

Abstract
A synthesis of C11-desmethoxy soraphen A1α is described that proceeds in just 14 steps from readily available starting materials. This natural product analogue was identified as a target of interest in a program aimed at identifying novel natural product-inspired inhibitors of acetyl-CoA carboxylase (ACC) as potential anticancer therapeutics. While describing the most efficient synthesis of a soraphen A1α analogue (total syntheses of the natural product have been reported that proceed in 25 to ≥40 linear steps), we also present data supporting the conclusion that C11-heteroatom functionality is a beneficial but unnecessary structural characteristic of soraphen A1α analogues for inhibiting ACC.
Keywords: Soraphen A, function-oriented synthesis, acetyl-CoA carboxylase (ACC), polyketide synthesis
Soraphen A1α (1) is a natural product that was first isolated from the soil bacterium Sorangium cellulosum (strain So ce26) and recognized as a potent antimicrobial agent (Figure 1A).1−3 Early enthusiasm for commercial development of this compound was centered on its use for the control of plant disease due to the inhibitory activity of 1 against several phytopathogenic fungi.3 Subsequent investigation led to discovery that the biological target of soraphen A1α (1) is acetyl coenzyme A carboxylase (ACC);4−7 ACCs play crucial roles in fatty acid metabolism and, as such, have risen as attractive therapeutic targets for diabetes.8 More recently, ACCs have become of interest to those seeking novel targeted cancer therapeutics,9 as several studies have established that cancer cells have an intrinsic dependence on de novo fatty acid synthesis and that inhibition of ACC, the rate-limiting step of biosynthesis, triggers apoptosis of cancer cells with little to no effect on normal cells.9−15 While soraphen A1α has been described as a unique tool to study altered fatty acid metabolism in cancer cells, its in vivo utility appears hampered by its poor solubility and bioavailability.8,9 Over 30 natural product family members have been isolated from Sorangium cellulosum and numerous synthetic analogues have been prepared, yet no analogues have been discovered with activity akin to 1. Although the natural product is readily available from fermentation (∼130 mg/L),16 the nature of structural changes that are easily accessible from the fully functionalized natural product are quite limited; such is often the case with natural product derivatization.17−20 Soraphen A1α (1) has been pursued as a target for total synthesis for ∼20 years, with two successful syntheses being reported.21−23 Unfortunately, these accomplishments call for synthetic sequences of 25 to ≥40 steps (longest linear sequence) and define a rather substantial barrier to the use of chemical synthesis for the discovery of novel soraphen analogues that may have better properties than 1. Here, we report a concise synthesis of C11-desmethoxy soraphen A1α (2), a natural product analogue that was targeted in a function-oriented synthesis24 pursuit after analysis of the ACC2–1 structure and an assessment of the challenges that have plagued previous syntheses of 1. Our synthesis proceeds in just 14 chemical steps from a readily available starting material (longest linear sequence) and employs only eight chromatographic purifications. Finally, we report that C11-desmethoxy soraphen A1α (2) has an ACC-inhibitory profile within 1 order of magnitude of the natural product.
Figure 1.
(A) Structure of soraphen A1α. (B) A structural motif within soraphen A1α that impairs efficient synthesis of the natural product.
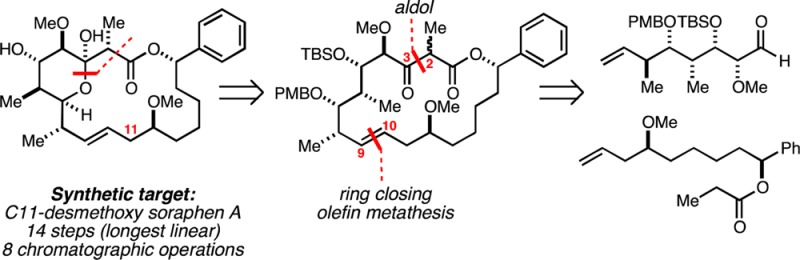
While a cursory inspection of the structure of soraphen A1α may lead one to suggest that the challenge associated with preparing this target may lie in the stereochemically dense C1–C7 domain, the region of soraphen A1α that has had the most significant impact on efficiency associated with prior syntheses is C8–C12 (Figure 1B). While many modern chemical methods can be employed to establish this subunit, within the broader problem of completing the total synthesis of 1, successful solutions come with significant disadvantages; i.e., they require either multistep sequences to establish the disubstituted alkene and the C11–C12 anti-diol, and/or dictate the use of coupling partners that require numerous subsequent chemical transformations to establish the functionalized macrocyclic structure.21−23 Given these facts, we sought to define a soraphen A1α analogue that would have similar activity versus ACC but would harbor functionality in this C8–C12 region that would simplify efforts to accomplish de novo chemical synthesis.
