

Abstract
HPVs are the causative agents of cervical and other anogenital cancers. HPVs infect stratified epithelia and link their productive life cycles to cellular differentiation. Low levels of viral genomes are stably maintained in undifferentiated cells and productive replication or amplification is restricted to differentiated suprabasal cells. Amplification is dependent on the activation of the ATM DNA damage factors that are recruited to viral replication centers and inhibition of this pathway blocks productive replication. The STAT-5 protein appears to play a critical role in mediating activation of the ATM pathway in HPV-positive cells. While HPVs need to activate the DNA damage pathway for replication, cervical cancers contain many genomic alterations suggesting that this pathway is circumvented during progression to malignancy.
Keywords: amplification, ATM, CHK2, differentiation, DNA damage, papillomaviruses, replication foci, STAT-5
HPVs
HPVs are the causative agents of over 98% of cervical cancers, which are the second most common cancer in women worldwide [1]. Approximately 11,000 women are diagnosed with cervical cancer in the USA annually and a third die of this disease [2]. Moreover, HPVs also contribute to cancers of the vulva, vagina, anus and penis, as well as the oral cavity, identifying HPV as a risk factor for multiple human cancers. Over 120 types of HPV have been identified and approximately a third of these infect the squamous epithelia of the genital tract [3]. HPV infections in the genital tract can be divided into two groups depending on the association with the development of cancers. The low-risk types, including HPV6 and HPV11, cause benign proliferation of epithelial cells that can lead to the development of genital warts, but rarely result in cancers. By contrast, high-risk genital HPVs (including HPV16, 18, 31 and 35) are the causative agents of cervical and other genital cancers. Prophylactic vaccines have been developed by Merck (NJ, USA) and GlaxoSmithKline (PA, USA) that prevent the development of HPV-associated cancers by blocking the initial infection from high-risk HPV16 and 18 [4]. These vaccines primarily target two of the high-risk types that are responsible for approximately 70% of HPV-associated cancers, but have a limited effect on viral types responsible for the remaining 30% of cancers, although some cross-protection has been reported with other high-risk types [5]. Furthermore, there is no good treatment outside of surgery for those patients with HPV-induced cancers. Thus, it is important to understand the mechanisms of HPV viral replication and signaling pathways regulating malignant progression in order to develop new therapeutic drugs. The importance of HPV and cancer was highlighted by the awarding of the 2008 Nobel Prize to Haraldzur Hausen for his pioneering work in identifying high-risk HPVs as the causative agents of cervical cancer [6].
HPVs infect cells in the basal layer of stratified epithelia and link their life cycles to epithelial differentiation. During infection, HPVs escape host innate immune surveillance and the adaptive responses through mechanisms that are not fully understood to establish persistent infections. HPVs encode two major oncoproteins, E6 and E7, which play important roles in the regulation of the productive life cycle and immune evasion, as well as in the development of anogenital cancers (Figure 1) [3]. Both E6 and E7 promote proliferation of undifferentiated or differentiated suprabasal cells without inducing apoptosis, and block the action of tumor suppressors p53 and Rb [7]. This results in the accumulation of chromosomal alterations and mutations that eventually leads to the development of carcinoma. Recent work indicates that HPVs target the DNA damage pathways to promote viral replication, and this may also contribute to the development of cancers.
Figure 1. Structure of the HPV genome.
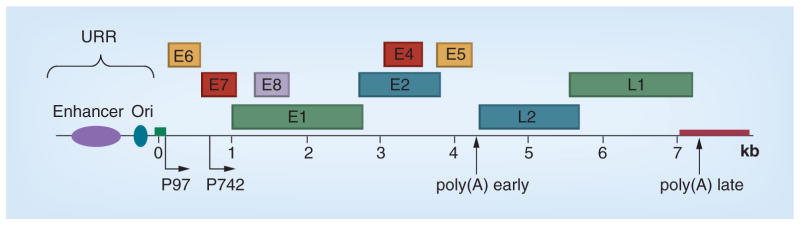
The linearized HPV31 genome is shown. The replication ori and enhancer are located in the URR. The viral early promoter, later promoter and two polyadenylation sites regulate the expression of three groups of viral genes. E6 and E7 immortalize the host cells and maintain replication competence. E1, E2, E4, E5 and E8 are involved in viral DNA replication, alternation in DNA damage response, suppression of immune response and other functions. L1 and L2 are the two capsid genes.
Ori: Origin; URR: Upstream regulatory region.
Persistent infection by high-risk HPVs is the greatest risk factor for the development of cervical cancer. Cervical cancer, like other virally induced malignancies, typically occurs several decades after initial infection. These cancers are clonal and have lost the ability to differentiate. In addition, viral genomes are often found integrated into the host chromosomes. In HPV16 cervical cancers, over 60% contain integrated copies of viral DNAs [8]. As a result of integration no virions are produced from HPV-positive cancers. HPV integration, usually occurs near or within known fragile sites, and often disrupts E2 expression [9–11]. E2 is a repressor of early gene expression that autoregulates its own expression, along with that of other early genes such as E6 and E7. Disruption of E2 results in increased levels of expression of E6 and E7, and this contributes to malignant progression [12]. E6 and E7 promote genomic instability, allowing mutations to accumulate in the cellular genome [13]. Since many more individuals are infected with high-risk HPVs than develop anogenital cancers, infection is necessary but not sufficient for malignant progression [14]. HPV must also induce genetic changes in cellular genes, as well as avoid immune surveillance, for cancers to develop. This review summarizes our current knowledge on the pathways utilized in the HPV life cycle and how these might contribute to tumorigenesis.
HPV life cycle
The HPV life cycle is tightly associated with epithelial differentiation. Many viruses produce the progeny viruses from the same cell that was initially infected. By contrast, HPVs infect undifferentiated basal cells, but restrict productive replication to one of the daughter cells that has undergone differentiation (Figure 2). Infection by HPVs is thought to occur in stem cells or transit amplifying cells in the basal layer of stratified epithelia that become exposed through micro-wounds. Following entry and establishment in the nucleus, viral genomes are replicated in basal cells, together with cellular chromosomes. Only a low level of expression of viral genes is observed in infected basal cells and significant levels of viral transcripts are seen in differentiated cells. Vegetative genome replication, a process that is also referred to as amplification, occurs together with late gene expression, and virion assembly in highly differentiated cells present in the uppermost epithelial layers.
Figure 2. Life cycle of HPV.
