

Abstract
The purine nucleoside adenosine is present in all cells in tightly regulated concentrations. It is released under a variety of physiological and pathophysiological conditions to facilitate protection and regeneration of tissues. Adenosine acts via specific GPCRs to either stimulate cyclic AMP formation, as exemplified by Gs-protein-coupled adenosine receptors (A2A and A2B), or inhibit AC activity, in the case of Gi/o-coupled adenosine receptors (A1 and A3). Recent advances in our understanding of GPCR structure have provided insights into the conformational changes that occur during receptor activation following binding of agonists to orthosteric (i.e. at the same binding site as an endogenous modulator) and allosteric regulators to allosteric sites (i.e. at a site that is topographically distinct from the endogenous modulator). Binding of drugs to allosteric sites may lead to changes in affinity or efficacy, and affords considerable potential for increased selectivity in new drug development. Herein, we provide an overview of the properties of selective allosteric regulators of the adenosine A1 and A3 receptors, focusing on the impact of receptor dimerization, mechanistic approaches to single-cell ligand-binding kinetics and the effects of A1- and A3-receptor allosteric modulators on in vivo pharmacology.
Linked ArticlesThis article is part of a themed section on Molecular Pharmacology of GPCRs. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2014.171.issue-5
Keywords: adenosine, allosterism, receptor, PCR, dimerization, biased signalling
Introduction
Adenosine is a reactive metabolite that has a major role in regulating a number of physiological and pathological processes, including inflammation, pain, hypoxia and cardiovascular regulation (Fredholm et al., 2011). Adenosine acts via four specific GPCRs, which have been denoted as adenosine A1, A2A, A2B and A3 receptors (Alexander et al., 2013; Fredholm et al., 2011). The A1 and A3 receptors preferentially couple to Gi/o proteins and have an inhibitor action on AC activity, while the A2A and A2B receptors couple to Gs proteins and stimulate cyclic AMP formation (Jacobson, 2009; Fredholm et al., 2011; Muller and Jacobson, 2011). The crystal structure of the A2A receptor in both antagonist (Jaakola et al., 2008) and agonist (Xu et al., 2011) bound conformations has recently been solved. Numerous selective agonists and antagonists for each adenosine receptor subtype are now available for the study of receptor function (reviewed in Jacobson, 2009; Fredholm et al., 2011; Muller and Jacobson, 2011). In the case of the Gi/o-coupled adenosine receptors (A1 and A3) reviewed here, a number of compounds are undergoing evaluation for disease indications (Muller and Jacobson, 2011). These include A1-receptor partial agonists (e.g. capadenoson, selodenoson and tecadenoson) for paroxysmal supraventricular tachycardia, atrial fibrillation and angina pectoris, and A3-receptor agonists (e.g. CF101, CF102) for inflammatory disease, glaucoma and cancer (Bar-Yehuda et al., 2011; Cohen et al., 2011; Muller and Jacobson, 2011; Albrecht-Kupper et al., 2012; Tendera et al., 2012).
Activation of cell surface adenosine receptors by endogenous adenosine requires it to be available at the extracellular surface of cells. Extracellular adenosine can rise as a consequence of several pathways (Fredholm et al., 2011). It can be formed intracellularly following various metabolic processes and be exported from cells via membrane transporters, or it can be formed in the extracellular space from adenine nucleotides released from cells. Once ATP or ADP is released, the nucleotide is broken down by nucleoside triphosphate diphosphohydrolases (e.g. CD39) and then ecto-5′-nucleotidase (CD73) to adenosine (Fredholm et al., 2011; Knapp et al., 2012). It is well known that neurons and platelets can store and release ATP and ADP, respectively, in response nerve stimulation and platelet activation. However, more recently, there has been a growing awareness that the intracellular second messenger cyclic AMP may also be a source of extracellular adenosine in many cell types. Thus, intracellular cyclic AMP can be released from cells in response to receptor stimulation (McCrea and Hill, 1993; Baker et al., 2004) and it is known that extracellular cyclic AMP can be rapidly converted to adenosine via the action of ecto-phosphodiesterase and ecto-5′-nucleotidase (Dubey et al., 2001; Chiavegatta et al., 2008; Goedeke, 2008).
The intricacies of localized extracellular release of adenine nucleotides and subsequent production of adenosine following CD73 activity has recently provided insights into the role of adenosine A1 receptors in mediating localized analgesia in animals and humans (Goldman et al., 2010; Sowa et al., 2010; Street and Zylka, 2011). Thus, there is evidence that localized A1-receptor activation may underlie the antinociceptive effects of acupuncture as manual stimulation of acupuncture needles can result in localized release of adenine nucleotides and adenosine formation, leading to an analgesia that can be mimicked by an A1-receptor agonist (Goldman et al., 2010). Furthermore, intrathecal application of recombinant CD73 (to enhance formation of adenosine) produced a long-lasting antinociceptive effect that was not observed in adenosine A1-receptor knockout mice (Sowa et al., 2010). Similarly, recent work with human neutrophils has highlighted an autocrine role for ATP, which is released from the leading edge of neutrophils during chemotaxis (Chen et al., 2006; Corriden and Insel, 2012). ATP is then rapidly converted to adenosine, which then acts via adenosine A3 receptors (recruited to the leading edge of neutrophils) to promote cell migration (Chen et al., 2006; Corriden and Insel, 2012; Corriden et al., 2013). In addition, there is increasing evidence that adenosine A1 and A3 receptors may be involved in promoting angiogenesis and the release of VEGF in response to local hypoxia and neoplasia (Clark et al., 2007; Merighi et al., 2009).
The above studies suggest that localized regulation of adenosine production (e.g. by recombinant CD73), or its activity at its target adenosine receptor itself (e.g. A1 or A3), may have important therapeutic implications. One way in which the activity of endogenous adenosine can be subtly regulated at the level of its target receptor is via drugs that bind to an allosteric site on the receptor and act as allosteric modulators to enhance or inhibit the binding and/or function of adenosine. Here, we review the properties of various small-molecule allosteric regulators of the adenosine A1 and A3 receptors focusing on the impact of receptor dimerization, mechanistic approaches to single-cell ligand-binding kinetics and the effects of A1- and A3-receptor allosteric modulators on in vivo pharmacology.
Allosteric regulation of GPCRs
GPCRs comprise the largest family of transmembrane proteins and represent major targets for drug discovery (Williams and Hill, 2009; Roth and Marshall, 2012). Considerable advances in our knowledge of GPCR structure have been made recently (Jaakola et al., 2008; Chien et al., 2010; Chung et al., 2011; Rasmussen et al., 2011) and this has led to significant insights into the conformational changes that occur during receptor activation in response to agonists that act at the same site (orthosteric) as the endogenous hormone or neurotransmitter (Chung et al., 2011; de Graaf et al., 2011; Rasmussen et al., 2011). However, over the past decade, there has been an increasing acceptance that drugs can also bind to a topographically distinct site (allosteric) on the GPCR protein and elicit a conformational change that can lead to a change in the affinity or efficacy of a ligand occupying the classical orthosteric binding site (Figure 1A; May et al., 2007; Kenakin, 2009, 2012; Keov et al., 2010). This suggests that GPCRs are able to bind more than one ligand simultaneously (i.e. both an allosteric and an orthosteric ligand; May et al., 2007; Kenakin, 2009, 2012; Keov et al., 2010). Various mathematical models have been developed to explain these phenomena, but key features of an allosteric mechanism of action are that the effect is saturable, can depend on the specific ligand occupying the orthosteric site (probe dependence) and provides scope for both positive and negative effects on ligand binding and/or function (May et al., 2007; Kenakin, 2009, 2012; Keov et al., 2010).
Figure 1.

Schematic representation of allosteric regulation of GPCRs by (A) allosteric ligands, (B) signalling proteins or (C) GPCR dimerization. See text for further explanation.
Some of the earliest allosteric modulators were discovered for the adenosine A1 receptor (Bruns and Fergus, 1990; Bruns et al., 1990; Göblyös and Ijzerman, 2011; Kimatrai-Salvador et al., 2012). PD 81,723 has become a reference allosteric enhancer for the A1 receptor. Early studies demonstrated that PD 81,723, which has A1-receptor antagonist properties at high concentrations, was able to increase the binding of an orthosteric agonist radioligand at lower concentrations of PD 81,723 to enhance the functional activation of the A1 receptor in the brain (Janusz and Berman, 1993) and cardiovascular tissues (Amoah-Apraku et al., 1993) and to slow down the dissociation of the agonist radioligand from the A1 receptor (Bruns and Fergus, 1990; Bruns et al., 1990); the latter effect being indicative of an allosteric mechanism of action (see below; May et al., 2007; Keov et al., 2010; Göblyös and Ijzerman, 2011). Furthermore, recent studies using site-directed mutagenesis have indicated that the allosteric binding site for PD 81,723 may reside within extracellular loop 2 of the adenosine A1 receptor (Peeters et al., 2012).