With this goal in mind, we considered the structure of the human ACC2–soraphen A1α complex (Figure 2A).7 While the C11 and C12 methyl ethers of 1 have been proposed to hydrogen bond to Arg277 of human ACC2, we speculated that interaction with the C11 methyl ether may be less important in the complex. In fact, it has been shown that a C11-ester-linked BODIPY analogue of soraphen A1α potently binds to the BC domain of ACC (<5 nM).25 Given that the ester functionality at C11 of this BODIPY-conjugate effectively decreases electron density at the C11 oxygen atom and diminishes its character as a hydrogen bond acceptor, we speculated that C11-desmethoxy soraphen A1α (2) may have anti-ACC properties similar to the natural product. Further, this molecular deletion in the otherwise challenging C8–C12 domain was reasoned to have a substantial and beneficial impact on the efficiency of potential routes for chemical synthesis.
Figure 2.
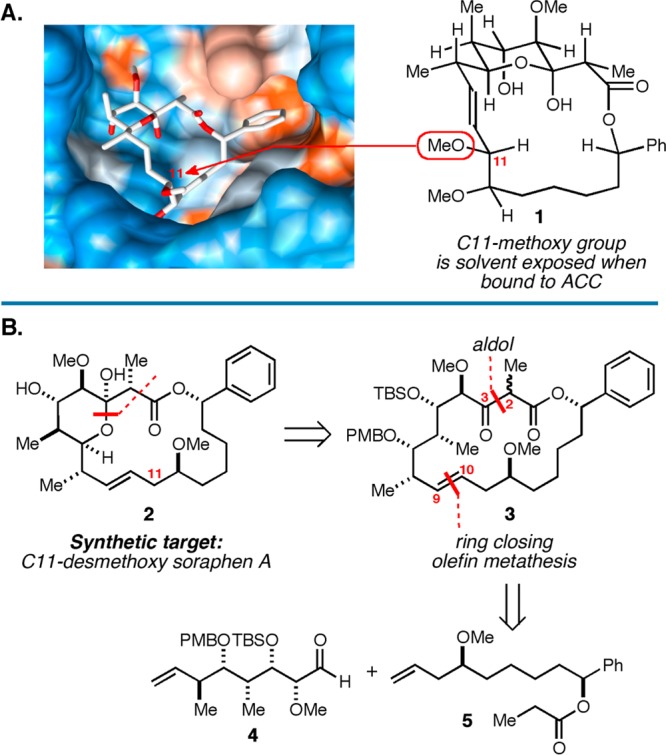
Targeting C11-desmethoxy soraphen A1α. (A) Structure of soraphen A1α bound to human ACC2. (B) Retrosynthesis of C11-desmethoxy soraphen A1α.
Our efforts began by adopting a retrosynthetic strategy for 2 based on late stage hemiketal formation (3 → 2; Figure 2B), and two key C–C bond forming reactions: aldol between the Li-enolate of 5 and aldehyde 4 (to forge the C2–C3 bond), and ring-closing metathesis (RCM) to establish the macrocyclic skeleton. Given the structure of the natural product and the numerous examples of RCM en route to macrocyclic lactones, this strategy for synthesis of 2 is not particularly surprising. In fact, these very bond constructions were targeted by Ciufolini in his studies directed toward a total synthesis of 1.26 Interestingly, these efforts were not successful as an entry to the natural product, as the fully functionalized precursor 6 (that bears the C11-methoxy group; Figure 3) could not be efficiently converted to macrocycle 9. Here, ring-closing metathesis required 30 mol % of the second generation Hoveyda–Grubbs catalyst and delivered macrocycle 9as a minor product; the major product of this reaction sequence was ethyl ketone 10 (derived from initial isomerization of the alkene to the enol ether and subsequent hydrolysis). Despite a number of attempts to optimize the ring-closing process, the authors failed to find a substrate/catalyst combination that provided better conversion to 9, and the study was concluded without completing a synthesis of soraphen A1α.26
Figure 3.