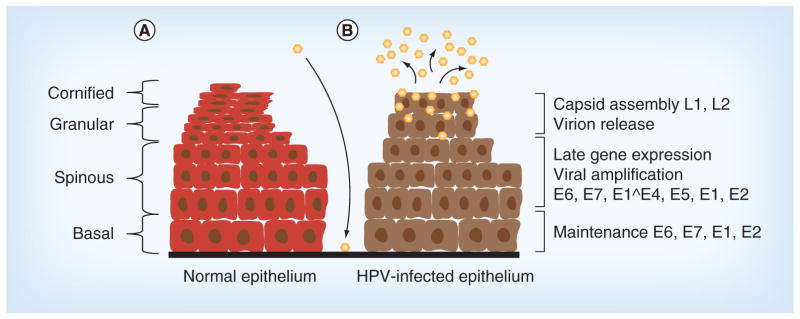
HPVs infect keratinocytes in the basal layer of stratified epithelia following exposure through microwounds. (A) A normal uninfected epithelium and (B) a HPV-infected epithelium. Following entry and establishment in the nucleus, viral genomes are replicated in basal cells together with cellular chromosomes. Only a low level of expression of viral genes is observed in infected basal cells and significant levels of viral transcripts are seen in differentiated cells. Upon differentiation, HPV late gene expression and amplification is activated. This is followed by virion assembly, and the newly synthesized virions are shed from the top layers of epithelium.
Viral gene expression is regulated through two viral promoters and is active at defined times during the differentiation-dependent viral life cycle [15–17]. The early promoter (p97 for HPV31 and 16; p105 for HPV18) is located in the upstream regulatory region adjacent to the E6 open reading frame (ORF) and is active early in infection, prior to productive replication. This promoter directs expression of the E1 and E2 replication factors that leads to establishment of viral genomes as stable episomes at 50–100 copies per cell. It also controls the expression of the viral oncoproteins E6 and E7, which regulate cell cycle progression. As HPV-infected cells divide and differentiate, the late HPV promoter located in the middle of the E7 ORF is activated, leading to expression of late gene products such as E1^E4, E5, L1 and L2, as well as increased levels of E1 and E2. This results in genome amplification, virion assembly and release from suprabasal cells [18]. While E1^E4 and E5 are expressed on early polycistronic transcripts, they are usually the third and fourth ORFs and unlikely to be translated. By contrast, upon differentiation, these two ORFs are the first and second ORFs on late transcripts, indicating that they are synthesized primarily during the late phase. How the viral life cycle is regulated in the differentiation-dependent phase is not well understood and significant efforts are focused on identifying the host cellular factors that regulate differentiation-dependent HPV genome amplification. Recent studies have demonstrated an important role for the ATM DNA damage responses (DDRs) in this processs [19–21].
The circular double-stranded DNA genomes of HPV encode approximately eight ORFs that consist of the early genes E1, E2, E4, E5, E6 and E7 and the two late capsid genes L1 and L2 (Figure 1). Most HPV early gene products contribute to the activation of late viral functions upon differentiation. The E1 protein possesses DNA helicase and ATPase activities that catalyze the unwinding of DNA and recruits cellular replication machinery to viral origins [22,23]. E2 is a DNA-binding protein that helps to load E1 onto origins and tethers the viral DNA to the host chromosome during segregation [24–26]. Increased expression of E1 and E2 occurs upon differentiation and is necessary for genome amplification. The E6 and E7 proteins provide functions necessary for the differentiation-dependent life cycle of HPVs in addition to their roles in immortalization. These two HPV proteins also play critical roles in modulating immune evasion. The high-risk E6 proteins bind to p53 in complexes with the cellular E3 ubiquitin ligase E6AP, which leads to the rapid degradation of p53 [22–27]. p53 function is further inhibited through E6 interactions with p300, leading to reduced levels of p53 acetylation [27,28]. Additional functions of E6 include activation of expression of the catalytic subunit of telomerase hTert and the binding of PDZ domain-containing proteins [29–31]. The E7 protein is necessary to maintain differentiating cells active in the cell cycle and modulates late viral events through its targeted degradation of pRb family members, leading to the constitutive activation of E2F family members [32,33]. E7 has also been shown to interact with class I histone deacetylases to increase the level of E2F2-mediated transcription [34]. E5 is expressed in differentiating cells and its binding to BAP31 suggests a potential regulatory role in the trafficking of MHC class I proteins[35]. During virion assembly, L1 and L2 capsid proteins are first synthesized in the cytoplasm and then trafficked to the nucleus for viral chromatin packaging and virion assembly [36,37].
Regulation of differentiation-dependent late gene expression
The HPV early promoters are located upstream of the E6 ORF and are activated by multiple cellular transcriptional factors, including YY-1 [38], TBP [39], AP-1 [40], Oct-1 [41] and Sp1 [42]. The early promoter is active in undifferentiated as well as differentiated cells. The E2 protein is the major HPV-encoded transcriptional regulator and functions as a repressor of early promoter transcription by binding to E2 binding sites proximal to the start site of early transcription [43–45]. At low levels, E2 can activate early gene expression, while at high levels, it acts as a repressor, leading to the hypothesis that E2 regulates its own expression to control the copy number in undifferentiated cells.
Upon differentiation, the HPV late promoter, which is located in the middle of the E7 ORF, is activated in suprabasal cells. In HPV31, this promoter is referred to as p742, while in HPV16, it is called p670. The factors regulating this promoter are primarily cellular factors, which are just now being elucidated. The primary identified regulators of the late promoter are C/EBPβ isoforms, LIP and LAP [26,46]. The late promoter also activates expression of transcripts encoding the L1 and L2 capsid. Expression of L1 and L2 requires changes in polyadenylation site recognition, along with alternative splicing mechanisms.
Additional regulators of the HPV life cycle are miRNAs. HPVs do not encode their own miRNAs, but rather modulate the expression of a number of cellular miRNAs [47]. One of these cellular miRNAs is miRNA-203, which is a primary regulator of p63 in differentiated cells. The expression of miRNA-203 from heterologous promoters in HPV-positive cells blocks HPV genome amplification by suppressing p63 levels in differentiating cells, which are needed to both maintain cell cycle competence [48] and activate the ATM DDR [49]. The expression of a number of other cellular miRNAs is modulated by HPVs [47,50], and these also likely contribute to regulation of the HPV life cycle [50].
HPVs activate or suppress a number of cellular pathways that play critical roles in regulating the HPV life cycle. One important signaling pathway that regulates the innate immune response is the JAK–STAT pathway. STAT-1 is suppressed in HPV-positive cells at the transcriptional level, and this is necessary for HPV genome amplification [51]. One target of STAT-1 is IFIT1. This protein is also referred to as p56 or ISG56, and can inhibit HPV DNA replication [52,53]. Another member of the JAK–STAT pathway targeted by HPV proteins is the STAT-5 transcriptional activator. However, instead of being repressed, STAT-5 is activated [54]. STAT-5 shares some similarity with STAT-1, but activates a different spectrum of downstream genes and responds to cytokines as well as growth factors. As we discuss below, activation of STAT-5 by HPV is necessary for the induction of the ATM DNA damage pathway, which is critical for differentiation-dependent genome amplification.
