

Abstract
BACKGROUND AND PURPOSE
Retinal neurodegeneration is an early and critical event in several diseases associated with blindness. Clinically, therapies that target neurodegeneration fail. We aimed to elucidate the multiple roles by which thioredoxin-interacting protein (TXNIP) contributes to initial and sustained retinal neurodegeneration.
EXPERIMENTAL APPROACH
Neurotoxicity was induced by intravitreal injection of NMDA into wild-type (WT) and TXNIP-knockout (TKO) mice. The expression of apoptotic and inflammatory markers was assessed by immunohistochemistry, elisa and Western blot. Microvascular degeneration was assessed by periodic acid-Schiff and haematoxylin staining and retinal function by electroretinogram.
KEY RESULTS
NMDA induced early (1 day) and significant retinal PARP activation, a threefold increase in TUNEL-positive nuclei and 40% neuronal loss in ganglion cell layer (GCL); and vascular permeability in WT but not TKO mice. NMDA induced glial activation, expression of TNF-α and IL-1β that co-localized with Müller cells in WT but not TKO mice. In parallel, NMDA triggered the expression of NOD-like receptor protein (NLRP3), activation of caspase-1, and release of IL-1β and TNF-α in primary WT but not TKO Müller cultures. After 14 days, NMDA induced 1.9-fold microvascular degeneration, 60% neuronal loss in GCL and increased TUNEL-labelled cells in the GCL and inner nuclear layer in WT but not TKO mice. Electroretinogram analysis showed more significant reductions in b-wave amplitudes in WT than in TKO mice.
CONCLUSION AND IMPLICATIONS
Targeting TXNIP expression prevented early retinal ganglion cell death, glial activation, retinal inflammation and secondary neuro/microvascular degeneration and preserved retinal function. TXNIP is a promising new therapeutic target for retinal neurodegenerative diseases.
Keywords: retina, NMDA, TXNIP, neurotoxicity, apoptosis, Müller cell, TNF-α, IL-1β, acellular capillary, vascular permeability, ERG
Introduction
Retinal ganglion cell (RGC) death is a hallmark of multiple retinal diseases, including traumatic optic neuropathy, diabetic retinopathy and glaucoma (Schmidt et al., 2008; Whitmire et al., 2011). NMDA is an analogue of glutamate, the primary excitatory neurotransmitter in the retina. NMDA overstimulation of retinal neurons triggers RGC death as a result of its excitatory neurotoxic effect (Lam et al., 1999). We and others have used an NMDA-induced neurotoxicity model in an attempt to elucidate the possible molecular mechanisms of RGC death and develop therapeutic targets (Al-Gayyar et al., 2010,2011; Bessero et al., 2010; Shimazawa et al., 2010; Koseki et al., 2012). Interestingly, clinical trials using memantine to block NMDA receptor activity in glaucoma patients did not achieve the anticipated outcome (Osborne, 2009); therefore, identifying downstream signals secondary to RGC death can lead to devising better therapeutic targets than targeting the NMDA receptor itself.
In response to RGC death, glial cells become activated and release cytokines and inflammatory mediators that can adversely affect other retina cell types including neurons and vasculature. In previous studies, using the NMDA model, we identified thioredoxin-interacting protein (TXNIP) as a pro-oxidative and pro-apoptotic stress sensor that plays a pivotal role in retinal neuronal death (Al-Gayyar et al., 2010,2011). However, the exact role of TXNIP in mediating glial inflammation in neurotoxicity models has not been explored. TXNIP has been recently identified as an activator of the NOD-like receptor protein (NLRP3) inflammasome, the most established multi-protein complex responsible for instigating sterile innate inflammatory responses. Inflammasome activators can induce the dissociation of TXNIP from thioredoxin in a reactive oxygen species-sensitive manner, allowing it to bind to NLRP3. NLRP3 then oligomerizes with the ASC (apoptosis-associated speck-like) adaptor protein that recruits procaspase-1, allowing its auto-cleavage and activation. Activated caspase-1 enzyme in turn cleaves up-regulated premature pro-inflammatory cytokines IL-1β and IL-18 before their release (Schroder et al., 2010,2012). A recent study showed that hyperglycaemia can trigger TXNIP-mediated NLRP3 inflammasome activation and the release of IL-1β in retinal Müller cells (Devi et al., 2012). Therefore, we were particularly interested in examining whether the NLRP3 inflammasome also plays an important role in the NMDA-induced stress response. The current study was undertaken to assess the neuroprotective effect of TXNIP deletion on both the short-and long-term effects of NMDA-induced retinal neurotoxicity. Wild-type (WT) and TXNIP-null mice received intravitreal injections of NMDA to determine the effect of TXNIP on inflammation, vascular integrity and retinal function. The results indicate that TXNIP deletion protected against retinal vascular degeneration and preserved retinal function.
