresi RE, Shaiu CW, Chen CS, Chatterjee VK, Waite KA, Eng C. Increased PTEN expression due to transcriptional activation of PPARgamma by lovastatin and rosiglitazone. Int J Cancer. 2006;118:2390–2398. doi: 10.1002/ijc.21799. [DOI] [PubMed] [Google Scholar]
- To KK, Polgar O, Huff LM, Morisaki K, Bates SE. Histone modifications at the ABCG2 promoter following treatment with histone deacetylase inhibitor mirror those in multidrug-resistant cells. Mol Cancer Res. 2008;6:151–164. doi: 10.1158/1541-7786.MCR-07-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To KK, Yu L, Liu S, Fu J, Cho CH. Constitutive AhR activation leads to concomitant ABCG2-mediated multidrug resistance in cisplatin-resistant esophageal carcinoma cells. Mol Carcinog. 2012;51:449–464. doi: 10.1002/mc.20810. [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Weiss J, Sauer A, Divac N, Herzog M, Schwedhelm E, Boger RH, et al. Interaction of angiotensin receptor type 1 blockers with ATP-binding cassette transporters. Biopharm Drug Dispos. 2010;31:150–161. doi: 10.1002/bdd.699. [DOI] [PubMed] [Google Scholar]
- Yap TA, Garrett MD, Walton MI, Raynaud F, Bono de JS, Workman P. Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmacol. 2008;8:393–412. doi: 10.1016/j.coph.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Yuan B, Latek R, Hossbach M, Tuschi T, Lewitter F. siRNA Selection Server: an automated siRNA oligonucleotide prediction server. Nucleic Acids Res. 2004;32:W130–W134. doi: 10.1093/nar/gkh366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Berger J, Zhou G, Elbrecht A, Biswas S, White-Carrington S, et al. Insulin- and mitogen-activated protein kinase-mediated phosphorylation and activation of peroxisome proliferator-activated receptor γ. J Biol Chem. 1996;271:31771–31774. doi: 10.1074/jbc.271.50.31771. [DOI] [PubMed] [Google Scholar]
- Zhang PY, Wong IL, Yan CS, Zhang XY, Jiang T, Chow LM, et al. Design and synthesis of permethyl ningalin B analogues: potent multidrug resistance (MDR) reversal agents of cancer cells. J Med Chem. 2010;53:5108–5120. doi: 10.1021/jm100035c. [DOI] [PubMed] [Google Scholar]
- Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side population phenotype. Nat Med. 2001;7:1028–1034. doi: 10.1038/nm0901-1028. [DOI] [PubMed] [Google Scholar]
- Zong Y, Zhou S, Fatima S, Sorrentino BP. Expression of mouse Abcg2 mRNA during hematopoiesis is regulated by alternative use of multiple leader exons and promoters. J Biol Chem. 2006;281:29625–29632. doi: 10.1074/jbc.M606314200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Cell proliferation data showing that GW9662 alone did not affect cell viability but it abolished the circumvention of ABCG2-mediated resistance in MCF-7 FLV1000 by the PPARγ agonists tested. Data are presented as means ± SDs from at least three independent experiments performed in quadruplicate. The fold reversal of resistance was calculated by dividing the IC50 for mitoxantrone in the absence of PPARγ agonist by that obtained in the presence of PPARγ agonist. *P < 0.05, versus that obtained in mitoxantrone alone in the same cell line.
Figure S1 Immunofluorescence analyses of the translocation of ABCG2 in MCF-7 FLV1000 cells before and after treatment with PPARγ agonists. Confocal microscopy was performed as described in Figure 8. In the presence of the chemical PPARγ antagonist (GW9662, 200 nM), the predominant cell surface expression of ABCG2 was restored in PPARγ agonists-treated MCF-7 FLV1000 cells. Scale bar, 50 μm.
Figure S2 Immunoblot analysis showing that other angiotensin II receptor blockers [ARBs; including losartan (Lo), valsartan (Val) and irbesartan (Irbe)], which have minimal PPARγ agonist effect, did not correct PTEN loss and affect Akt phosphorylation in MCF-7 FLV1000 cells. MCF-7 FLV1000 cells were treated for 24 h with these three ARBs at 50 μM before harvested for immunoblot analysis. Representative results from three independent and reproducible experiments are shown.
Figure S3 Immunofluorescence analyses of the plasma membrane localization of ABCG2 in MCF-7 FLV1000 cells before and after treatment with a few ARBs (losartan, valsartan, and irbesartan; at 50 μM for 24 h) with minimal PPARγ agonist effect. Confocal microscopy was performed as described in Figure 6. (A) Representative images taken from three independent experiments are shown. Predominant cell surface expression of ABCG2 was still observed after treatment with these ARBs. Scale bar, 50 μm. (B) Cell surface 5D3 staining of MCF-7 FLV1000 cells after the indicated treatments was performed as in Figure 4. Mean ± SD from three independent experiments is shown. There is no remarkable change in ABCG2 cell surface expression.
Figure S4 Flow cytometric PhA efflux analysis showing that losartan, valsartan and irbesartan did not directly compete for ABCG2-mediated PhA efflux. The assay was performed as described in Figure 1 in MCF-7 FLV1000 or HEK293/ABCG2 cells incubated for 30 min with the indicated concentrations of the three ARBs tested. PhA fluorescence retention in the cells after a 1-h PhA-free efflux was measured by flow cytometry. Representative histograms from three independent experiments are shown.
Figure S5 Quantitative PCR analysis showing that the tested PPARγ agonists did not affect expression levels of other ABC transporters [(A) MDR-1/P-gp and (B) MRP-1] commonly associated with multidrug resistance. Parental MCF-7 and resistant MCF-7 FLV1000 cells were treated with the three tested PPARγ agonists for 24 h [at concentrations found to inhibit ABCG2-mediated transport; telmisartan (Tel): 10 μM; pioglitazone (Pgz): 5 μM; rosiglitazone (Rgz): 25 μM]. Total RNA was then harvested for RT-PCR analysis. Relative MDR- 1/P-gp and MRP-1 mRNA levels were expressed relative to that in the untreated parental MCF-7 cells, after normalization with GAPDH. It is noted that the basal expression of both MDR-1/P-gp and MRP-1 are lower in the resistant MCF-7 FLV1000 than in the parental MCF-7 cells. The PPARγ agonists tested did not significantly affect MDR-1/P-gp and MRP-1 expression in MCF-7 FLV1000 cells. Mean ± SD from three independent experiments is shown.