DNA damage response
Following exposure to damaging agents, such as reactive oxygen species, UV or x-rays, cells mount an elaborate DDR to repair double- and single-strand breaks (SSBs) to maintain genome integrity [55]. The DDR is a complex signaling network initiated by lesion recognition and amplified by multiple mediators that ultimately activate downstream factors to repair DNA. The DDR controls cell cycle checkpoints, regulates gene expression, induces DNA repair machinery and controls cell fate decisions, such as apoptosis and senescence [56]. Evidence suggests that the DDR is a barrier to the development of malignancies that can be circumvented through mutation of genes in the DDR pathway [57–61]. DNA is continually exposed to various damaging agents that induce a range of lesions, including SSBs, double-strand breaks (DSBs), base mismatches, bulk adducts and base alkylation. In eukaryotes, DSBs are repaired via two mechanisms: nonhomologous end-joining and homologous recombination repair (HRR). SSBs are repaired through base excision repair, while other bulk single-strand lesions are processed by nucleotide excision repair.
The DDR is activated by three PI3K-like protein kinases: ATM, ATR and DNA PK. All three play central roles in the DDR. For instance, DSB-initiated nonhomologous end-joining is mediated by DNA PK, whereas ATM–CHK2 and ATR–CHK1 signaling pathways activate the HRR. Historically, the ATM and ATR pathways were thought to function in parallel with overlapping functions activated by DSBs and SSBs, respectively. However, recent studies indicate that these two pathways are interdependent [61]. ATM is required for strand resection and downstream activation of ATR–CHK1 in response to DSBs [62]. This model was supported by several observations:
ATM is required for rapid activation of ATR in response to DSBs [63,64];
ATR is activated by radiation-stimulated DSBs and induces a S/G2 phase arrest that is a characteristic of ATM signaling [65,66];
The MRE11/RAD50/NBS1 (MRN) complex that is responsible for inducing autophosphorylation of ATM is also required for rapid ATR activation [64,67].
This suggests that the two DNA damage pathways are interlinked.
The ATM pathway consists of several arms that induce cell cycle arrest at different checkpoints. The p53/MDM2 arm induces a G1/S arrest, while the CHK2/Cdc25 and NBS1/BRCA1/SMC1 arm induces a S/G2 or G2/M arrest. ATM directly phosphorylates p53 and regulates its stability [68,69]. The interaction between both proteins is involved in the regulation of G1 cell cycle arrest [70,71]. In the second arm, ATM activation leads to a S/G2 arrest by inhibiting Cdc25 phosphatase [72]. CHK2 activation results in Cdc25C phosphorylation, inducing a G2/M arrest [73]. Another branch of S-phase checkpoint control involves NBS1, SMC1 and BRCA1 [74,75]. DSBs are recognized by the MRN protein complex that then recruits ATM and induces its autophosphorylation. The activation of ATM is also dependent upon acetylation by TIP60 [76]. Following the activation of ATM, a number of substrates are recruited to the sites of damage, including γH2AX [77], the cohesin factor SMC-1 [78] and the downstream effector kinase CHK2 [58]. This results in activation of CHK2, which then phosphorylates additional substrates, such as BRCA1 tumor suppressor [79], p53 tumor suppressor [80] and the Cdc25 family of phosphatases [81]. By contrast, ATR activation is dependent upon ATRIP [82], claspin [83] and TopBP1 [84]. TopBP1 is recruited to the ssDNA lesion together with the RAD9/RAD1/HUS1 (9–1–1) complex. Claspin, which associates with active replication forks during normal replication, is also recruited to these sites. The activation of ATR leads to the phosphorylation of BRCA1, MCM proteins and CHK1. Once activated, CHK1 phosphorylates Cdc25A [85], Wee1 kinase [86] and RAD51 [87]. Activation of ATM and ATR pathways leads to the induction of a large signaling network, including factors involved in regulating cell cycle checkpoints (e.g., FancD2 and RAD9), transcriptional regulation (e.g., E2F1, BRCA1 and p53), DNA repair (e.g., NBS1 and BLM1) and apoptosis (e.g., Mdm2), as well as chromatin remodeling (e.g., Tlk1/2).
HPV & DDR
Infection by a number of DNA viruses induces a DDR. Viruses escape immune surveillance by inactivating the innate or adaptive immune responses to establish persistent infection, but at the same time utilize components of the host defense machinery for their productive life cycles. One of the mechanisms hijacked by several DNA viruses is the ATM signaling pathway. The ATM pathway has been shown to be a critical regulator of HPV vegetative replication in differentiated keratinocytes [19,20], as well as the lytic replication of a number of other DNA viruses.
HPVs activate the ATM pathway in both undifferentiated and differentiated cells, but utilize this pathway primarily for differentiation-dependent genome amplification. Multiple HPV proteins can activate the ATM pathway by themselves or in collaboration. High levels of expression of HPV E1 helicase activates the ATM DDR, and E2 can modulate this activity by binding to E1 [88,89]. The high-level expression of E1 and E2 in cells with integrated HPV genomes has been shown to induce ‘onion-skin’ replication, resulting in activation of the ATM pathway through stalled replication forks [21]. In transient replication assays in undifferentiated cells expressing complete viral genomes, E1 appears to be a major activator of the ATM response. High-risk E7 proteins can also activate the ATM pathway and may be the primary physiological activator of this pathway in HPV-infected cells upon differentiation. In raft cultures of cells containing complete HPV18 genomes or expressing just E7, activation of ATM, CHK2 and CHK1 was observed [20], and similar effects were seen in HPV31 organotypic cultures [19]. ATM activation by high-risk E7 appears to be mediated by several mechanisms [19,20]. E7 binding to Rb and subsequent activation of E2F family members contribute to the induction of the ATM pathway [90]. Furthermore, recent studies have shown that E7 also activates the transcription factor STAT-5 and this appears to be an equally important regulator of this pathway [54]. The role of STAT-5 activation in HPV replication will be discussed in detail in the next section. Interestingly, E7 also forms complexes with phosphorylated ATM proteins, but not the unphosphorylated form, suggesting that this interaction may direct ATM to novel substrates [19].