Methods
Animals
Experiments were approved by the Institutional Committee for Animal Use in Research and Education at Charlie Norwood VA Medical Center (ACORP #04-12-043) and conformed to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. All experiments were performed using age-matched WT C57Bl/6 mice (Jackson Laboratory, Bar Harbor, ME, USA) and TXNIP-knockout (TKO) mice (Hui et al., 2008) that were a kind gift from Dr AJ Lusis and Dr ST Hui at the BioSciences Center, San Diego State University, San Diego, CA, USA. TKO mice were similar in weight and activity to WT littermates, with no differences in food consumption or litter sizes. Genotyping was performed as described previously by our group (Abdelsaid et al., 2013). The mice were kept at 20°C, maximum five mice in a single cage, with a 12 h day/12 h night cycle and had free access to water and food (standard diet). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). Neurotoxicity was induced by intravitreal injection of 2 μL NMDA (40 nmol ≈ 5.9 μg dissolved in normal saline; Sigma-Aldrich, St. Louis, MO, USA) in deeply anaesthetized animals and an equal concentration of N-methyl-L-aspartate (NMLA); the inactive stereo-isomer of NMDA (Santa Cruz Biotech, Santa Cruz, CA, USA) served as the control, as previously described by our group (El-Remessy et al., 2003; Al-Gayyar et al., 2010). Mice were anaesthetized with an i.p. injection of 1% avertin (2,2,2-tribromoethanol) initially dissolved in tert-amyl alcohol then final dilution (100 mL) in buffered saline then injected at 25 μL·g−1 of body weight. Depth of anaesthesia was evident by loss of reflex response to forceps. The dose of NMDA was selected based on previous studies (Bonhomme-Faivre et al., 2002; Al-Gayyar et al., 2010). A total of 80 mice were divided into a total of four groups: two WT groups received intravitreal injections of either NMDA (WT-NMDA) in both eyes, or NMLA (WT-NMLA) as the control group, and two TKO groups were given the same intravitreal injections of either NMDA (TKO-NMDA) or NMLA (TKO-NMLA) as a control. Mice were killed using the CO2 chamber and the eyes were immediately enucleated after either 1 day or 14–21 days after injection for studying the acute short-term and the long-term effects, respectively, of NMDA toxicity in both WT and TKO mice.
Tissue culture studies
Primary mouse Müller cells were isolated and used for experiments as described previously (Mysona et al., 2009). Briefly, Müller cells were isolated from 6-to 7-day-old mice and were grown to confluence in complete medium (5 mM glucose DMEM supplemented with 10% FBS and 1% penicillin/streptomycin). For the experiments, Müller cells were switched to serum free media and were treated with 10 μM NMDA overnight. The rat retinal Müller cell line (rMC-1), a kind gift from Dr Vijay Sarthy, was utilized to establish the NMDA dose needed before performing the primary Müller cell experiments.
elisa assay of inflammatory mediators
TNF-α and IL-1β in cell-conditioned media were detected by TNF-α and IL-1β elisa sensitive kits (R&D Systems, Minneapolis, MN, USA). Equal volumes of conditioned media for each treatment group were concentrated using Amicon 10 K concentration columns (Millipore, Temecula, CA, USA) and then the elisa was performed by following the manufacturer's protocol. The levels of TNF-α and IL-1β are expressed as pg·mL−1 of cell-conditioned media.