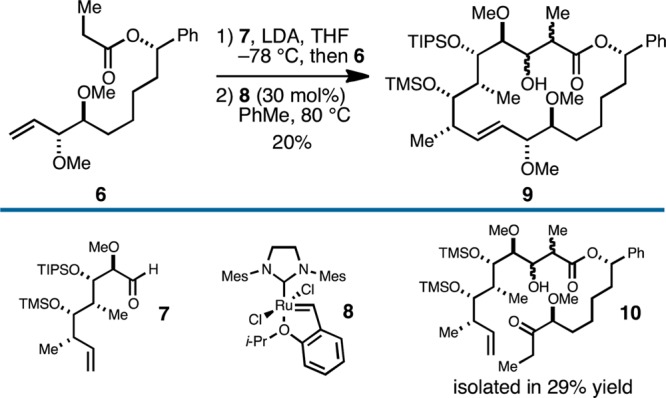
Aldol/RCM-based strategy of Ciufolini15 to generate the macrocyclic framework of soraphen A1α.
The challenges encountered by Ciufilini and co-workers were undoubtedly due to the rate of competing reactions made available by the C11-methyl ether, a functionality that is not present in our designed analogue (2; Figure 2B). As such, we moved forward with a plan to prepare C11-desmethoxy soraphen A1α (2) by a synthetic pathway that embraced sequential convergent aldol/ring-closing metathesis, despite the established failure of this approach in efforts to target 1.
Synthesis of coupling partner 5 follows a simple sequence from 1-phenyl cyclohexene26 as illustrated in Figure 4A. With aldehyde 12 in hand, diastereoselective allylboration28 followed by methylation delivered the propionate ester subunit 5 in 35% yield over the 6-step sequence. Moving forward, synthesis of the more stereochemically complex fragment began from the readily available aldehyde 13 (Figure 4B).27 Diastereoselective crotylboration,29 desilylation, and acid-mediated reaction with the dimethyl acetal of p-anisaldehyde provided the cyclic acetal 14 in 46% yield. Regioselective opening of the PMP acetal,30,31 followed by oxidation of the resulting primary alcohol to the aldehyde,32 asymmetric Evans’ aldol reaction,33−35 silylation, and reduction delivered the stereochemically dense primary alcohol 16 in 38% yield over the five-step sequence. Conversion to the β-ketoester 17 was then accomplished by a simple three-step sequence: oxidation to the aldehyde (DMP, DCM), aldol reaction with the Li-enolate of 5, and oxidation to the ketone (DMP, DCM).
Figure 4.
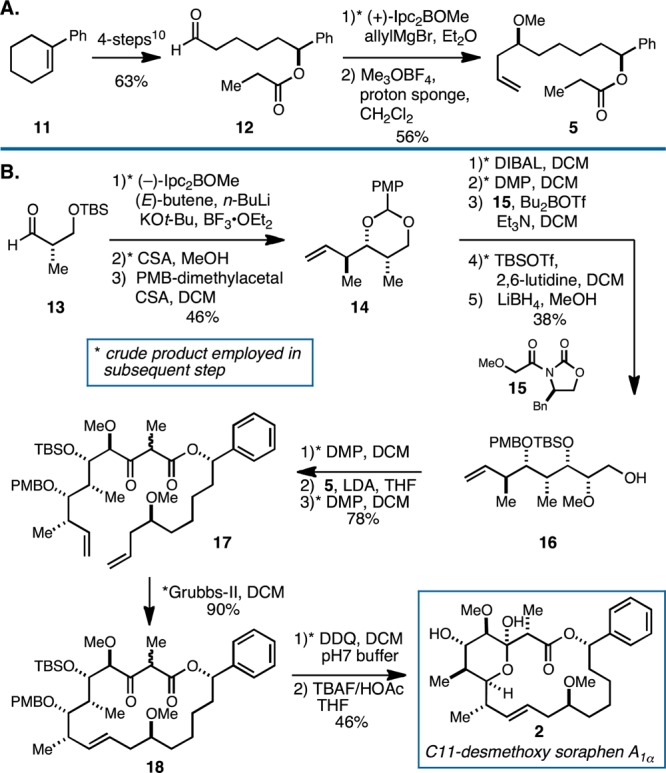
Synthesis of C11-desmethoxy soraphen A1α. (A) Synthesis of the proponiate ester-containing coupling partner 5. (B) Completion of the synthesis of 2.