While inhibitors of ATM activation have no effect on stable maintenance replication in undifferentiated keratinocytes, they block differentiation-dependent genome amplification. Upon epithelial differentiation, several crucial members of the ATM DDR pathway are activated in HPV-positive cells, including pATM, pCHK2, pNBS1 and γH2AX [19]. These factors are recruited into distinct foci in the nuclei of HPV-positive cells and colocalize with HPV genomes undergoing amplification. Chromatin immunoprecipitation studies show that γH2AX directly associates with viral genomes, and it is likely that other ATM factors form complexes on viral sequences. Interestingly, the homologous recombination factors Rad51 and BRCA1 are also recruited to viral replication centers, suggesting that this process may be important for amplification. BRCA1 forms complexes with RAD51, and this is critical for DNA strand exchange. A role for HRR in mediating HPV amplification would be consistent with the finding that both occur in G2/M. Whether DNA repair polymerases are mediating viral replication remains to be determined. In undifferentiated cells, HPVs replicate in synchrony with chromosomal replication through bidirectional θ-structures. The mechanism of differentiation-dependent amplification remains controversial, as one report suggested that it is mediated through unidirectional rolling circle mechanisms, and this needs further study [91,92]. HRR is one of the major DSB repair mechanisms, and two HRR factors, RAD51 and γH2AX, have recently been shown to be recruited to HPV replication foci [93]. It is still not clear how HPV manipulates HRR during amplification, but an understanding of this process may suggest novel therapeutic targets to treat HPV infection. Interestingly, HPV31 proteins also activate low levels of caspase 3 and 7 upon differentiation, and this is dependent upon induction of the ATM pathway [94]. One target of these caspases is the E1 replication protein, and blocking cleavage impairs genome amplification [94].
In addition to ATM factors, HPV also utilizes the ATR pathway for replication [95,96]. ATR activates CHK1 kinases, which leads to p53 phosphorylation and formation of complexes consisting of TopBP1, BLM and γH2AX. The phosphorylation of ATR and CHK1 is activated in both undifferentiated and differentiated cells, and members of this pathway are localized to HPV replication centers [95]. Recent studies have shown that inhibition of ATR reduces the number of HPV episomes that are stably maintained in keratinocyte cell lines, suggesting a role for this pathway in HPV replication in undifferentiated cells. Whether ATR has any role in genome amplification remains to be determined, but clearly both ATR and ATM pathways play critical roles in the differentiation-dependent life cycle of high-risk HPV.
STAT-5 activation by HPV E7 proteins induces the ATM DDR
Significant cross-talk exists between the DNA damage pathway and other signaling networks, such as those controlling apoptosis [97] and immune recognition [98]. Members of the JAK–STAT family are important regulators of the innate immune response, and HPV targets members of this pathway to establish persistent infections. While STAT-1 expression is suppressed in HPV infections, recent studies indicate that STAT-5 is activated in HPV31-positive cells, and that inhibition of STAT-5 activation by either inhibitors or shRNA knockdowns blocks HPV genome amplification [54]. Importantly, STAT-5 activation regulates genome amplification through activation of the ATM DDR (Figure 3). Inhibition of STAT-5 suppresses the phosphorylation of ATM, CHK2 and BRCA1, as well as the level of RAD51, but not BRCA2 or SMC-1. One downstream target of STAT-5, PPARγ, has been shown to play a role in activating the ATM DDR. Importantly, blocking PPARγ activation through the use of inhibitors reduces genome amplification. Both STAT-5 and PPARγ are transcription factors, indicating that additional intermediary proteins must be responsible for activation of ATM phosphorylation. Potential targets include members of the MRN complex or TIP60, an acetyltransferase, whose activity is needed for ATM. Future studies are needed to fully elucidate all the members of this important pathway. The E7 protein is responsible for enhanced phosphorylation of STAT-5 and binds the active phosphorylated form of ATM. It is possible that E7 redirects ATM kinase activity to novel viral or cellular proteins, and that this complements its role in activating STAT-5 downstream effectors of the ATM pathway.
Figure 3. HPV uses STAT-5 signaling to activate the ATM kinase DNA damage response.

STAT-5 has important roles in cell proliferation, apoptosis and differentiation. Normally, the cytoplasmic forms of STAT-5 are phosphorylated upon stimulation with cytokines, such as GM-CSF and IL-6. The activated form of STAT-5 translocates into the nucleus to turn on downstream genes, such as PPARγ. Upon HPV infection, HPV activates STAT-5 in the absence of exogenous cytokines. The phosphorylated STAT-5 induces increased levels of PPARγ in HPV-positive cells, which activates the ATM DNA damage pathway. The ATM DNA damage is necessary for HPV genome amplification. E7 induces activation of ATM, which then turns on downstream effectors, such as BRCA1, CHK2 and p53, leading to cell cycle arrest and HPV amplification.
P: Phosphate.
Previous studies have shown that both Kaposi sarcoma herpes viruses [99] and human T-cell lymphotropic virusI [100] activate STAT-5 phosphorylation and, interestingly, both viruses also induce the ATM DDR. This suggests that STAT-5-mediated regulation of the ATM DDR may be a common feature of multiple viruses.
Other viruses & DNA damage
A number of other DNA viruses use the DDR as part of their replication schemes. For example, SV40 activates the ATM DDR, which may be essential for resolving concatemeric replication intermediates [101,102]. The induction of lytic replication from cells latently infected with EBV depends on the ATM DNA damage pathway [103]. Interestingly, EBV activation of DDR in latently infected cells acts to inhibit virally mediated cell proliferation [104]. Another DNA virus that activates the DDR is the B19 parvovirus that depends on this pathway for its replication [105]. Overall, many DNA viruses activate the ATM or ATR pathways, and use this as part of their replication mechanisms, suggesting common mechanisms for regulating the replication of DNA viruses.
Conclusion & future perspective
The productive replication of a number of DNA viruses is dependent upon the cellular DDR pathways. The ATM and ATR pathways normally play critical roles in mediating repair of double- or single-strand DNA breaks. Viruses associated with human cancers such as HPV and EBV have hijacked these pathways to facilitate viral replication. HPVs link their productive life cycles to differentiation and activate the ATM DNA damage pathway in both undifferentiated and differentiated cells. Interestingly, HPVs only use the ATM pathway for productive replication, but not stable maintenance of episomes. It is unclear how ATM factors specifically contribute to replication, and this is an important area for future studies. It is possible that the primary function of ATM factors is to help resolve concatemers of viral DNA. An alternate explanation is that viral replication is mediated by DNA repair polymerases and ligases. Future studies should investigate whether the DNA repair polymerases and ligases are directly involved through the use of knockdowns.
The demonstration that viruses associated with human malignancies, such as HPV and EBV, activate the DNA damage pathways for replication raises questions about what role this activation plays in cancer development. The DNA repair pathway is a mechanism that cells use to protect themselves from the accumulation of DNA damage. HPV-induced cervical cancers are highly aneuploid and contain many chromosomal rearrangements, as well as point mutations. If HPV proteins activate the ATM DNA damage pathway, then are not these cancers resistant to mutation? Mutations in the ATM pathway are common in other cancers [106] and it has been suggested that activation of the ATM pathway facilitates the selection of mutations in ATM factors. In this model, only a small number of cells would acquire mutations in the ATM pathway. Cervical cancers are clonal in origin, which would be consistent with this model. An important area for future research would be the sequencing of multiple cervical cancers to determine whether any members of the ATM or ATR pathways have acquired mutations that block the ability to repair DNA lesions. The linkage of DNA damage to viral replication and malignant progression can also identify potential targets for therapeutic intervention.