Selective allosteric enhancers of agonist binding have also been described for the adenosine A3 receptor (Gao et al., 2001; Heitman et al., 2009; Göblyös and Ijzerman, 2011). The impact of an allosteric modulator is not, however, restricted to the binding and function of orthosteric agonists. For example, the food dye Brilliant Black BN is able to act allosterically to reduce the affinity of particular adenosine A1- and A3-receptor antagonists (e.g. xanthine amine congener) for the orthosteric site without altering the ability of agonists to interact with these two receptors (May et al., 2010a). This is a good example of probe dependence where the effect observed differs, depending on the nature of the ligand occupying the orthosteric site. In addition to small molecules exerting allosteric influences on GPCRs, there is considerable evidence that sodium ions can also mediate allosteric effects on a range of GPCRs, including both the adenosine A1 and A3 receptors (Liu et al., 2012). In the case of A1 and A3 receptors, a highly conserved aspartate residue in transmembrane region 2 of each receptor (Asp2,50) has been implicated in the allosteric actions of sodium ions. Mutation of this residue to alanine or asparagine largely abolishes the effect (Barbhaiya et al., 1996; Gao et al., 2003). In a recent high-resolution crystal structure of the adenosine A2A receptor, the precise location of the sodium ion and its associated water cluster has been identified and shown to interact with Asp2,50 (Liu et al., 2012).
Recent studies with imidazoquinolinamine allosteric enhancers (e.g. LUF5999, LUF6000 and LUF6001) of the adenosine A3 receptor have shown that they have differing effects on the affinity and efficacy of a selective A3-agonist Cl-IB-MECA (Gao et al., 2011). This illustrates the independence of allosteric actions on binding affinity and efficacy. Furthermore, the allosteric modulation of orthosteric agonist efficacy was dependent on the intracellular signalling response being measured. This suggests that the allosteric modulation of agonist efficacy may be functionally biased (Gao et al., 2011). Functional selectivity of orthosteric and allosteric ligands has also been investigated for the adenosine A1 receptor (Cordeaux et al., 2004; Valant et al., 2010; Langemeijer et al., 2013). The magnitude of positive allosteric modulation of the 2-amino-3-benzoylthiophene adenosine A1-receptor allosteric enhancer, VCP520, varied between pathways (Valant et al., 2010). This is an example of an allosteric modulation engendering functional selectivity in the actions of orthosteric ligands (Valant et al., 2010). These studies highlight the ability of allosteric ligands to further ‘fine-tune’ orthosteric ligand responses. Signalling bias from GPCRs is a concept that has developed considerably over the last few years as knowledge that GPCRs can regulate signalling pathways independently of heterotrimeric G protein has become available (e.g. β-arrestin pathways; Kenakin, 2012; Whalen et al., 2011). Thus, it is clear that activation of β-arrestin pathways are not only associated with desensitization and receptor internalization but can also change the signalling pathways that are activated. Furthermore, specific agonists appear to be able to direct signalling to different pathways via the same cell surface receptor. Some of the best evidence for this has come from the β2-adenoceptor field, where certain β-blockers (e.g. propranolol) can have an inverse agonist effect of Gs-mediated signalling pathways, but an agonist action on MAP kinase (Azzi et al., 2003; Baker et al., 2003). The concept of biased signalling, however, is a natural extension of allosterism (Figure 1B). The intracellular signalling proteins (e.g. heterotrimeric G proteins of β-arrestin) bind to the GPCR at a site distinct from the orthosteric binding site. As a consequence, they can be considered as allosteric regulators (in this case, proteins) that can have a reciprocal effect on ligand binding (or coupling in the case of the protein) and lead to altered affinity and efficacy for particular agonists (Kenakin, 2012). In many ways, therefore, biased signalling is a natural consequence of a key feature of allosterism, namely probe dependence (Figure 1B).
The ability of receptor-associated proteins to act as allosteric modulators of ligand binding and efficacy can be extended to neighbouring receptors that form homo- or heterodimers or higher order oligomers (Figure 1C). For example, we have recently provided evidence for negative cooperativity across the dimer interface of an adenosine A3-receptor homodimer (May et al., 2011). In this case, binding of an orthosteric ligand to one protomer (monomeric component) of the homomeric complex can markedly alter the affinity of a ligand binding to the second protomer (May et al., 2011). Evidence is also accumulating that the adenosine A1 receptor can form heterodimers with P2Y receptors, adenosine A2A receptors, β1- and β2-adrenoceptors to influence orthosteric ligand binding and/or intracellular signalling (Suzuki et al., 2006; Chandrasekera et al., 2013; Cristovao-Ferreira et al., 2013; Franco et al., 2013). In addition to partner receptors within oligomeric complexes, other extracellular proteins can also bind to GPCRs and mediate allosteric influences. For example, adenosine deaminase (ADA), which is a key enzyme catalysing the deamination of adenosine, can be released from cells and bind to cell surface proteins and act as an ectoenzyme (Gracia et al., 2013). One of the proteins that bind ADA is the adenosine A1 receptor (Ciruela et al., 1996; Gracia et al., 2013). The association of ADA with the adenosine A1 receptor can lead to enhanced agonist affinity and efficacy (Gracia et al., 2008; 2013). The consequence of a metabolic enzyme for the endogenous activator being associated with the cell surface adenosine receptor, which can enhance affinity and efficacy, is therefore likely to amplify local signalling while limiting the duration of action (and spread of activity) due to its local metabolic activity.
Overview of small-molecule allosteric regulators acting on the adenosine A1 and A3 receptors
The therapeutic potential of allosteric regulators that can amplify or modulate the local actions of adenosine is clear and efforts are in progress to develop these reagents for a wide range of GPCRs. The development of allosteric modulators targeted at the adenosine receptor family has recently been comprehensively reviewed (Göblyös and Ijzerman, 2011; Jacobson et al., 2011). For brevity, we will focus our discussion on the medicinal chemistry of allosteric modulators specifically mentioned within this review (Figure 2). As stated earlier, PD 81,723 was one of the key compounds originally described in back-to-back papers from the Parke-Davis Pharmaceutical Research Division (Bruns and Fergus, 1990; Bruns et al., 1990) recounting the identification of the first allosteric regulators of adenosine A1-receptor binding. The chemical series was originally identified from the Parke-Davis compound bank via a 300-ligand adenosine A1 binding screen. While the 2-amino-3-benzoylthiophene chemical scaffolds had been originally synthesized as intermediates for benzodiazepine-like compounds (Tinney et al., 1974), recognition of their adenosine antagonist activity prompted a more thorough analysis of this privileged chemical template (Bruns and Fergus, 1990). From this medicinal chemistry study, second-generation compounds were unexpectedly found to increase the specific binding of [3H]N6-cyclohexyladenosine to rat brain membranes. This, in turn, resulted in the synthesis of further compounds to identify pertinent structure–activity relationships and, in so doing, identified PD 81,723 as a key analogue displaying a significantly improved allosteric profile (Bruns et al., 1990). This core structure has been further modified by a number of groups thereby developing a robust structure activity relationship profile and a series of ligands with comparable or more favourable allosteric activity (van der Klein et al., 1999; Kourounakis et al., 2000; Baraldi et al., 2003, 2004; Nikolakopoulos et al., 2006; Romagnoli et al., 2008; Valant et al., 2010). One particularly successful manipulation centred on removal of the 4- and 5-methyl groups and installation of substituted phenyl rings back into these positions of PD 81,723 (Aurelio et al., 2008). It was interesting to note that the most efficacious compound possessed no substituent in the 5-position and this lead to further exploitation of this observation through the synthesis and evaluation of the next generation of ligands, which identified VCP520 as a potent allosteric enhancer of A1-receptor-mediated signalling (Aurelio et al., 2009).
Figure 2.
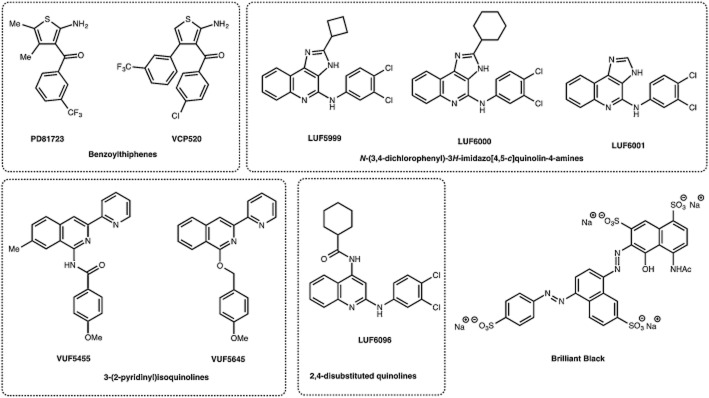
A selection of adenosine receptor allosteric modulators.