Gratifyingly, exposure of 17 to the Grubbs second-generation catalyst resulted in efficient ring-closing metathesis to deliver the macrocyclic lactone 18 as a mixture of isomeric products in 90% yield.36,37 At this juncture, we did not determine the stereoselection of the RCM due to the inseparable nature of the product mixture (C2-stereochemistry). Thus, we moved forward with the planned sequence by oxidative deprotection of the PMB ether (DDQ, DCM, pH 7 buffer) and subsequent desilylation with TBAF. Remarkably, the C11-desmethoxy soraphen A1α analogue 2 was produced as a single isomer in 46% yield over the final two-step sequence. Notably, the absence of the C11 stereocenter apparently had a marked effect on the efficiency of macrocyclization and enabled the conclusion of a synthesis of C11-desmethoxy soraphen A1α (2) in only 14 steps from a readily available starting material.
To test our hypothesis that the lack of C11-heteroatom substitution has little effect on the ACC-inhibitory profile of 2, we initiated biological evaluation of this analogue. The inhibitory mechanism of action of 1 is to bind the biotin carboxylase (BC) domain of ACC in the dimer/oligomer interface and disrupt the protein–protein interactions critical for the full-length (active) ACC oligomer to occur.38,39 In cells, treatment with 1 results in significant reductions in de novo fatty acid biosynthesis.9 If 2 has related activity to 1, then one would predict a similar decrease in fatty acid synthesis following treatment. Indeed, a 4 h treatment of two independent B-lymphoma cell lines (SCR1163 and SCR1693; derived from Eμ-Myc transgenic mice) with 2 resulted in a 59% and 48% reduction in 14C-acetate incorporation into fatty acid (Figure 5A), reflecting a potency that is within an order of magnitude of the natural product (1 μM vs 100 nM). Dose response experiments with 2 also showed that it impaired lymphoma cell proliferation at all concentrations and time points tested (Figure 5B), albeit being ca. 10-fold less active than 1.
Figure 5.
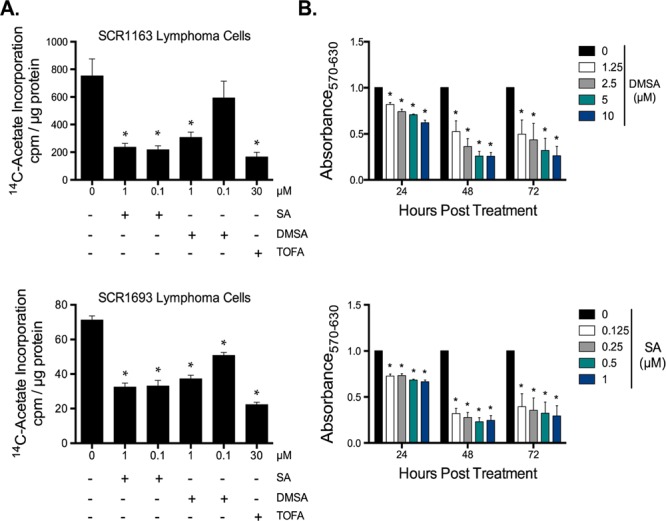
Biological activity of C11-desmethoxy soraphen A1α. (A) Measurement of nascent fatty acid production by 14C-acetate incorporation in SCR1163 and SCR1693 Eμ-Myc B-lymphoma cells. (B) MTT proliferation assays in SCR1163 lymphoma cells treated with C11-desmethoxy soraphen A1α (DMSA), soraphen A1α (SA), or the ACC inhibitor, TOFA, at the indicated concentrations for 24, 48, and 72 h. Values were expressed relative to control. All graphs represent the mean of 3 independent experiments. Error bars represent standard error of the mean. *p ≤ 0.05.
It has been reported that mutations in yeast ACC near the site where soraphen A1α’s C11–C12 bismethyl ether resides (residues surrounding R76, including S77 and K73) abolish or greatly reduce the binding of 1 to the modified enzyme.5 Similarly, Cho et al. demonstrated that R277 of the human ACC2 BC domain participates in hydrogen bonding to both the C11 and C12 methoxy groups of soraphen A1α.38 Given the promising potency of 2 in comparison to the natural product, our data suggest that the hydrogen bonding interaction of yeast ACC R76 and of human ACC2 R277 to the C11 methoxy group of 1 is not essential for the biological activity of soraphen A1α. Given the increased potency of the natural product, optimization of C11-desmethoxy soraphen A1α may be possible by including functionality at C12 that can project electron density into the position where the C11 methyl ether of the natural product resides within ACC2, such that additional opportunities for H-bonding with R277 of the human BC domain can be achieved without the necessity of C11 oxygenation. The ease with which C11-desmethoxy soraphen A1α (2) can now be synthesized40 and the promising activity that has been identified for this natural product analogue supports the growth of a program aimed at synthesizing natural product-inspired agents based on soraphen A1α to fuel the discovery of novel targeted cancer therapeutics. Results associated with these pursuits will be reported in due course.