Executive summary.
HPVs
Over 120 types of HPV have been identified and each specifically targets epithelial tissues at different body locations.
A third of HPV types infect the genital tract and are sexually transmitted. A subset of these are referred to as high risk and are the causative agents of cervical, as well as other anogenital, cancers.
Cervical cancer is the second most common cancer in women worldwide, and approximately 11,000 cases are diagnosed annually in the USA.
Prophylactic vaccines against two high-risk HPV types, HPV16 and 18, block initial infection, but have no effect on existing lesions. These vaccines effectively prevent the establishment of HPV16 and 18 infections, and prevent the development of cervical cancers.
HPV life cycle
HPVs infect cells in the basal layer of stratified epithelia and establish their genomes as low-copy episomes that replicate in synchrony with cellular chromosomes.
Productive replication of HPVs occurs upon differentiation in suprabasal layers in cells that have entered G2/M. This process is referred to as amplification and occurs together with activation of the late viral promoter and virion assembly.
Amplification of HPV genomes upon differentiation is dependent upon the ATM DNA damage pathway.
In cervical cancers, the ability to differentiate is lost and no progeny virions are produced. Viral genomes are often found integrated into host chromosomes.
Regulation of differentiation-dependent late promoter
HPV late promoters are activated upon differentiation and provide increased expression of the viral replication proteins E1 and E2, as well as the capsid proteins L1 and L2.
Cellular factors, such as C/EBPβ, are responsible for activation of the late promoter.
Differentiation-dependent changes in polyadenylation site usage together with alternative splicing contribute to regulation of late gene expression.
HPVs also alter expression of cellular miRNAs to regulate late gene expression.
DNA damage response
The ATM pathway is activated by dsDNA breaks and induces phosphorylation of downstream effectors, such as CHK2, p53 and NBS1.
The ATM- and RAD3-related kinase pathway is activated by single-strand breaks, resulting in phosphorylation of CHK1.
HPV & DNA damage response
HPV proteins activate the ATM DNA damage response in both undifferentiated and differentiated cells.
ATM activation has no effect on stable maintenance replication in undifferentiated cells, but is required for amplification in differentiated cells.
ATM factors including CHK2 and γH2AX are recruited to replicating viral genomes located in multiple nuclear foci in differentiating cells.
STAT-5 activation of the DNA damage response
HPV E7 activates STAT-5 phosphorylation without altering total levels.
Inhibition of STAT-5 phosphorylation blocks genome amplification, as well as activation of the ATM pathway.
PPARγ is a downstream mediator of STAT-5 action and may contribute to regulating HPV genome amplification.
Other viruses & DNA damage
SV40 activates the ATM DNA damage response, and this may be essential for resolving concatemers.
Lytic reactivation of latently infected EBV-positive cells requires activation of the ATM pathway.
B19 parvoviruses activate DNA damage response for viral replication.
Footnotes
Financial & competing interests disclosure
LA Laimins and S Hong were supported by grants from the NIH (R37 CA74202, RO1CA59655 and RO1CA142861). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
▪ of interest
- 1.Zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2(5):342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 2.Markowitz LE, Dunne EF, Saraiya M, Lawson HW, Chesson H, Unger ER. Quadrivalent human papillomavirus vaccine: recommendations of the Advisory Committee on Immunization Practices (ACIP) MMWR Recomm Rep. 2007;56(RR-2):1–24. [PubMed] [Google Scholar]
- 3▪.Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10(8):550–560. doi: 10.1038/nrc2886. Describes the critical role of the ATM DNA damage pathway in HPV genome amplification. [DOI] [PubMed] [Google Scholar]
- 4.Harper DM, Franco EL, Wheeler CM, et al. Sustained efficacy up to 4.5 years of a bivalent L1 virus-like particle vaccine against human papillomavirus types 16 and 18: follow-up from a randomised control trial. Lancet. 2006;367(9518):1247–1255. doi: 10.1016/S0140-6736(06)68439-0. [DOI] [PubMed] [Google Scholar]
- 5.Wheeler CM, Castellsague X, Garland SM, et al. Cross-protective efficacy of HPV-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by non-vaccine oncogenic HPV types: 4-year end-of-study analysis of the randomised, double-blind PATRICIA trial. Lancet Oncol. 2012;13(1):100–110. doi: 10.1016/S1470-2045(11)70287-X. [DOI] [PubMed] [Google Scholar]
- 6.zur Hausen H. The search for infectious causes of human cancers: where and why (Nobel Lecture) Angew Chem Int Ed. 2009;48:5798–5808. doi: 10.1002/anie.200901917. [DOI] [PubMed] [Google Scholar]
- 7.Bodily J, Laimins LA. Persistence of human papillomavirus infection: keys to malignant progression. Trends Microbiol. 2011;19(1):33–39. doi: 10.1016/j.tim.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arias-Pulido H, Peyton CL, Joste NE, Vargas H, Wheeler CM. Human papillomavirus type 16 integration in cervical carcinoma in situ and in invasive cervical cancer. J Clin Microbiol. 2006;44(5):1755–1762. doi: 10.1128/JCM.44.5.1755-1762.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schneider-Maunoury S, Croissant O, Orth G. Integration of human papillomavirus type 16 DNA sequences: a possible early event in the progression of genital tumors. J Virol. 1987;61(10):3295–3298. doi: 10.1128/jvi.61.10.3295-3298.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith PP, Friedman CL, Bryant EM, McDougall JK. Viral integration and fragile sites in human papillomavirus-immortalized human keratinocyte cell lines. Genes Chromosomes Cancer. 1992;5(2):150–157. doi: 10.1002/gcc.2870050209. [DOI] [PubMed] [Google Scholar]
- 11.Thorland EC, Myers SL, Gostout BS, Smith DI. Common fragile sites are preferential targets for HPV16 integrations in cervical tumors. Oncogene. 2003;22(8):1225–1237. doi: 10.1038/sj.onc.1206170. [DOI] [PubMed] [Google Scholar]
- 12.Romanczuk H, Howley PM. Disruption of either the E1 or the E2 regulatory gene of human papillomavirus type 16 increases viral immortalization capacity. Proc Natl Acad Sci USA. 1992;89(7):3159–3163. doi: 10.1073/pnas.89.7.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duensing S, Munger K. Mechanisms of genomic instability in human cancer: insights from studies with human papillomavirus oncoproteins. Int J Cancer. 2004;109(2):157–162. doi: 10.1002/ijc.11691. [DOI] [PubMed] [Google Scholar]