Evaluation of neuronal cell death in mouse retina
TUNEL was performed using the ApopTAG in situ apoptosis detection kit (Millipore, Billerica, MA, USA), following the manufacturer's instructions. Briefly, optimum cutting temperature (OCT)-frozen eye sections were fixed and incubated with terminal deoxynucleotidyl transferase enzyme followed by incubation with anti-digoxigenin conjugate. Propidium iodide (1 μg·mL−1) was added as a nuclear counterstain and coverslips were applied using Vectashield mounting medium for fluorescence (Vector Laboratories, Burlingame, CA, USA). Four mice from each group and six sections for each animal were used. Each section was systematically scanned and counted for green fluorescent TUNEL-positive cells in retinal layers, which indicates apoptosis. Images were obtained using an AxioObserver.Z1 Microscope (Carl Zeiss, Jena, Germany) with 200× magnification.
Counting number of neuronal cells in ganglion cell layer (GCL)
OCT-frozen retinal sections were stained with haematoxylin and eosin (H&E) for light microscopy. The nuclei in the GCL, excluding nuclei in the vessels, were counted in four locations in each retina [both sides of the optic nerve (central) and mid-retina (peripheral)] in a masked manner as described previously (Zheng et al., 2007; Matragoon et al., 2012). Of note, multiple fields count resulted in a single data point for each animal and four to six animals from each group were used. Retinas were imaged with an AxioObserver.Z1 Microscope.
Western blot analysis
Retinas and cultured Müller cells were homogenized in RIPA and processed for Western blotting to examine the expression of various proteins as described previously by our group (Ali et al., 2008; Al-Gayyar et al., 2010). The total amount of protein was determined by protein assay (DC protein assay; Bio-Rad, CA, USA). Samples (30 μg protein) were separated by SDS-PAGE and transferred to a nitrocellulose membrane. The membranes were blocked in PBS, 0.02% Tween 20 (PBS-T), containing 5% non-fat milk. Antibodies for TXNIP (Santa Cruz Biotechnology), PARP (BD Bioscience Pharmingen, San Diego, CA, USA), NLRP3 (Enzo Life Sciences, Farmingdale, NY, USA) and cleaved caspase-1 (Enzo Life Sciences) were used in 1:500 dilutions in PBS-T containing 5% non-fat milk overnight at 4°C. Membranes were re-probed with 1:2000 β-actin (Sigma-Aldrich) in PBS-T containing 5% non-fat milk for 2 h to confirm equal amounts of protein were loaded. Primary antibody was detected by 1:5000 dilutions of HRP-conjugated sheep anti-mouse or anti-rabbit antibodies in PBS-T and enhanced chemiluminescence (GE Healthcare, Piscataway, NJ, USA). The band intensity was quantified using densitometry software (Alpha Innotech, Santa Clara, CA, USA) and expressed as relative optical density.
Determination of thioredoxin reductase (Trx-R) activity
Trx-R activity was measured using a colorimetric assay kit (Sigma-Aldrich) as previously described by our group (Al-Gayyar et al., 2011). Briefly, retinal samples were homogenized in assay buffer followed by the addition of 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) with NADPH. Reduction of DTNB produced a strong yellow colour that was measured colorimetrically at 412 nm. TRX-R activity was measured by the difference between DTNB measurement of sample and sample plus selective TRX-R inhibitor and expressed as unit g-1min-1.
Measurement of nitrotyrosine
Slot blot analysis was performed as described previously (Ali et al., 2008; Abdelsaid et al., 2010). Retinal homogenates were immobilized onto a nitrocellulose membrane. After being blocked, membranes were incubated with a 1:500 dilution of nitrotyrosine (Calbiochem, San Diego, CA, USA) antibody followed by HRP-conjugated sheep anti-rabbit antibody and enhanced chemiluminescence (GE Healthcare). The optical density of various samples was quantified using densitometry software (Alpha Innotech).
Immunolocalization studies
OCT-frozen sections (10 μm) were fixed using 2% paraformaldehyde, blocked using 5% goat serum dissolved in 1% BSA in 0.3% Triton X-100 in PBS and then incubated with the following antibodies overnight: polyclonal anti-glial fibrillary acidic protein (GFAP) as a marker of glial cell activation (Santa Cruz Biotechnology), polyclonal anti-IL-1β and monoclonal anti-TNF-α (Santa Cruz Biotechnology). All the primary antibodies were used in 1:100 dilutions in 1% BSA in 0.3% Triton X-100 in PBS. Then, the sections were incubated with 1:500 dilutions of Texas-red or Oregon-green-conjugated goat anti-mouse or goat anti-rabbit antibodies (Invitrogen, Grand Island, NY, USA) for 2 h. Images (n = 4 in each group) were collected using an AxioObserver.Z1 Microscope.