Glossary
Abbreviations
- ACC
acetyl-CoA carboxylase
- BODIPY
boron-dipyrromethene
- DCM
dichloromethane
- DDQ
(2,3-dichloro-5,6-dicyanobenzo-quinone)
- DMP
Dess–Martin periodinane
- DMSA
C11-desmethoxy soraphen A1α
- PBM
p-methoxy-benzyl ether
- PMP
p-methoxyphenyl
- RCM
ring-closing metathesis
- SA
soraphen A1α
- TBAF
tetrabutylammonium fluoride
- TOFA
5-(tetradecyloxy)-2-furoic acid
Supporting Information Available
Experimental procedures and tabulated spectroscopic data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∥ Department of Chemistry, Dartmouth College, Hanover, New Hampshire 03755, United States.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
We gratefully acknowledge financial support of this work by the Scripps Research Institute, the National Institutes of Health (GM080266 to G.C.M.; CA076379 to J.L.C.), the Jane and Leonard Korman Family Foundation (postdoctoral fellowship to K.E.N.S.), the Japan Society for the Promotion of Science (JSPS, postdoctoral fellowship to O.K.), and funds from the State of Florida to Scripps Florida.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Reichenbach H.; Höfle G.; Augustiniak H.; Bedorf N.; Forche E.; Gerth K.; Irschik H.; Jansen R.; Kunze B.; Sasse F.; Steinmetz H.; Trowitzch-Kienast W.; Pachlatko J. P.. EP 282455A2, 1988.
- Bedorf N.; Schomburg D.; Gerth K.; Reichenbach H.; Höfle G. Antibiotics from gliding bacteria, LIV. Isolation and structure elucidation of soraphen A1α, a novel antifungal macrolide from Sorangium cellulosum. Liebigs Ann. Chem. 1993, 1017–1021. [Google Scholar]
- Gerth K.; Bedorf N.; Irschik H.; Höfle G.; Reichenbach H. The Soraphens: A family of novel antifungal compounds from sorangium-cellulosum (myxobacteria). 1. Soraphen A(1-alpha): fermentation, isolation, biological properties. J. Antibiot. 1994, 47, 23–31. [DOI] [PubMed] [Google Scholar]
- Vahlensieck H. F.; Pridzun L.; Reichenbach H.; Hinnen A. Identification of the yeast ACC1 gene-product (acetyl-CoA carboxylase) as the target of the polyketide fungicide soraphen-A. Curr. Genet. 1994, 25, 95–100. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Volrath S. L.; Weatherly S. C.; Elich T. D.; Tong L. A mechanism for the potent inhibition of eukaryotic acetyl-coenzyme a carboxylase by soraphe A, a macrocyclic polyketide natural product. Mol. Cell 2004, 16, 881–891. [DOI] [PubMed] [Google Scholar]
- Cho Y. S.; Lee J. I.; Shin D.; Kim H. T.; Cheon Y. H.; Seo C. I.; Kim Y. E.; Hyun Y.; Lee Y. S.; Sugiyama K.; Park S.; Ro S.; Cho J. M.; Lee T. G.; Heo Y. Crystal structure of the biotin carboxylase domain of human acetyl-CoA carboxylase 2. Proteins 2008, 70, 268–272. [DOI] [PubMed] [Google Scholar]
- Cho Y. S.; Lee J. I.; Shin D.; Kim H. T.; Jung H. Y.; Lee T. G.; Kang L.; Ahn Y.; Cho H.; Heo Y. Molecular mechanism for the regulation of human ACC2 through phosphorylation by AMPK. Biochem. Biophys. Res. Commun. 2010, 391, 187–192. [DOI] [PubMed] [Google Scholar]
- Harwood H. J. Jr. Treating the metabolic syndrome: acetyl-CoA carboxylase inhibition. Expert Opin. Ther. Targets 2005, 9, 267–281. [DOI] [PubMed] [Google Scholar]
- Beckers A.; Organe S.; Timmermans L.; Scheys K.; Peeters A.; Brusselmans K.; Verhoeven G.; Swinnen J. V. Chemical inhibition of Acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res. 2007, 67, 8180–8187. [DOI] [PubMed] [Google Scholar]