In a similar fashion as the discovery of PD 81,723, the lead compounds recognized as allosteric modulators of the adenosine A3 receptor were identified from screening diverse chemical libraries in binding assays at this receptor subtype (Jacobson et al., 2011). In this instance, certain lead molecules were shown to increase the level of binding of [125I]AB-MECA (Gao et al., 2001; 2002). Key molecular scaffolds that supported allosteric modulation at the adenosine A3 receptor were identified as 3-(2-pyridinyl)isoquinolines (e.g. VUF5455) and 3H-imidazo-[4,5-c]quinolin-4-amines (e.g. LUF5999, LUF6000 and LUF6001). With regard to the former, further exploration of the 3-(2-pyridinyl)isoquinoline scaffold revealed a complex situation where some members were pure antagonists of the orthosteric binding site, for example, VUF5455 itself (Heitman et al., 2009), consequently rendering them not particularly useful as future therapeutics.
However, the imidazoquinolinamines fared better in this respect; the original molecule DU124183 (Gao et al., 2002) was further modified at the 2- and 4-positions and numerous resultant derivatives displayed potentiation of the maximum efficacy of Cl-IB-MECA at the A3 receptor (Göblyös et al., 2006). Indeed, LUF6000 was shown to enhance agonist efficacy in a functional assay and decrease agonist dissociation rate without influencing agonist potency. This was postulated to be a result of its experimentally observed decreased interaction with the orthosteric binding site on the adenosine A3 receptor. As previously mentioned, a more thorough analysis of this and related ligands (LUF5999 and LUF6001) identified that these imidazoquinolinamine allosteric enhancers displayed differing effects on the affinity and efficacy of Cl-IB-MECA at the A3 receptor (Gao et al., 2011). In a related study, with the intention of overcoming the issues associated with the orthosteric antagonism shown by the 3-(2-pyridinyl)isoquinolines, a series of ring opened imidazoquinolinamines were synthesized to afford a range of 2,4-disubstituted quinolines as a new class of allosteric enhancers at the A3 receptor (Heitman et al., 2009). Rewardingly, the best compound (LUF6096) was not only able to allosterically enhance the binding of Cl-IB-MECA to a similar level as LUF6000 but it also displayed negligible orthosteric affinity for any of the adenosine receptor subtypes. These compounds have begun to be used in mechanistic studies to identify the basis of these allosteric effects on efficacy and affinity and the extent to which these two effects are related.
Mechanistic insights from single-cell ligand-binding kinetics
Allosteric interactions are a mode of communication between distal binding sites. Intra- and intermolecular GPCR allosterism with transmembrane proteins and allosteric small molecules can generate a unique spectrum of resting and/or active distribution of GPCR conformations, which, in turn, can significantly influence the pharmacology of orthosteric and/or allosteric ligands. Typically, GPCR allosterism changes the properties of conformationally linked binding sites and therefore the association and/or dissociation kinetics of the cognate orthosteric ligands (May et al., 2007; Smith and Milligan, 2010). Orthosteric ligand affinity is described by the ratio of the association to dissociation rates, and as such, an allosteric interaction that alters orthosteric ligand affinity does so by mediating a change in one or both of these parameters. Dissociation kinetic assays can be used as a powerful mechanism to validate an allosteric mechanism of action of a ligand since orthosteric and allosteric ligands must interact with the receptor simultaneously to change the dissociation kinetics of a labelled orthosteric ligand. Plotting the dissociation rate of labelled orthosteric ligand in the presence of a range of interacting ligand concentrations provides a concentration–response relationship of a purely allosteric effect (Kostenis and Mohr, 1996). Furthermore, the midpoint of this curve provides an estimate of affinity of the orthosteric ligand for the allosteric modulator occupied receptor.
Typically, dissociation kinetic studies investigating intramolecular allosterism use isotopic dilution. That is, the influence of an allosteric ligand on the dissociation kinetics of an orthosteric radiolabelled probe is assessed in the presence of a saturating concentration of a second competitive orthosteric ligand (Bruns and Fergus, 1990; Ellis et al., 1991; Lee and el-Fakahany, 1991; Lazareno and Birdsall, 1995; Christopoulos et al., 1997; Gao et al., 2001; Avlani et al., 2004; Dowling and Charlton, 2006). A key assumption required for interpreting such dissociation kinetic studies, however, is that the second ligand does not alter the rate of radioligand dissociation. This assumption is consistent within a theoretical framework describing competitive interactions between compounds at a monomeric receptor; however, more complex interactions resulting from multistep ligand binding (Swaminath et al., 2004; Ilien et al., 2009) or receptor dimerization (Christopoulos and Kenakin, 2002; Springael et al., 2006; Han et al., 2009; May et al., 2011) could lead to a change in the radioligand dissociation rate. Recently, the binding kinetics of a fluorescent adenosine derivative was determined in the absence and presence of allosteric modulators at the adenosine A1 and A3 receptor in live single cells (May et al., 2010b). Importantly, these studies were performed using a closed perfusion system that enabled rapid removal of free ligand (May et al., 2010a) and therefore assessed the dissociation kinetics under ‘infinite dilution’ conditions in the absence of a saturating concentration of competitive orthosteric ligand. Similar to the previous studies, which used isotopic dilution to promote orthosteric radioligand dissociation, PD 81,723 significantly retarded the dissociation of the fluorescent adenosine derivate from the adenosine A1 receptor (May et al., 2010b). In contrast, VUF5455, which has previously been demonstrated to decrease the rate of agonist dissociation from the adenosine A3 receptor (Gao et al., 2001), was found to significantly enhance the fluorescent agonist dissociation rate (May et al., 2010b). This discrepancy may reflect the different orthosteric agonist probes used in the different studies and therefore the ability of allosteric modulators to be highly probe-dependent (May et al., 2007). Alternatively, the difference could reflect a more complex receptor arrangement than a non-interacting monomer, that is, a dimer or higher order oligomer (see below).
Traditionally, GPCRs have been considered to exist and function as monomeric proteins. However, it is now known that GPCRs can form homodimers, heterodimers and/or higher order oligomers (Smith and Milligan, 2010). Non-visual GPCRs can be classified into three families, A–C. Family C GPCRs are known to function as obligate dimers (May et al., 2007; Smith and Milligan, 2010). In contrast, the extent, stability and physiological consequence of dimerization remains highly controversial for family A GPCRs, which are the largest family and include the well-characterized adrenoceptors, dopamine receptors, adenosine receptors and muscarinic ACh receptors (Smith and Milligan, 2010). Monomeric family A GPCRs reconstituted in high-density lipoprotein phospholipid bilayer particles can couple to G proteins, suggesting that this family does not need to function as obligate dimers (Kuszak et al., 2009). However, evidence suggests that cell surface complexes of family A GPCRs may display a distinct profile of functional properties relative to their monomeric counterparts. For example, dimerization and/or oligomerization may influence signal transduction efficiency, receptor desensitization and/or the ligand preference for coupling to particular downstream signalling cascades (May et al., 2007; Smith and Milligan, 2010; Franco et al., 2013). Furthermore, a recent study provided evidence for communication between simultaneously bound orthosteric sites on homodimeric dopamine D2 receptors (Urizar et al., 2011). As such, a ligand bound to one protomer can modulate ligand function and/or affinity at a second interacting protomer. This may lead to complex pharmacology and/or the potential for dimeric species to elicit specific signalling events with unique pharmacological properties.