- 14.Munger K, Baldwin A, Edwards KM, et al. Mechanisms of human papillomavirus-induced oncogenesis. J Virol. 2004;78(21):11451–11460. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geisen C, Kahn T. Promoter activity of sequences located upstream of the human papillomavirus types of 16 and 18 late regions. J Gen Virol. 1996;77(Pt 9):2193–2200. doi: 10.1099/0022-1317-77-9-2193. [DOI] [PubMed] [Google Scholar]
- 16.Ozbun MA, Meyers C. Temporal usage of multiple promoters during the life cycle of human papillomavirus type 31b. J Virol. 1998;72(4):2715–2722. doi: 10.1128/jvi.72.4.2715-2722.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Braunstein TH, Madsen BS, Gavnholt B, Rosenstierne MW, Johnsen CK, Norrild B. Identification of a new promoter in the early region of the human papillomavirus type 16 genome. J Gen Virol. 1999;80(Pt 12):3241–3250. doi: 10.1099/0022-1317-80-12-3241. [DOI] [PubMed] [Google Scholar]
- 18.Hebner CM, Laimins LA. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity. Rev Med Virol. 2006;16(2):83–97. doi: 10.1002/rmv.488. [DOI] [PubMed] [Google Scholar]
- 19.Moody CA, Laimins LA. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009;5(10):e1000605. doi: 10.1371/journal.ppat.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banerjee NS, Wang HK, Broker TR, Chow LT. Human papillomavirus (HPV) E7 induces prolonged G2 following S phase reentry in differentiated human keratinocytes. J Biol Chem. 2011;286(17):15473–15482. doi: 10.1074/jbc.M110.197574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kadaja M, Isok-Paas H, Laos T, Ustav E, Ustav M. Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog. 2009;5(4):e1000397. doi: 10.1371/journal.ppat.1000397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hughes FJ, Romanos MA. E1 protein of human papillomavirus is a DNA helicase/ATPase. Nucleic Acids Res. 1993;21(25):5817–5823. doi: 10.1093/nar/21.25.5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Conger KL, Liu JS, Kuo SR, Chow LT, Wang TS. Human papillomavirus DNA replication. Interactions between the viral E1 protein and two subunits of human DNA polymerase alpha/primase. J Biol Chem. 1999;274(5):2696–2705. doi: 10.1074/jbc.274.5.2696. [DOI] [PubMed] [Google Scholar]
- 24.Oliveira JG, Colf LA, McBride AA. Variations in the association of papillomavirus E2 proteins with mitotic chromosomes. Proc Natl Acad Sci USA. 2006;103(4):1047–1052. doi: 10.1073/pnas.0507624103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poddar A, Reed SC, McPhillips MG, Spindler JE, McBride AA. The human papillomavirus type 8 E2 tethering protein targets the ribosomal DNA loci of host mitotic chromosomes. J Virol. 2009;83(2):640–650. doi: 10.1128/JVI.01936-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kukimoto I, Takeuchi T, Kanda T. CCAAT/enhancer binding protein beta binds to and activates the P670 promoter of human papillomavirus type 16. Virology. 2006;346(1):98–107. doi: 10.1016/j.virol.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 27.Patel D, Huang SM, Baglia LA, McCance DJ. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J. 1999;18(18):5061–5072. doi: 10.1093/emboj/18.18.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hebner C, Beglin M, Laimins LA. Human papillomavirus E6 proteins mediate resistance to interferon-induced growth arrest through inhibition of p53 acetylation. J Virol. 2007;81(23):12740–12747. doi: 10.1128/JVI.00987-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu X, Dakic A, Zhang Y, Dai Y, Chen R, Schlegel R. HPV E6 protein interacts physically and functionally with the cellular telomerase complex. Proc Natl Acad Sci USA. 2009;106(44):18780–18785. doi: 10.1073/pnas.0906357106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wise-Draper TM, Wells SI. Papillomavirus E6 and E7 proteins and their cellular targets. Front Biosci. 2008;13:1003–1017. doi: 10.2741/2739. [DOI] [PubMed] [Google Scholar]
- 31.Howie HL, Katzenellenbogen RA, Galloway DA. Papillomavirus E6 proteins. Virology. 2009;384(2):324–334. doi: 10.1016/j.virol.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243(4893):934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 33.Munger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989;8(13):4099–4105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Longworth MS, Wilson R, Laimins LA. HPV31 E7 facilitates replication by activating E2F2 transcription through its interaction with HDACs. EMBO J. 2005;24(10):1821–1830. doi: 10.1038/sj.emboj.7600651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Regan JA, Laimins LA. Bap31 is a novel target of the human papillomavirus E5 protein. J Virol. 2008;82(20):10042–10051. doi: 10.1128/JVI.01240-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nelson LM, Rose RC, Moroianu J. Nuclear import strategies of high risk HPV16 L1 major capsid protein. J Biol Chem. 2002;277(26):23958–23964. doi: 10.1074/jbc.M200724200. [DOI] [PubMed] [Google Scholar]
- 37.Darshan MS, Lucchi J, Harding E, Moroianu J. The l2 minor capsid protein of human papillomavirus type 16 interacts with a network of nuclear import receptors. J Virol. 2004;78(22):12179–12188. doi: 10.1128/JVI.78.22.12179-12188.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ai W, Narahari J, Roman A. Yin yang 1 negatively regulates the differentiation-specific E1 promoter of human papillomavirus type 6. J Virol. 2000;74(11):5198–5205. doi: 10.1128/jvi.74.11.5198-5205.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hartley KA, Alexander KA. Human TATA binding protein inhibits human papillomavirus type 11 DNA replication by antagonizing E1–E2 protein complex formation on the viral origin of replication. J Virol. 2002;76(10):5014–5023. doi: 10.1128/JVI.76.10.5014-5023.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Offord EA, Beard P. A member of the activator protein 1 family found in keratinocytes but not in fibroblasts required for transcription from a human papillomavirus type 18 promoter. J Virol. 1990;64(10):4792–4798. doi: 10.1128/jvi.64.10.4792-4798.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Connor M, Bernard HU. Oct-1 activates the epithelial-specific enhancer of human papillomavirus type 16 via a synergistic interaction with NFI at a conserved composite regulatory element. Virology. 1995;207(1):77–88. doi: 10.1006/viro.1995.1053. [DOI] [PubMed] [Google Scholar]