Determination of blood-retinal barrier (BRB) breakdown
BRB breakdown was measured as previously described by our group (Ali et al., 2011) by i.v. injection of BSA-fluorescein (20 mg·kg−1) in all groups. Animals were killed 30 min after injection, and plasma and retinal lysates were collected. Plasma and retinal lysate samples were assayed for fluorescein concentration using a plate reader (excitation 370 nm, emission 460 nm; Bio-Tek, Winooski, VT, USA). A standard curve was established using BSA-fluorescein in normal mouse serum. The fluorescein intensity of retinal lysate was normalized to the fluorescein intensity of plasma samples.
Determination of degenerated (acellular) capillaries
The retinal vasculature was isolated as described previously (Stitt et al., 1997; Zheng et al., 2007; Al-Gayyar et al., 2010). Briefly, fresh enucleated eyes were fixed with 2% paraformaldehyde overnight. Retina cups were dissected from underlying retina pigment epithelium/choroid/sclera, washed in PBS and then incubated with 3% crude Difco trypsin250 (BD Diagnostic System, Franklin Lakes, NJ, USA) in 20 nM Tris buffer, pH 8, at 37°C for 2 h. The vitreous was gently removed from the retinal vasculature. The vasculature was soaked in several washes of 0.5% Triton X-100 to get rid of neuronal portions of retina. The transparent vasculature was laid out on slides and stained with periodic acid-Schiff and haematoxylin for the examination of acellular capillaries. A microscope (400×) was used to identify acellular capillaries, which are capillary-sized vessels containing no nuclei anywhere along their length. Six animals were used for each group and the number of acellular capillaries was counted in a masked manner in 10 fields of different areas of the mid-retina and calculated as the number mm-2 of retinal area.
Electroretinogram analysis
Mice were dark-adapted overnight and anaesthetized with 4% isoflurane in oxygen, lowered to 1.5% isoflurane during testing. Drops of proparacaine were placed in each eye for topical anaesthesia, and pupils were dilated with tropicamide and phenylephrine. Needle electrodes were placed in the tail (ground) and cheeks (references), Dawson–Trick–Litzkow electrodes were gently positioned on the eyes, and a drop of hypromellose was added to ensure conductivity and lubricate the eye.
A wide variety of stimuli were generated, including 5 ms flashes, either from darkness (scotopic) or from a background luminance level (photopic). Other stimuli include pseudorandom noise sequences, where the luminance varies over the trial and the response is correlated with the stimulus to obtain kernels describing the retinal mechanisms that transform stimulus to response.
Responses to brief flashes were analysed primarily by measuring the amplitudes of the a-and b-waves. The a-wave is the initial negative response following within about 10 ms of the flash onset, and originating largely from photoreceptor and OFF-bipolar cell hyperpolarization. The b-wave is a large positive signal that breaks into the a-wave and peaks between about 30 and 200 ms, arising largely from ON-bipolar activity. We also analysed the ratio of the b-wave amplitude to the a-wave amplitude.
Post-mortem eye specimens
Human eyes that were obtained from the Georgia Eye Bank (Atlanta, GA, USA) had the following selection criteria: either open-angle glaucoma or no glaucoma, and no life support measures. The eyes were enucleated on average 5.7 ± 0.8 h after death. Retinas were preserved by immediate freezing, followed by homogenization and analysis of protein expression using Western blot, and vitreous samples were collected and subjected to elisa as described previously. Patients' characteristics are shown in Supporting Information Table S1.
Statistical analysis
The results are expressed as mean ± SEM. Differences among experimental groups were evaluated by anova followed by Tukey–Kramer multiple comparison test, unless noted otherwise. Significance was defined as P < 0.05.