The fundamental premise of intra- and intermolecular allosteric modulation is based on conformational rearrangements; therefore, a wealth of information can be gained through assessing ligand-binding kinetics under different conditions. Dissociation kinetic studies provided the first evidence for homodimerization of a family A GPCR, the β-adrenergic receptor. This study used an ‘infinite dilution’ approach to detect a change in the dissociation kinetics of the radiolabelled orthosteric ligand, [3H](-)alprenolol, in the absence and presence of unlabelled (-)alprenolol. The increased dissociation rate in the presence of unlabelled orthosteric ligand, (-)alprenolol, was suggestive of negatively cooperative interactions across a β-adrenergic homodimeric interface (Limbird et al., 1975). Intermolecular cooperativity between orthosteric binding sites has since been established for a number of additional GPCRs, including adenosine and muscarinic ACh receptor subtypes (Briddon et al., 2008; Casadó et al., 2010; Hern et al., 2010; Pisterzi et al., 2010; Hu et al., 2012; May et al., 2011). At the adenosine A1 and A3receptors, dissociation kinetic analysis has been employed as a powerful method to detect intermolecular allosterism, that is, cooperative interactions across a homodimeric interface (May et al., 2011). In contrast to the adenosine A1 receptor, highly cooperative interactions were observed under ‘infinite dilution’ conditions between the fluorescent adenosine derivative and orthosteric agonists and antagonists at the adenosine A3 receptor. Figure 3 shows an example of the effect of increasing concentrations of the endogenous orthosteric ligand adenosine on the dissociation kinetics of a fluorescent adenosine analogue from the human adenosine A3 receptor. In marked contrast, adenosine had a much less marked effect on the dissociation kinetics of the fluorescent ligand from the human adenosine A1 receptor (May et al., 2011). Importantly, the intermolecular allosterism was significantly decreased upon co-expression of a non-binding adenosine A3-receptor mutant, supporting the suggestion of cooperative interactions across the dimeric interface of cell surface adenosine A3 receptors (May et al., 2011). These studies add strength to the suggestion that the discrepancy observed between the influence of the allosteric modulator, VUF5455, in dissociation kinetic studies using isotopic dilution as compared to infinite dilution may reflect the ability of adenosine A3 receptors to form interacting homodimers and/or higher order oligomers. In keeping with this hypothesis, if a non-fluorescent orthosteric ligand (e.g. MRS1220) is added simultaneously with a derivative of VUF5455 (VUF5645), then this allosteric compound then produces a slowing down of the dissociation kinetics of the fluorescent adenosine analogue (Figure 4).
Figure 3.
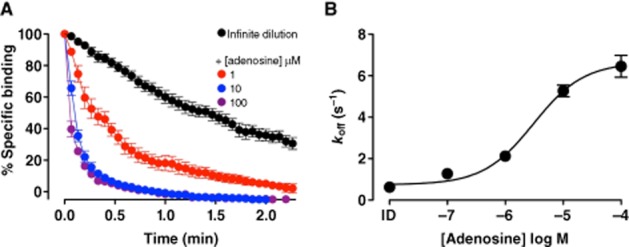
Adenosine mediates a significant enhancement in the dissociation of 30 nM ABA-X-BY630 from the human adenosine A3 receptor. (A) Dissociation of a fluorescent adenosine analogue, ABA-X-BY630 (30 nM), from CHO-A3 cells in the absence or presence of adenosine (1 μM; 10 μM; 100 μM). (B) Concentration dependence of the changes in koff of 30 nM ABA-X-BY630 from CHO-A3 cells in the absence and presence of adenosine. Data points are expressed as mean ± SEM from 3–11 separate experiments; each replicate represents the average fluorescence at the plasma membrane of 10 individual cells. Data taken from May et al. (2011).
Figure 4.
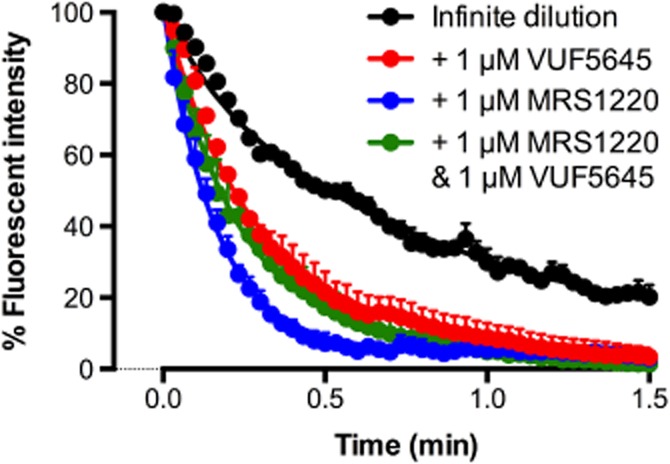
The influence of the competitive antagonist, MRS1220, and/or the allosteric ligand, VUF5645, on the dissociation kinetics of the fluorescent adenosine derivative, ABA-X-BY630. ABA-X-BY630 (30 nM) dissociation in the absence and presence of a 1 μM MRS1220, 1 μM VUF5645, or 1 μM MRS1220 and 1 μM VUF5645. Representative data performed in duplicate; each replicate represent the average fluorescence at the plasma membrane of 10 individual cells.
Impact of A1- and A3-receptor allosteric modulators on in vivo pharmacology
Adenosine A1- and A3-receptor ligands (both agonists and antagonists) have been developed for a number of potential therapeutic indications (Muller and Jacobson, 2011). These are summarized in Table 1. The best developed indications appear to be for agonists where A1-receptor agonism may have utility in angina, neuropathic pain, paroxysmal supraventricular tachycardia and ischaemia (Griffin et al., 2003; Morrison et al., 2006; Albrecht-Kupper et al., 2012; Tendera et al., 2012), and A3-receptor agonists may have benefit in liver cancer, rheumatoid arthritis, autoimmune inflammatory disease, dry eye and cardiac ischaemia (Table 1; Bar-Yehuda et al., 2011; Cohen et al., 2011; Fishman et al., 2012). With regard to cancer, it is interesting that A3 receptors appear to be overexpressed in certain cancers (e.g. breast and colon cancer) compared to normal cells (Gessi et al., 2004; Madi et al., 2004; Bar-Yehuda et al., 1990; Fishman et al., 2012).
Table 1.
Therapeutic indications for selective A1 and A3 adenosine receptor ligands
| Adenosine receptor | Changes following genetic deficit or overexpression | Potential therapeutic indications | Example drugs | |
|---|---|---|---|---|
| A1 | Analgesic effects of adenosine abolished in A1-KO mice. Overexpression of cardiac A1 receptors caused increased resistance to ischaemia. | Agonist | Atrial fibrillation, angina, hyperlipidaemia, neuropathic pain, paroxysmal supraventricular tachycardia, cardiac ischaemia | Capadenoson, Tecadenoson, RPR749, GR79236 |
| Antagonist | Acute renal failure, heart failure (renal function) | FK-453, SLC320 | ||
| A3 | Enhanced antigen-stimulated mast cell degranulation by A3-agonists lost in A3-KO mice. Studies with congenic A3-/- mice indicate a cadioprotective effect of A3-receptor activation. | Agonist | Liver cancer, rheumatoid arthritis, autoimmune inflammatory disease, dry eye, cardiac ischaemia, dry eye | Cl-IB-MECA (CF102) MRS3558 (CF502) IB-MECA (CF101) |
| Antagonist | Asthma, glaucoma | KF26777, OT-7999 | ||
KO, knockout.
Studies with genetically altered mice have also suggested a role of A1 receptors in pain (Sowa et al., 2010) and ischaemia (Matherne et al., 1997) and for A3 receptors in cardiac ischaemia (Ge et al., 2006) and mast cell degranulation (Salvatore et al., 2000). In the latter case, it is worth pointing out that functional A3 receptors appear to be absent from human mast cells (Fredholm et al., 2011). However, although selective A3-receptor activation is cardio-protective in wild-type mice and those overexpressing the A1 receptor, adenosine A3-receptor gene deletion generates an ischaemia-tolerant phenotype that might be indicative of compensatory changes (Harrison et al., 2002).
As mentioned in the introductory remarks, the ubiquitous distribution of adenosine receptors and the potential for serious side effects via the target receptor in a different organ or cell type can limit their utility. For example, in many non-cardiac therapeutic applications of A1-receptor agonists, the potential for major side effects due to A1-receptor actions in the heart will be seriously limiting. This may be particularly true in the case of adenosine A1-receptor agonists that may have potential utility in the treatment of CNS diseases, such as epilepsy (Mares, 2010; Klaft et al., 2012). This has led to the development of partial agonists (e.g. capadenoson; Albrecht-Kupper et al., 2012; Tendera et al., 2012) that may have less severe off-target profiles and are less prone to receptor desensitization. The potential to overcome these limitations with allosteric enhancers is obvious, particularly if the advantages offered by instilling bias into the final signalling outcome can be exploited (as a consequence of the allosteric impact of partner receptor-interacting proteins). However, although in vitro studies have provided convincing evidence for allosteric mechanisms of action for a number of ligands, therapeutic application of these mechanisms relies on their successful translation into whole animal physiology. Indeed, the in vivo actions of allosteric regulators have not been extensively investigated and there is a need to evaluate the potential for these small molecules to augment specific actions of adenosine in particular organs and cell types in a whole animal setting.