- 42.Stunkel W, Bernard HU. The chromatin structure of the long control region of human papillomavirus type 16 represses viral oncoprotein expression. J Virol. 1999;73(3):1918–1930. doi: 10.1128/jvi.73.3.1918-1930.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stubenrauch F, Lim HB, Laimins LA. Differential requirements for conserved E2 binding sites in the life cycle of oncogenic human papillomavirus type 31. J Virol. 1998;72(2):1071–1077. doi: 10.1128/jvi.72.2.1071-1077.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steger G, Corbach S. Dose-dependent regulation of the early promoter of human papillomavirus type 18 by the viral E2 protein. J Virol. 1997;71(1):50–58. doi: 10.1128/jvi.71.1.50-58.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thierry F. Transcriptional regulation of the papillomavirus oncogenes by cellular and viral transcription factors in cervical carcinoma. Virology. 2009;384(2):375–379. doi: 10.1016/j.virol.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 46.Gunasekharan V, Hache G, Laimins L. Differentiation-dependent changes in levels of C/EBPbeta repressors and activators regulate human papillomavirus type 31 late gene expression. J Virol. 2012;86(9):5393–5398. doi: 10.1128/JVI.07239-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zheng ZM, Wang X. Regulation of cellular miRNA expression by human papillomaviruses. Biochem Biophys Acta. 2011;1809(11–12):668–677. doi: 10.1016/j.bbagrm.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Melar-New M, Laimins LA. Human papillomaviruses modulate expression of microRNA 203 upon epithelial differentiation to control levels of p63 proteins. J Virol. 2010;84(10):5212–5221. doi: 10.1128/JVI.00078-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mighty KK, Laimins LA. p63 is necessary for the activation of human papillomavirus late viral functions upon epithelial differentiation. J Virol. 2011;85(17):8863–8869. doi: 10.1128/JVI.00750-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang X, Tang S, Le SY, et al. Aberrant expression of oncogenic and tumor-suppressive microRNAs in cervical cancer is required for cancer cell growth. PLoS ONE. 2008;3(7):e2557. doi: 10.1371/journal.pone.0002557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hong S, Mehta KP, Laimins LA. Suppression of STAT-1 expression by human papillomaviruses is necessary for differentiation-dependent genome amplification and plasmid maintenance. J Virol. 2011;85(18):9486–9494. doi: 10.1128/JVI.05007-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Terenzi F, Saikia P, Sen GC. Interferon-inducible protein, P56, inhibits HPV DNA replication by binding to the viral protein E1. EMBO J. 2008;27(24):3311–3321. doi: 10.1038/emboj.2008.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saikia P, Fensterl V, Sen GC. The inhibitory action of P56 on select functions of E1 mediates interferon’s effect on human papillomavirus DNA replication. J Virol. 2010;84(24):13036–13039. doi: 10.1128/JVI.01194-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54▪.Hong S, Laimins LA. The JAK–STAT transcriptional regulator, STAT-5, activates the ATM DNA damage pathway to induce HPV 31 genome amplification upon epithelial differentiation. PLoS Pathog. 2013;9(4):e1003295. doi: 10.1371/journal.ppat.1003295. Identifies STAT-5 as an important activator of the ATM DNA damage response and consequently a regulator of HPV differentiation-dependent genome amplification. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Price BD, D’Andrea AD. Chromatin remodeling at DNA double-strand breaks. Cell. 2013;152(6):1344–1354. doi: 10.1016/j.cell.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40(2):179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kastan MB. DNA damage responses: mechanisms and roles in human disease: 2007 GHA. Clowes Memorial Award Lecture. Mol Cancer Res. 2008;6(4):517–524. doi: 10.1158/1541-7786.MCR-08-0020. [DOI] [PubMed] [Google Scholar]
- 58.Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer. 2012;12(9):587–598. doi: 10.1038/nrc3342. [DOI] [PubMed] [Google Scholar]
- 59.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481(7381):287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 60.Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12(12):801–817. doi: 10.1038/nrc3399. [DOI] [PubMed] [Google Scholar]
- 61.Smith J, Tho LM, Xu N, Gillespie DA. The ATM–Chk2 and ATR–Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 62.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432(7015):316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 63.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19(9):1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Myers JS, Cortez D. Rapid activation of ATR by ionizing radiation requires ATM and Mre11. J Biol Chem. 2006;281(14):9346–9350. doi: 10.1074/jbc.M513265200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jazayeri A, Falck J, Lukas C, et al. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8(1):37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 66.Walker M, Black EJ, Oehler V, Gillespie DA, Scott MT. Chk1 C-terminal regulatory phosphorylation mediates checkpoint activation by de-repression of Chk1 catalytic activity. Oncogene. 2009;28(24):2314–2323. doi: 10.1038/onc.2009.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yarden RI, Pardo-Reoyo S, Sgagias M, Cowan KH, Brody LC. BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat Genet. 2002;30(3):285–289. doi: 10.1038/ng837. [DOI] [PubMed] [Google Scholar]
- 68.Banin S, Moyal L, Shieh S, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281(5383):1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 69.Maya R, Balass M, Kim ST, et al. ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev. 2001;15(9):1067–1077. doi: 10.1101/gad.886901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Canman CE, Wolff AC, Chen CY, Fornace AJ, Jr, Kastan MB. The p53-dependent G1 cell cycle checkpoint pathway and ataxia-telangiectasia. Cancer Res. 1994;54(19):5054–5058. [PubMed] [Google Scholar]
- 71.Chen CY, Oliner JD, Zhan Q, Fornace AJ, Jr, Vogelstein B, Kastan MB. Interactions between p53 and MDM2 in a mammalian cell cycle checkpoint pathway. Proc Natl Acad Sci USA. 1994;91(7):2684–2688. doi: 10.1073/pnas.91.7.2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282(5395):1893–1897. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- 73.Zhou BB, Chaturvedi P, Spring K, et al. Caffeine abolishes the mammalian G(2)/M DNA damage checkpoint by inhibiting ataxia-telangiectasia-mutated kinase activity. J Biol Chem. 2000;275(14):10342–10348. doi: 10.1074/jbc.275.14.10342. [DOI] [PubMed] [Google Scholar]
- 74.Gatei M, Zhou BB, Hobson K, Scott S, Young D, Khanna KK. Ataxia telangiectasia mutated (ATM) kinase and ATM and Rad3 related kinase mediate phosphorylation of BRCA1 at distinct and overlapping sites. In vivo assessment using phospho-specific antibodies. J Biol Chem. 2001;276(20):17276–17280. doi: 10.1074/jbc.M011681200. [DOI] [PubMed] [Google Scholar]
- 75.D’Amours D, Jackson SP. The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat Rev Mol Cell Biol. 2002;3(5):317–327. doi: 10.1038/nrm805. [DOI] [PubMed] [Google Scholar]