- Brusselmans K.; De Schrijver E.; Verhoeven G.; Swinnen J. V. RNA interference-mediated silencing of the Acetyl-CoA-Carboxylase-α gene induces growth inhibition and apoptosis of prostate cancer cells. Cancer Res. 2005, 65, 6719–6725. [DOI] [PubMed] [Google Scholar]
- Chajes V.; Cambot M.; Moreau K.; Lenoir G. M.; Joulin V. Acetyl-CoA carboxylase α is essential to breast cancer cell survival. Cancer Res. 2006, 66, 5287–5294. [DOI] [PubMed] [Google Scholar]
- Menendez J. A.; Colomer R.; Lupu R. Why does tumor-associated fatty acid synthase (oncogenic antigen-519) ignore dietary fatty acids?. Med. Hypotheses 2005, 64, 342–349. [DOI] [PubMed] [Google Scholar]
- Menendez J. A.; Mehmi I.; Atlas E.; Colomer R.; Lupu R. Novel signaling molecules implicated in tumor-associated fatty acid synthase-dependent breast cancer cell proliferation and survival: Role of exogenous dietary fatty acids, p53-p21(WAF1/CIP1), ERK1/2 MAPK, p27(KIP1), and NF-kappa B. Int. J. Oncol. 2004, 24, 591–608. [PubMed] [Google Scholar]
- Wang C.; Xu C.; Sun M.; Luo D.; Liao D-F.; Cao D. Acetyl-CoA carboxylase-alpha inhibitor TOFA induces human cancer cell apoptosis. Biochem. Biophys. Res. Commun. 2009, 385, 302–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guseva N. V.; Rokhlin O. W.; Glover R. A.; Cohen M. B. TOFA (5-tetradecyl-oxy-2-furoic acid) reduces fatty acid synthesis, inhibits expression of AR, neuropilin-1, and Mcl-1 and kills prostate cancer cells independent of p53 status. Canc. Biol. Ther. 2011, 12, 80–85. [DOI] [PubMed] [Google Scholar]
- The production of soraphen A1α reported in reference (2) proceeds on a 5500-L scale and delivers 700 g of the natural product (determined by HPLC analysis; 300 g of 1 were obtained after recrystallization).
- Schummer D.; Jahn T.; Höfle G. Antibiotics from Gliding Bacteria. 65. Synthesis of soraphen analogs by substitution of the phenyl-C-17 ring segment of soraphen A(1-alpha). Liebigs Ann. Chem. 1995, 803–816. [Google Scholar]
- Kiffe M.; Schummer D.; Höfle G. Chemical modification of the antifungal macrolide soraphen A(1 alpha): Deoxygenation in the south-east ring segment. Liebigs Ann. Chem. 1997, 245–252. [Google Scholar]
- Höfle G.; O’Sullivan A. C.; Rihs G.; Sutter M.; Winkler T. The stereoselective derivatization of the Re or Si faces of the delta(9,10)-double bond of soraphen-A. Tetrahedron 1995, 51, 3159–3174. [Google Scholar]
- Hill A. M.; Thompson B. L. Novel soraphens from precursor directed biosynthesis. Chem. Commun. 2003, 1360–1361. [DOI] [PubMed] [Google Scholar]
- Abel S.; Faber D.; Hüter O.; Giese B. Total synthesis of soraphen A(1-alpha). Angew. Chem., Int. Ed. 1994, 33, 2466–2468. [Google Scholar]
- Abel S.; Faber D.; Hüter O.; Giese G. Total synthesis of soraphen A(1 alpha). Synthesis 1999, 188–197. [Google Scholar]
- Trost B. M.; Sieber J.; Qian W.; Dhawan R.; Ball Z. T. Asymmetric total synthesis of soraphen A: A flexible alkyne strategy. Angew. Chem., Int. Ed. 2009, 48, 5478–5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wender P. A.; Verma V. A.; Paxton T. J.; Pillow T. H. Function-oriented synthesis, step-economy, and drug design. Acc. Chem. Res. 2008, 41, 40–49. [DOI] [PubMed] [Google Scholar]
- Raymer B.; Kavana M.; Price A.; Wang B.; Corcoran L.; Kulathila R.; Groarke J.; Mann T. Synthesis and characterization of a BODIPY-labeled derivative of Soraphen A that binds to acetyl-CoA carboxylase. Bioorg. Med. Chem. Lett. 2009, 19, 2804–2807. [DOI] [PubMed] [Google Scholar]