Some success has been achieved in vivo with two allosteric ligands. Adenosine receptor activation has been implicated in the mechanism of ischaemic pre-conditioning (Carr et al., 1997; Uematsu et al., 1998). Ischaemic pre-conditioning is where an organ (normally the heart) is subjected to brief periods of ischaemia and reperfusion, resulting in a resistance to infarction. For example, in human atrial muscle, both adenosine A1- and A3-receptor activation can mimic ischaemic pre-conditioning (Carr et al., 1997). Activation of adenosine A1 receptors has been shown to protect against renal ischaemia/reperfusion injury (Lee and Emala, 2000; Lee et al., 2004). However, extra-renal side effects (e.g. bradycardia, hypotension) may limit the use of A1-receptor agonist therapy for acute ischaemic kidney injury (Park et al., 2012). Interestingly, the A1-receptor allosteric enhancer PD 81,723 produced a dose-dependent protection against ischaemia/reperfusion injury in the kidneys of wild-type mice but not in adenosine A1-receptor-deficient mice (Park et al., 2012). This was achieved in the absence of significant effects on heart rate and BP, which suggests that renal A1-receptor selectivity had been achieved with PD 81,723 as a consequence of amplifying the restricted increase in adenosine in the kidney following local ischaemia (Park et al., 2012). A similar outcome has been reported in the CNS where administration of PD 81,723 can lead to a reduction in hippocampal injury following hyperglycaemic ischaemia in the rat (Meno et al., 2003).
A positive allosteric modulator of the adenosine A3 receptor (LUF6096) has also been shown to have benefit in an in vivo model of myocardial ischaemia/reperfusion injury in the dog (Du et al., 2012). Thus, LUF6096 had no effect on baseline haemodynamic parameters, but pre-treatment with LUF6096 prior to coronary occlusion and during reperfusion produced a marked reduction in infarct size (ca. 50% reduction; Du et al., 2012). An equivalent reduction in the infarct size could also be demonstrated if LUF6096 was administered immediately before reperfusion (Du et al., 2012). These studies collectively indicate that allosteric enhancers of the adenosine A1 and A3 receptors may have great utility as therapeutic strategies to provide selective augmentation of the actions of adenosine released locally in conditions of disease and stress.
Concluding remarks
It is clear that allosteric mechanisms of action provide unique ways to regulate receptor function at a local level to ‘fine-tune’ intracellular signalling. This can be achieved by small molecules (allosteric regulators) or by protein–protein interactions involving signalling proteins (leading to biased signalling) or oligomeric partners (e.g. as a consequence dimerization). In all cases, these mechanisms provide the potential to exploit the unique pharmacology provided by allosterism to achieve both better cell and tissue selectivity of drug treatments and also interventions with more physiologically relevant kinetic profiles. Novel fluorescent techniques have been able to unravel some of the intricacies involved at the single cell level. However, the therapeutic potential of these actions awaits the clear demonstration of these mechanisms in an in vivo setting.
Acknowledgments
We thank the Medical Research Council (G0800006) and NHMRC for the financial support and Prof Rob Leurs (VU University, Amsterdam) for supplying VUF5645.
Glossary
- ADA
adenosine deaminase
Conflict of interest
None.
References
- Albrecht-Kupper BE, Leineweber K, Nell PG. Partial adenosine A1-receptor agonists for cardiovascular therapies. Purinergic Signal. 2012;8:S91–S99. doi: 10.1007/s11302-011-9274-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, Spedding M, Peters JA, Harmar AJ CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amoah-Apraku B, Xu J, Lu JY, Pelleg A, Bruns RF, Belardinelli L. Selective potentiation by an A1 adenosine receptor enhancer of the negative dromotropic action of adenosine in the guinea-pig heart. J Pharm Exp Ther. 1993;266:611–617. [PubMed] [Google Scholar]
- Aurelio L, Figler H, Flynn BL, Linden J, Scammells PJ. 5-Substituted 2-aminothiophenes as A1 adenosine receptor allosteric enhancers. Bioorg Med Chem. 2008;16:1319–1327. doi: 10.1016/j.bmc.2007.10.065. [DOI] [PubMed] [Google Scholar]
- Aurelio L, Valant C, Flynn BL, Sexton PM, Christopoulos A, Scammells PJ. Allosteric modulators of the adenosine A1 receptor: synthesis and pharmacological evaluation of 4-substituted 2-amino-3-benzoylthiophenes. J Med Chem. 2009;52:4543–4547. doi: 10.1021/jm9002582. [DOI] [PubMed] [Google Scholar]
- Avlani V, May LT, Sexton PM, Christopoulos A. Application of a kinetic model to the apparently complex behavior of negative and positive allosteric modulators of muscarinic acetylcholine receptors. J Pharmacol Exp Ther. 2004;308:1062–1072. doi: 10.1124/jpet.103.059840. [DOI] [PubMed] [Google Scholar]
- Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, et al. Beta-arrestin-mediated activation of MPAK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci U S A. 2003;100:11406–11411. doi: 10.1073/pnas.1936664100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JG, Hall IP, Hill SJ. Agonist and inverse agonist actions of ‘β-blockers’ at the human β2-adrenoceptor provide evidence for agonist-directed signalling. Mol Pharmacol. 2003;64:1357–1369. doi: 10.1124/mol.64.6.1357. [DOI] [PubMed] [Google Scholar]
- Baker JG, Hall IP, Hill SJ. Temporal characteristics of CRE-mediated gene transcription: requirement for sustained cAMP production. Mol Pharmacol. 2004;65:986–998. doi: 10.1124/mol.65.4.986. [DOI] [PubMed] [Google Scholar]
- Baraldi PG, Romagnoli R, Pavani MG Nunez MC, Tabrizi MA, Shryock JC, et al. Synthesis and biological effects of novel 2-amino-3-naphthoylthiophenes as allosteric enhancers of the A1 adenosine receptor. J Med Chem. 2003;46:794–809. doi: 10.1021/jm0210212. [DOI] [PubMed] [Google Scholar]
- Baraldi PG, Pavani MG, Shryock JC, Moorman AR, Iannatta V, Borea PA, et al. Synthesis of 2-amino-3-heteroaroylthiophenes and evaluation of their activity as potential allosteric enhancers at the human A1 receptor. Eur J Med Chem. 2004;39:855–865. doi: 10.1016/j.ejmech.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Barbhaiya H, McClain R, Ijzerman A, Rivkees SA. Site-directed mutagenesis of the human A1 adenosine receptor: influences of acidic and hydroxyl residues in the first four transmembrane domains on ligand binding. Mol Pharmacol. 1996;50:1635–1642. [PubMed] [Google Scholar]
- Bar-Yehuda S, Stemmer SM, Madi L, Castel D, Ochaion A, Cohen S, et al. The A3 adenosine receptor agonist CF102 induces apoptosis of hepatocellular carcinoma via de-regulation of the Wnt and NF-kB signal transduction pathways. Int J Oncol. 2008;33:287–295. [PubMed] [Google Scholar]
- Bar-Yehuda S, Luger D, Ochaion A, Cohen S, Patokaa R, Zozulya G, et al. Inhibition of experimental auto-immune uveitis by the A3 adenosine receptor agonists CF101. Int J Mol Med. 2011;28:727–731. doi: 10.3892/ijmm.2011.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briddon SJ, Gandia J, Amaral OB, Ferre S, Lluis C, Franco R, et al. Plasma membrane diffusion of G protein-coupled receptor oligomers. Biochim Biophys Acta. 2008;1783:2262–2268. doi: 10.1016/j.bbamcr.2008.07.006. [DOI] [PubMed] [Google Scholar]
- Bruns RF, Fergus JH. Allosteric enhancement of adenosine-A1-receptor binding and function by 2-amino-3-benzoylthiophenes. Mol Pharmacol. 1990;38:939–949. [PubMed] [Google Scholar]
- Bruns RF, Fergus JH, Coughenour LL, Courtland GG, Pugsley TA, Dodd JH, et al. Structure-activity relationships for enhancement of adenosine A1-receptor binding by 2-amino-3-benzoylthiophenes. Mol Pharmacol. 1990;38:950–958. [PubMed] [Google Scholar]
- Carr CS, Hill RJ, Masamune H, Kennedy SP, Knight DR, Tracey WR, et al. Evidence for a role for both the adenosine A1 and A3 receptors in protection of isolated human atrial muscle against simulated ischaemia. Cardiovasc Res. 1997;36:52–59. doi: 10.1016/s0008-6363(97)00160-0. [DOI] [PubMed] [Google Scholar]