Results
Deletion of TXNIP expression prevents NMDA-induced apoptosis and neuronal loss
Our previous analyses using intravitreal injection of NMDA demonstrated significant up-regulation of TXNIP expression in rat retina that was associated with RGC death (Al-Gayyar et al., 2010,2011). Here, we examine the neuro-and vascular protective effect of genetic deletion of TXNIP in response to NMDA. As shown in Figure 1A–C, 24 h after NMDA injection, there was a massive increase in neuronal cell death as indicated by ∼3-fold increase of TUNEL-positive nuclei (white arrowheads) in the GCL of WT NMDA compared with WT-NMLA-control mice. Multiple TUNEL-positive nuclei were observed also in the inner nuclear layer (INL) (Figure 1A, white arrows). Retinas from TKO mice were protected from NMDA-induced neuronal death. In parallel, the protective effect of TXNIP deletion in NMDA-induced neuronal damage was assessed histologically by counting the number of nuclei in the GCL in retinal sections stained with H&E (Figure 1B–D). Intravitreal NMDA injections resulted in ∼40% reduction in the GCL of the central and peripheral regions of the retina. TXNIP deletion preserved GCL count in TKO mice to a level comparable to NMLA-control mice. Of note, NMDA induced a marked increase in number of TUNEL nuclei in the INL in WT but not in TKO mice. To confirm that the observed cell death is apoptosis, we also examined the expression of cleaved PARP by Western blot. NMDA injection resulted in ∼2.5-fold increase in cleaved-PARP expression in WT mice compared with WT-NMLA controls but not in TKO mice (Figure 1E).
Figure 1.
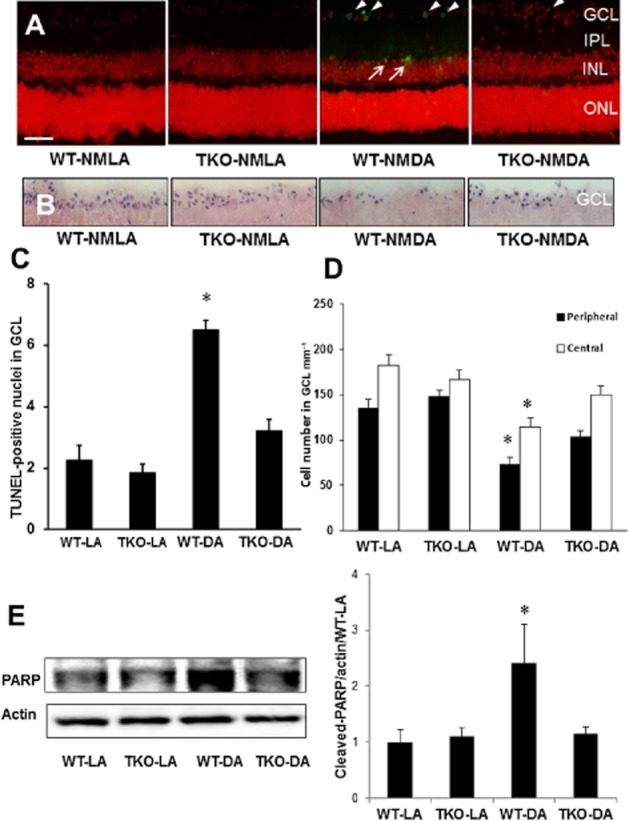
Deletion of TXNIP expression prevents apoptosis and neuronal loss. (A, C) Representative images and statistical analysis indicating that intravitreal injection of NMDA induced RGC death as seen by ∼3-fold increase of TUNEL-positive-labelled cells mainly in the GCL (white arrowheads) and INL (white arrows) of the mouse retina compared with NMLA controls (n = 5 per group). TKO mice subjected to intravitreal NMDA injections showed significantly fewer TUNEL-positive cells than WT-NMDA mice. (B, D) Representative images and statistical analysis showing the number of nuclei in the GCL of retinal sections stained with H&E. Intravitreal NMDA injections resulted in 40% cell loss in the GCL in the posterior and central regions of the WT retina which was prevented in the TKO retina (n = 6 per group). (E) Western blot analysis indicated ∼2.5-fold increase in cleaved-PARP expression in NMDA-injected retinas compared with NMLA controls (n = 4–6 per group). Enhanced cleaved-PARP expression was not observed in TKO-NMDA mice. Data are expressed as fold value of control and represented as mean ± SEM. *Significant difference as compared with WT-LA at P < 0.05. IPL, inner plexiform layer; ONL, outer nuclear layer; ROD, relative optical density (200× magnification, scale bar is 20 μm).