- Vincent G.; Mansfield D. J.; Vors J.; Ciufolini M. A. Studies toward soraphen A: An aldol-metathesis avenue to the macrocyclic framework. Org. Lett. 2006, 8, 2791–2794. [DOI] [PubMed] [Google Scholar]
- Park S. H.; Lee H. W. Synthetic studies on soraphen A: Synthesis of the C1-C9 hemiketal segment. Bull. Korean Chem. Soc. 2008, 29, 1445–1446. [Google Scholar]
- Brown H. C.; Jadhav P. K. Asymmetric carbon–carbon bond formation via beta-allyldiisopinocampheylborane: simple synthesis of secondary homoallylic alcohols with excellent enantiomeric purities. J. Am. Chem. Soc. 1983, 105, 2092–2093. [Google Scholar]
- Brown H. C.; Bhat K. S. Chiral synthesis via organoboranes. 7. Diastereoselective and enantioselective synthesis of erythro-beta-methylhomoallyl and threo-beta-methylhomoallyl alcohols via enantiomeric (Z)-crotylborane and (E)-crotylborane. J. Am. Chem. Soc. 1986, 108, 5919–5923. [DOI] [PubMed] [Google Scholar]
- Eliel E. L.; Rerick M. Reduction of acetals to ethers by means of lithium aluminum hydride-aluminum chloride. J. Org. Chem. 1958, 23, 1088. [Google Scholar]
- Takano S.; Akiyama M.; Sato S.; Ogasawara K. A facile cleavage of benzylidene acetals with diisobutylaluminum hydride. Chem. Lett. 1983, 1593–1596. [Google Scholar]
- Dess D. B.; Martin J. C. Readily accessible 12-I-5 oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones. J. Org. Chem. 1983, 48, 4155–4156. [Google Scholar]
- Evans D. A.; Bartroli J.; Shih T. L. Enantioselective aldol condensations. 2. Erythro-selective chiral aldol condensations via boron enolates. J. Am. Chem. Soc. 1981, 103, 2127–2129. [Google Scholar]
- For examples with glycolate-based imides, see:Ku T. W.; Kondrad K. H.; Gleason J. G. Stereospecific synthesis of leukotriene antagonists. J. Org. Chem. 1989, 54, 3487–3491. [Google Scholar]
- Maier M. E.; Schöffling B. Synthesis of the cyclohexyl fragment of FK-506 by intramolecular Ene-reaction. Tetrahedron Lett. 1990, 31, 3007–3010. [Google Scholar]
- Scholl M.; Ding S.; Lee C. W.; Grubbs R. H. Synthesis and activity of a new generation of ruthenium-based olefin metathesis catalysts coordinated with 1,3-dimesityl-4,5-dihydroimidazol-2-ylidene ligands. Org. Lett. 1999, 1, 953–956. [DOI] [PubMed] [Google Scholar]
- Grubbs R. H. Olefin metathesis. Tetrahedron 2004, 60, 7117–7140. [Google Scholar]
- Cho Y. S.; Lee J. I.; Shin D.; Kim H. T.; Jung H. Y.; Lee T. G.; Kang L.-W.; Ahn Y.-J.; Cho H.-S.; Heo Y.-S. Molecular mechanism for the regulation of human ACC2 through phosphorylation by AMPK. Biochem. Biophys. Res. Commun. 2010, 391, 187–192. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Volrath S. L.; Weatherly S. C.; Elich T. D.; Tong L. A mechanism for the potent inhibition of eukaryotic acetyl-coenzyme A carboxylase by sorpahen A, a macrocyclic polyketide natural product. Mol. Cell 2004, 16, 881–91. [DOI] [PubMed] [Google Scholar]
- For a recent example where chemical synthesis has risen as an enabling technology to drive exploration of ingenol (an exquisitely comple natural product), see:Jørgensen L.; McKerrall S. J.; Kuttruff C. A.; Ungeheuer F.; Felding J.; Baran P. S. 14-Step synthesis of (+)-ingenol from (+)-2-carene. Science 2013, 341, 878–882. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.