- Casadó V, Barrondo S, Spasic M, Callado LF, Mallol J, Canela E, et al. Gi protein coupling to adenosine A1-A2A receptor heteromers in human brain caudate nucleus. J Neurochem. 2010;114:972–980. doi: 10.1111/j.1471-4159.2010.06810.x. [DOI] [PubMed] [Google Scholar]
- Chandrasekera PC, Wan TC, Gizewski ET, Auchampach JA, Lasley R. Adenosine A1 receptors heterodimerize with β1- and β2-adrenergic receptor complexes with altered G protein coupling and signalling. Cell Signal. 2013;25:736–742. doi: 10.1016/j.cellsig.2012.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, et al. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science. 2006;314:1792–1795. doi: 10.1126/science.1132559. [DOI] [PubMed] [Google Scholar]
- Chiavegatta T, Cost JVL, Araujo MS, Godinho RO. Skeletal muscle expresses the extracellular cyclic AMP-adenosine pathway. Br J Pharmacol. 2008;153:1331–1340. doi: 10.1038/sj.bjp.0707648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54:323–374. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Lanzafame A, Ziegler A, Mitchelson F. Kinetic studies of cooperativity at atrial muscarinic M2 receptors with an ‘infinite dilution’ procedure. Biochem Pharmacol. 1997;53:795–800. doi: 10.1016/s0006-2952(96)00814-3. [DOI] [PubMed] [Google Scholar]
- Chung KY, Rasmussen SGF, Liu T, Li S, DeVree BT, Chae PS, et al. Conformational changes in the G protein Gs induced by the â2-adrenergic receptor. Nature. 2011;477:611–615. doi: 10.1038/nature10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciruela F, Saura C, Canela EI, Mallol J, Lluis C, Franco R. Adenosine deaminase affects ligand-induced signalling by interacting with cell surface adenosine receptors. FEBS Lett. 1996;380:219–223. doi: 10.1016/0014-5793(96)00023-3. [DOI] [PubMed] [Google Scholar]
- Clark AN, Youkey R, Liu X, Jia L, Blatt R, Day YJ, et al. A1 adenosine receptor activation promotes angiogenesis and release of VEGF from monocytes. Circ Res. 2007;101:1130–1138. doi: 10.1161/CIRCRESAHA.107.150110. [DOI] [PubMed] [Google Scholar]
- Cohen S, Stemmer SM, Zozulya G, Ochaion A, Patoka R, Barer F, et al. CF102 an A3 adenosine receptor agonist mediates anti-tumor and anti-inflammatory effects in the liver. J Cell Physiol. 2011;226:2438–2447. doi: 10.1002/jcp.22593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeaux Y, Ijzerman AP, Hill SJ. Coupling of the human A1 adenosine receptor to different heterotrimeric G-proteins: evidence for agonist-specific G-protein activation. Br J Pharmacol. 2004;143:705–714. doi: 10.1038/sj.bjp.0705925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corriden R, Insel PA. New insights regarding the regulation of chemotaxis by nucleotides, adenosine and their receptors. Purineric Signal. 2012;8:587–598. doi: 10.1007/s11302-012-9311-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corriden R, Self T, Akong-Moore K, Nizet V, Kellam B, Briddon SJ, et al. Adenosine A3-receptors in neutrophil microdomains promote the formation of bacteria-tethering cytonemes. EMBO Rep. 2013;14:726–732. doi: 10.1038/embor.2013.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristovao-Ferreira S, Navarro G, Brugarolas M, Perez-Capote K, Vaz SH, Fattorina G, et al. A1-R-A2A-R heteromers coupled to Gs and Gi/o proteins modulate GABA transport into astrocytes. Purinergic Signal. 2013;9:433–449. doi: 10.1007/s11302-013-9364-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling MR, Charlton SJ. Quantifying the association and dissociation rates of unlabelled antagonists at the muscarinic M3 receptor. Br J Pharmacol. 2006;148:927–937. doi: 10.1038/sj.bjp.0706819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L, Gao ZG, Nithipatikom K, Ijzerman AP, van Veldhoven JPD, Jacobson KA, et al. Protection from myocardial ischaemia/reperfusion injury by a positive allosteric modulator of the A3 adenosine receptor. J Pharmacol Exp Ther. 2012;340:210–217. doi: 10.1124/jpet.111.187559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey RK, Gillespie DG, Mi Z, Jackson EK. Endogenous cyclic AMP-adenosine pathway regulates cardiac fibroblast growth. Hypertension. 2001;37:1095–1100. doi: 10.1161/01.hyp.37.4.1095. [DOI] [PubMed] [Google Scholar]
- Ellis J, Huyler J, Brann MR. Allosteric regulation of cloned M1-M5 muscarinic receptor subtypes. Biochem Pharmacol. 1991;42:1927–1932. doi: 10.1016/0006-2952(91)90591-r. [DOI] [PubMed] [Google Scholar]
- Fishman P, Bar-Yehuda S, Liang BT, Jacobson KA. Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug Discov Today. 2012;17:359–366. doi: 10.1016/j.drudis.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Martinez-Pinilla E, Ricobaraza A, McCormick PJ. Challenges in the development of heteromer-GPCR-based drugs. Prog Mol Biol Transl Sci. 2013;117:143–162. doi: 10.1016/B978-0-12-386931-9.00006-4. [DOI] [PubMed] [Google Scholar]
- Fredholm BF, Ijzerman AP, Jacobson KA, Linden J, Muller CE. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors – an update. Pharmacol Rev. 2011;63:1–34. doi: 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao GZ, van Muijlwijk-Koezen JE, Chen A, Müller CE, IJzerman AP, Jacobson KA. Allosteric modulation of A3 adenosine receptors by a series of 3-(2-pyridinyl)isoquinoline derivatives. Mol Pharmacol. 2001;60:1057–1063. [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Kim SG, Soltysiak KA, Melman N, Ijzerman AP, Jacobson KA. Selective allosteric enhancement of agonist binding and function at human A(3) adenosine receptors by a series of imidazoquinoline derivatives. Mol Pharmacol. 2002;62:81–89. doi: 10.1124/mol.62.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Kim SK, Gross AS, Chen A, Balustein JB, Jacobson KA. Identification of essential residues involved in the allosteric modulation of the human A3 adenosine receptor. Mol Pharmacol. 2003;63:1021–1031. doi: 10.1124/mol.63.5.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Verziji D, Zweemer A, Ye K, Goblyos A, Ijzerman AP, et al. Functionally biased modulation of A3- adenosine receptor agonist efficacy and potency by imidazoquinolinamine allosteric enhancers. Biochem Pharmacol. 2011;82:658–668. doi: 10.1016/j.bcp.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge ZD, Peart JN, Kreckler LM, Wan TC, Jacobson MA, Gross GJ, et al. Cl-IB-MECA[2-chloro-N6-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide] reduces ischemia/reperfusion injury in mice by activating the A3 adenosine receptor. J Pharmacol Exp Ther. 2006;319:1200–1210. doi: 10.1124/jpet.106.111351. [DOI] [PubMed] [Google Scholar]
- Gessi S, Cattabriga E, Avitabile A, Gafa R, Lanza G, Cavazzini L, et al. Elevated expression of A3 adenosine receptors in human colorectal cancer is reflected in peripheral blood cells. Clin Cancer Res. 2004;210:5895–5901. doi: 10.1158/1078-0432.CCR-1134-03. [DOI] [PubMed] [Google Scholar]
- Göblyös A, Ijzerman AP. Allosteric modulation of adenosine receptors. Biochim Biophys Acta. 2011;1808:1309–1318. doi: 10.1016/j.bbamem.2010.06.013. [DOI] [PubMed] [Google Scholar]
- Göblyös A, Gao ZG, Brussee J, Connestari R, Santiago SN, Ye K, et al. Structure-activity relationships of new 1H-imidazo[4,5-c]quinolin-4-amine derivatives as allosteric enhancers of the A3 adenosine receptor. J Med Chem. 2006;49:3354–3361. doi: 10.1021/jm060086s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedeke A. cAMP: fuel for extracellular adenosine formation. Br J Pharmacol. 2008;153:1087–1089. doi: 10.1038/bjp.2008.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman N, Chen M, Fujita T, Xu Q, Peng W, Liu W, et al. Adenosine A1 receptors mediate local anti-nociceptive effects of acupuncture. Nature Neurosci. 2010;13:883–888. doi: 10.1038/nn.2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf C, Kooistra AJ, Vischer HF, Katritch V, Kuijer M, Shiroishi M, et al. Crystal structure-based virtual screening for fragment-like ligands of the human histamine H1 receptor. J Med Chem. 2011;54:8195–8206. doi: 10.1021/jm2011589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracia E, Cortes A, Meana JJ, Garcia-Sevilla J, Hershfield MS, Canela EI, et al. Human adenosine deaminase as an allosteric modulator of human A1 adenosine receptor: abolishment of negative cooperativity for 3H-R-PIA binding to the caudate nucleus. J Neurochem. 2008;107:161–170. doi: 10.1111/j.1471-4159.2008.05602.x. [DOI] [PubMed] [Google Scholar]