- 76.Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11–Rad50–Nbs1 complex. Science. 2005;308(5721):551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 77.Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair. 2004;3(8–9):959–967. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 78.Kitagawa R, Bakkenist CJ, McKinnon PJ, Kastan MB. Phosphorylation of SMC1 is a critical downstream event in the ATM–NBS1–BRCA1 pathway. Genes Dev. 2004;18(12):1423–1438. doi: 10.1101/gad.1200304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of BRCA1 in the DNA damage response to double-strand breaks. Science. 1999;286(5442):1162–1166. doi: 10.1126/science.286.5442.1162. [DOI] [PubMed] [Google Scholar]
- 80.Chen L, Gilkes DM, Pan Y, Lane WS, Chen J. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J. 2005;24(19):3411–3422. doi: 10.1038/sj.emboj.7600812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Blasina A, De Weyer IV, Laus MC, Luyten WH, Parker AE, McGowan CH. A human homologue of the checkpoint kinase Cds1 directly inhibits Cdc25 phosphatase. Curr Biol. 1999;9(1):1–10. doi: 10.1016/s0960-9822(99)80041-4. [DOI] [PubMed] [Google Scholar]
- 82.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA–ssDNA complexes. Science. 2003;300(5625):1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 83.Kumagai A, Dunphy WG. Repeated phosphopeptide motifs in Claspin mediate the regulated binding of Chk1. Nat Cell Biol. 2003;5(2):161–165. doi: 10.1038/ncb921. [DOI] [PubMed] [Google Scholar]
- 84.Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR–ATRIP complex. Cell. 2006;124(5):943–955. doi: 10.1016/j.cell.2005.12.041. [DOI] [PubMed] [Google Scholar]
- 85.Falck J, Petrini JH, Williams BR, Lukas J, Bartek J. The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat Genet. 2002;30(3):290–294. doi: 10.1038/ng845. [DOI] [PubMed] [Google Scholar]
- 86.Lee J, Kumagai A, Dunphy WG. Positive regulation of Wee1 by Chk1 and 14–13–3 proteins. Mol Biol Cell. 2001;12(3):551–563. doi: 10.1091/mbc.12.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sorensen CS, Hansen LT, Dziegielewski J, et al. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 2005;7(2):195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- 88.Sakakibara N, Mitra R, McBride AA. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J Virol. 2011;85(17):8981–8995. doi: 10.1128/JVI.00541-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fradet-Turcotte A, Bergeron-Labrecque F, Moody CA, Lehoux M, Laimins LA, Archambault J. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J Virol. 2011;85(17):8996–9012. doi: 10.1128/JVI.00542-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rogoff HA, Pickering MT, Frame FM, et al. Apoptosis associated with deregulated E2F activity is dependent on E2F1 and Atm/Nbs1/Chk2. Mol Cell Biol. 2004;24(7):2968–2977. doi: 10.1128/MCB.24.7.2968-2977.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Flores ER, Lambert PF. Evidence for a switch in the mode of human papillomavirus type 16 DNA replication during the viral life cycle. J Virol. 1997;71(10):7167–7179. doi: 10.1128/jvi.71.10.7167-7179.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dasgupta S, Zabielski J, Simonsson M, Burnett S. Rolling-circle replication of a high-copy BPV-1 plasmid. J Mol Biol. 1992;228(1):1–6. doi: 10.1016/0022-2836(92)90485-3. [DOI] [PubMed] [Google Scholar]
- 93▪.Gillespie KA, Mehta KP, Laimins LA, Moody CA. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J Virol. 2012;86(17):9520–9526. doi: 10.1128/JVI.00247-12. Demonstrates that the members of the homologous DNA recombination pathway along with ATM DNA damage factors are recruited to HPV replication foci to facilitate genome amplification. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Moody CA, Fradet-Turcotte A, Archambault J, Laimins LA. Human papillomaviruses activate caspases upon epithelial differentiation to induce viral genome amplification. Proc Natl Acad Sci USA. 2007;104(49):19541–19546. doi: 10.1073/pnas.0707947104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Reinson T, Toots M, Kadaja M, et al. Engagement of the ATR-dependent DNA damage response at the human papillomavirus 18 replication centers during the initial amplification. J Virol. 2013;87(2):951–964. doi: 10.1128/JVI.01943-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Edwards TG, Helmus MJ, Koeller K, Bashkin JK, Fisher C. Human papillomavirus episome stability is reduced by aphidicolin and controlled by DNA damage response pathways. J Virol. 2013;87(7):3979–3989. doi: 10.1128/JVI.03473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ozaki T, Wu D, Sugimoto H, Nagase H, Nakagawara A. Runt-related transcription factor 2 (RUNX2) inhibits p53-dependent apoptosis through the collaboration with HDAC6 in response to DNA damage. Cell Death Dis. 2013;4:e610. doi: 10.1038/cddis.2013.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Croxford JL, Tang ML, Pan MF, et al. ATM-dependent spontaneous regression of early Emu-myc-induced murine B-cell leukemia depends on natural killer and T cells. Blood. 2013;121(13):2512–2521. doi: 10.1182/blood-2012-08-449025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yoo J, Lee HN, Choi I, et al. Opposing regulation of PROX1 by interleukin-3 receptor and NOTCH directs differential host cell fate reprogramming by Kaposi sarcoma herpes virus. PLoS Pathog. 2012;8(6):e1002770. doi: 10.1371/journal.ppat.1002770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Migone TS, Lin JX, Cereseto A, et al. Constitutively activated Jak–STAT pathway in T cells transformed with HTLV-I. Science. 1995;269(5220):79–81. doi: 10.1126/science.7604283. [DOI] [PubMed] [Google Scholar]
- 101.Zhao X, Madden-Fuentes RJ, Lou BX, et al. Ataxia telangiectasia-mutated damage-signaling kinase- and proteasome-dependent destruction of Mre11–Rad50–Nbs1 subunits in Simian virus 40-infected primate cells. J Virol. 2008;82(11):5316–5328. doi: 10.1128/JVI.02677-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sowd GA, Li NY, Fanning E. ATM and ATR activities maintain replication fork integrity during SV40 chromatin replication. PLoS Pathog. 2013;9(4):e1003283. doi: 10.1371/journal.ppat.1003283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li R, Zhu J, Xie Z, et al. Conserved herpes virus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe. 2011;10(4):390–400. doi: 10.1016/j.chom.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.McFadden K, Luftig MA. Interplay between DNA tumor viruses and the host DNA damage response. Curr Top Microbiol Immunol. 2013;371:229–257. doi: 10.1007/978-3-642-37765-5_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Luo Y, Lou S, Deng X, et al. Parvovirus B19 infection of human primary erythroid progenitor cells triggers ATR–Chk1 signaling, which promotes B19 virus replication. J Virol. 2011;85(16):8046–8055. doi: 10.1128/JVI.00831-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability – an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11(3):220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]