- Gracia E, Farre D, Cortes A, Ferrer-Costa C, Orozco M, Mallol J, et al. The catalytic site structural gate of adenosine deaminase allosterically modulates ligand binding to adenosine receptors. FASEB J. 2013;27:1048–1061. doi: 10.1096/fj.12-212621. [DOI] [PubMed] [Google Scholar]
- Griffin NJ, Kowacs F, Libri V, Williams P, Goadsby PJ, Kaube H. Effect of the adenosine A1 receptor agonist GR79236 on trigeminal nociception with blink reflex recordings in healthy human subjects. Cephalagia. 2003;23:287–292. doi: 10.1046/j.1468-2982.2003.00511.x. [DOI] [PubMed] [Google Scholar]
- Han Y, Moreira IS, Urizar E, Weinstein H, Javitch JA. Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nat Chem Biol. 2009;5:688–695. doi: 10.1038/nchembio.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison GJ, Cerniway RJ, Peart J, Berr SS, Ashton K, Regan S, et al. Effects of A3 adenosine receptor activation and gene knock-out in ischemic-reperfused mouse heart. Cardiovasc Res. 2002;53:147–155. doi: 10.1016/s0008-6363(01)00424-2. [DOI] [PubMed] [Google Scholar]
- Heitman LH, Göblyös A, Zweemer AM, Bakker R, Mulder-Krieger T, van Veldhoven JPD, et al. A series of 2,4-disubstituted quinolones as a new class of allosteric enhancers of the adenosine A3 receptor. J Med Chem. 2009;52:926–931. doi: 10.1021/jm8014052. [DOI] [PubMed] [Google Scholar]
- Hern JA, Baig AH, Mashanov GI, Birdsall B, Corrie JE, Lazareno S, et al. Formation and dissociation of M1 muscarinic receptor dimers seen by total internal reflection fluorescence imaging of single molecules. Proc Natl Acad Sci U S A. 2010;107:2693–2698. doi: 10.1073/pnas.0907915107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Thor D, Zhou Y, Liu T, Wang Y, McMillin SM, et al. Structural aspects of M3 muscarinic acetylcholine receptor dimer formation and activation. FASEB J. 2012;26:604–616. doi: 10.1096/fj.11-191510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilien B, Glasser N, Clamme JP, Didier P, Piemont E, Chinnappan R, et al. Pirenzepine promotes the dimerization of muscarinic M1 receptors through a three-step binding process. J Biol Chem. 2009;284:19533–19543. doi: 10.1074/jbc.M109.017145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA. Introduction to adenosine receptors as therapeutic targets. Hand Exp Pharmacol. 2009;193:1–24. doi: 10.1007/978-3-540-89615-9_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Gao ZG, Göblyös A, Ijzerman AP. Allosteric modulation of purine and pyrimidine receptors. Adv Pharmacol. 2011;61:187–220. doi: 10.1016/B978-0-12-385526-8.00007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janusz CA, Berman RF. The adenosine binding enhancer, PD 81,723, inhibits epileptiform bursting in the hippocampal brain slice. Brain Res. 1993;619:131–136. doi: 10.1016/0006-8993(93)91604-q. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. 7TM receptor allostery: putting numbers to shapeshifting proteins. Trends Pharmacol Sci. 2009;30:460–469. doi: 10.1016/j.tips.2009.06.007. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. Biased signalling and allosteric machines: new vistas and challenges for drug discovery. Br J Pharmacol. 2012;165:1659–1669. doi: 10.1111/j.1476-5381.2011.01749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keov P, Sexton PN, Christopoulos A. Allosteric modulation of G protein-coupled receptors: a pharmacological perspective. Neuropharmacology. 2010;60:24–35. doi: 10.1016/j.neuropharm.2010.07.010. [DOI] [PubMed] [Google Scholar]
- Kimatrai-Salvador M, Baraldi PG, Romagnoli R. Allosteric modulation of A1-adenosine receptor: a review. Drug Discov Today: Technol. 2012;10:e285–e296. doi: 10.1016/j.ddtec.2012.08.005. [DOI] [PubMed] [Google Scholar]
- Klaft ZJ, Schulz SB, Masalarova A, Gabriel S, Heinemann U, Gerevich Z. Extracellular ATP differentially affects epileptiform activity via purinergic P2X7 and adenosine A1 receptors in naïve and chronic epileptic rats. Epilepsia. 2012;53:1978–1986. doi: 10.1111/j.1528-1167.2012.03724.x. [DOI] [PubMed] [Google Scholar]
- van der Klein PAM, Kourounakis AP, IJzerman AP. Allosteric modulation of the adenosine A1 receptor: synthesis and biological evaluation of novel 2-amino-3-benzoylthiophenes as allosteric enhancers of agonist binding. J Med Chem. 1999;42:3629–3635. doi: 10.1021/jm991051d. [DOI] [PubMed] [Google Scholar]
- Knapp K, Zebisch M, Pippel J, El-Tayeb A, Muller CE, Srater N. Crystal structure of the human ecto-5′-nucleotidase (CD73): insights into the regulation of purinergic signaling. Structure. 2012;20:2161–2173. doi: 10.1016/j.str.2012.10.001. [DOI] [PubMed] [Google Scholar]
- Kostenis E, Mohr K. Two-point kinetic experiments to quantify allosteric effects on radioligand dissociation. Trends Pharmacol Sci. 1996;17:280–283. doi: 10.1016/0165-6147(96)10034-1. [DOI] [PubMed] [Google Scholar]
- Kourounakis AP, van de Klein PAM, IJzerman AP. Elucidation of structure-activity relationships of 2-amino-3-benzoylthiophenes: study of their allosteric enhancing vs. antagonistic activity on adenosine A1 receptors. Drug Dev Res. 2000;49:227–237. [Google Scholar]
- Kuszak AJ, Pitchiaya S, Anand JP, Mosberg HI, Walter NG, Sunahara RK. Purification and functional reconstitution of monomeric mu-opioid receptors: allosteric modulation of agonist binding by Gi2. J Biol Chem. 2009;284:26732–26741. doi: 10.1074/jbc.M109.026922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langemeijer EV, Verzijl D, Dekker SJ, Ijzerman AP. Functional selectivity of adenosine A1 receptor ligands? Purinergic Signal. 2013;9:91–100. doi: 10.1007/s11302-012-9334-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazareno S, Birdsall NJ. Detection, quantitation, and verification of allosteric interactions of agents with labeled and unlabeled ligands at G protein-coupled receptors: interactions of strychnine and acetylcholine at muscarinic receptors. Mol Pharmacol. 1995;48:362–378. [PubMed] [Google Scholar]
- Lee HT, Emala CW. Protective effects of renal ischemic preconditioning and adenosine pretreatment: role of A1 and A3 receptors. Am J Physiol Renal Physiol. 2000;278:F380–F387. doi: 10.1152/ajprenal.2000.278.3.F380. [DOI] [PubMed] [Google Scholar]
- Lee HT, Gallos G, Nasr SH, Emala CW. A1 adenosine receptor activation inhibits inflammation, necrosis, and apoptosis after renal ischemia-reperfusion injury in mice. J Am Soc Nephrol. 2004;15:102–111. doi: 10.1097/01.asn.0000102474.68613.ae. [DOI] [PubMed] [Google Scholar]
- Lee NH, El-Fakahany EE. Allosteric antagonists of the muscarinic acetylcholine receptor. Biochem Pharmacol. 1991;42:199–205. doi: 10.1016/0006-2952(91)90703-8. [DOI] [PubMed] [Google Scholar]
- Limbird LE, Meyts PD, Lefkowitz RJ. Beta-adrenergic receptors: evidence for negative cooperativity. Biochem Biophys Res Commun. 1975;64:1160–1168. doi: 10.1016/0006-291x(75)90815-3. [DOI] [PubMed] [Google Scholar]
- Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Vet K. Structural basis for allosteric regulation of GPCRs by sodium ions. Science. 2012;337:232–236. doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrea KE, Hill SJ. Salmeterol, a long-acting b2-adrenoceptor agonist mediating cyclic AMP accumulation in a neuronal cell line. Br J Pharmacol. 1993;110:619–626. doi: 10.1111/j.1476-5381.1993.tb13856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madi L, Ochaion A, Rath-Wolfson L, Bar-Yehuda S, Erlanger A, Get O. The A3 adenosine receptor is highly expressed in tumor vs. normal cells: potential target for tumor growth inhibition. Clin Cancer Res. 2004;10:4472–4479. doi: 10.1158/1078-0432.CCR-03-0651. [DOI] [PubMed] [Google Scholar]
- Mares P. Anticonvulsant action of 2-chloroadenosine against pentetrazol-induced seizures in immature rats is due to activation of A1 adenosine receptors. J Neural Transm. 2010;117:1269–1277. doi: 10.1007/s00702-010-0465-9. [DOI] [PubMed] [Google Scholar]
- Matherne GP, Linden J, Byford AM, Gauthier NS, Headrick JP. Transgenic A1 adenosine receptor overexpression increases myocardial resistance to ischaemia. Proc Natl Acad Sci. 1997;94:6541–6546. doi: 10.1073/pnas.94.12.6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- May LT, Briddon SJ, Hill SJ. Antagonist selective modulation of adenosine A1 and A3 receptor pharmacology by the food dye Brilliant Black BN: evidence for allosteric interactions. Mol Pharmacol. 2010a;77:678–686. doi: 10.1124/mol.109.063065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May LT, Self TJ, Briddon SJ, Hill SJ. The effect of allosteric modulators on the kinetics of agonist-G protein-coupled receptor interactions in single living cells. Mol Pharmacol. 2010b;78:511–523. doi: 10.1124/mol.110.064493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May LT, Bridge LJ, Stoddart LA, Briddon SJ, Hill SJ. Allosteric interactions across native adenosine A3 receptor homodimers: quantification using single cell ligand binding kinetics. FASEB J. 2011;25:3465–3476. doi: 10.1096/fj.11-186296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meno JR, Higashi H, Cambray AJ, Zhou J, D'Ambrosio R, Winn HR. Hippocampal injury and neurobehavioural deficits are improved by PD 81723 following hyperglycemic cerebral ischemia. Exp Neurol. 2003;183:188–196. doi: 10.1016/s0014-4886(03)00162-6. [DOI] [PubMed] [Google Scholar]
- Merighi S, Simioni C, Gessi S, Varani K, Mirandola P, Tabrizi MA, et al. A2B and A3 adenosine receptors modulate vascular endothelial growth factor and interleukin-8 expression in human melanoma cells treated with etoposide and doxorubicin. Neoplasia. 2009;11:1064–1073. doi: 10.1593/neo.09768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison RR, Teng B, Oldenburg PJ, Katwa LC, Schnermann JB, Mustafa J. Effect of targeted deletion of A1 adenosine receptors on post-ischaemic cardiac function and expression of adenosine receptor subtypes. Am J Physiol Heart Circ Physiol. 2006;291:H1875–h1882. doi: 10.1152/ajpheart.00158.2005. [DOI] [PubMed] [Google Scholar]
- Muller CE, Jacobson KA. Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim Et Biophys Acta. 2011;1808:1290–1308. doi: 10.1016/j.bbamem.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolakopoulos G, Figler H, Linden J, Scammels PJ. 2-Aminothiophene-3-carboxylates and carboxamides as adenosine A1 receptor allosteric enhancers. Bioorg Med Chem. 2006;14:2358–2365. doi: 10.1016/j.bmc.2005.11.018. [DOI] [PubMed] [Google Scholar]
- Park SW, Kim JY, Ham A, Brown KM, Kim M, D'Agati VD, et al. A1 adenosine receptor allosteric enhancer PD-81723 protects against renal ischaemia-reperfusion injury. Am J Physiol Renal Physiol. 2012;303:F721–F732. doi: 10.1152/ajprenal.00157.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters MC, Wisse LE, Dinaj A, Vriend G, Ijzerman AP. The role of the second and third extracellular loops of the adenosine A1 receptor in activation and allosteric modulation. Biochem Pharmacol. 2012;84:76–87. doi: 10.1016/j.bcp.2012.03.008. [DOI] [PubMed] [Google Scholar]
- Pisterzi LF, Jansma DB, Georgiou J, Woodside MJ, Chou JT, Angers S, et al. Oligomeric size of the m2 muscarinic receptor in live cells as determined by quantitative fluorescence resonance energy transfer. J Biol Chem. 2010;285:16723–16738. doi: 10.1074/jbc.M109.069443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, et al. Crystal structure of the â2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnoli R, Baraldi PG, Carrion MD, Cara CL, Cruz-Lopez O, Iaconinoto MA, et al. Synthesis and biological evaluation of 2-amino-3-(4-chlorobenzoyl)-4-[N-(substituted)piperazin-1-yl]thiophenes as potent allosteric enhancers of the A1 adenosine receptor. J Med Chem. 2008;51:5875–5879. doi: 10.1021/jm800586p. [DOI] [PubMed] [Google Scholar]
- Roth BL, Marshall FH. NOBEL 2012 chemistry: studies of a ubiquitous receptor family. Nature. 2012;492:57. doi: 10.1038/492057a. [DOI] [PubMed] [Google Scholar]
- Salvatore CA, Tilley SL, Latour AM, Fletcher DS, Koller BH, Jacobson MA. Disruption of the A3 adenosine receptor gene in mice and its effect on stimulated inflammatory cells. J Biol Chem. 2000;275:4429–4434. doi: 10.1074/jbc.275.6.4429. [DOI] [PubMed] [Google Scholar]
- Smith NJ, Milligan G. Allostery at G protein-coupled receptor homo- and heteromers: uncharted pharmacological landscapes. Pharmacol Rev. 2010;62:701–725. doi: 10.1124/pr.110.002667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa NA, Voss MK, Zylka MJ. Recombinant ecto-5′-nucleotidase (CD73) has long lasting antinociceptive effects that are dependent on adenosine A1 receptor activation. Mol Pain. 2010;6:20. doi: 10.1186/1744-8069-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springael JY, Le Minh PN, Urizar E, Costagliola S, Vassart G, Parmentier M. Allosteric modulation of binding properties between units of chemokine receptor homo- and hetero-oligomers. Mol Pharmacol. 2006;69:1652–1661. doi: 10.1124/mol.105.019414. [DOI] [PubMed] [Google Scholar]
- Street SE, Zylka MJ. Emerging roles for ectonucleotidases in pain-sensing neurons. Neuropsychopharmacol. 2011;36:358. doi: 10.1038/npp.2010.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Namba K, Tsuga H, Nakata H. Regulation of pharmacology by hetero-oligomerization between A1 adenosine and P2Y2 receptor. Biochem Biophys Res Commun. 2006;351:559–565. doi: 10.1016/j.bbrc.2006.10.075. [DOI] [PubMed] [Google Scholar]
- Swaminath G, Xiang Y, Lee TW, Steenhuis J, Parnot C, Kobilka BK. Sequential binding of agonists to the beta2 adrenoceptor. Kinetic evidence for intermediate conformational states. J Biol Chem. 2004;279:686–691. doi: 10.1074/jbc.M310888200. [DOI] [PubMed] [Google Scholar]
- Tendera M, Gaszekewska-Zurek E, Parma Z, Ponikowski P, Jankowska E, Kawecka-Jaszcz M, et al. The new oral adenosine A1 receptor agonist capadenoson in male patients with stable angina. Clin Res Cardiol. 2012;101:585–591. doi: 10.1007/s00392-012-0430-8. [DOI] [PubMed] [Google Scholar]
- Tinney FJ, Sanchez JP, Nogas JA. Synthesis and pharmacological evaluation of 2,3-dihydro-1H-thieno[2,3-e][1,4]diazepines. J Med Chem. 1974;17:624–630. doi: 10.1021/jm00252a011. [DOI] [PubMed] [Google Scholar]
- Uematsu M, Gaudette GR, Laurikka JO, Levitsky S, McCully JD. Adenosine-enhanced ischaemic preconditioning decreases infarct size in the regional ischaemic sheep heart. Ann Thorac Surg. 1998;66:382–387. doi: 10.1016/s0003-4975(98)00501-3. [DOI] [PubMed] [Google Scholar]
- Urizar E, Yano H, Kolster R, Galés C, Lambert N, Javitch JA. CODA-RET reveals functional selectivity as a result of GPCR heteromerization. Nat Chem Biol. 2011;7:624–630. doi: 10.1038/nchembio.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valant C, Aurelio L, Urmaliya VB, White P, Scammells PJ, Sexton PM, et al. Delineating the mode of action of adenosine A1 receptorallosteric modulators. Mol Pharm. 2010;78:444–455. doi: 10.1124/mol.110.064568. [DOI] [PubMed] [Google Scholar]
- Whalen EJ, Rajagopal S, Lefkowitz RJ. Therapeutic potential of β-arrestin and G protein-biased agonists. Trends Mol Med. 2011;17:126–139. doi: 10.1016/j.molmed.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C, Hill SJ. GPCR signalling: understanding the pathway to successful drug discovery. Methods Mol Biol. 2009;552:39–50. doi: 10.1007/978-1-60327-317-6_3. [DOI] [PubMed] [Google Scholar]
- Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, et al. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]