Deletion of TXNIP reduces nitrotyrosine and increases Trx-R activity
In comparison to WT, TKO mice showed a 1.45-fold of retinal Trx-R activity at baseline level. Upon NMDA insult, Trx-R activity was reduced by 35% in WT compared with NMLA controls while Trx-R activity maintained a comparable level in TKO-NMDA compared with TKO-NMLA mice (Figure 2A). In addition, slot blot analysis of retinal nitrotyrosine formation (a marker of peroxynitrite) showed 0.7-fold reduction in TKO mice versus WT. Upon NMDA insult, retinal nitrotyrosine was increased by 1.3-fold in WT mice compared with NMLA controls while the NMDA-injected TKO mice maintained the same nitrotyrosine level as their NMLA controls (Figure 2B,C).
Figure 2.
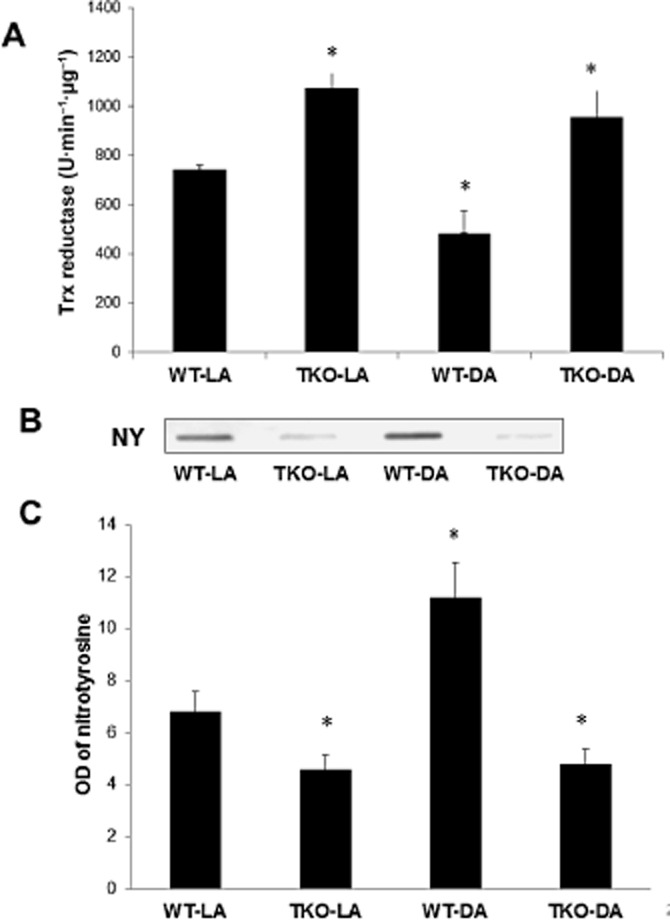
TXNIP deletion increases Trx-R reductase activity and reduces nitrative stress. (A) Statistical analysis of Trx-R activity showed that NMDA injection caused 35% reduction in WT compared with NMLA controls, and that TKO retinas showed a significant increase in Trx-R activity in both NMLA and NMDA groups (n = 8–10 per group). (B, C) Representative image and statistical analysis of retinal nitrotyrosine assessed by slot blot showed that NMDA injection induced 1.65-fold increase in peroxynitrite formation in WT compared with NMLA controls, and that deletion of TXNIP expression significantly decreased nitrotyrosine formation in both NMLA-and NMDA-injected mice (n = 6 per group). Data are expressed as mean ± SEM for all groups. *Significant difference as compared with WT-LA at P < 0.05.
Deletion of TXNIP inhibits Müller cell activation and prevents retinal inflammation
Next, we investigated the protective effect of TXNIP deletion on glial activation in WT versus TKO mice. As seen in Figure 3A, WT-NMDA mice showed an increased intensity of GFAP immunoreactivity in Müller cell filaments spanning from the nerve fibre layer to the INL of the retina as compared with the NMLA-control group. Immunohistochemical evaluation of retinal sections detected robust expression of TNF-α in the GCL and INL of NMDA-injected WT mice compared with their corresponding controls (Figure 3B–D). Co-localization of GFAP and TNF-α was observed particularly in the nerve fibre layer and GCL (Supporting Information Figure S1A). In Figure 3C–E, we detected abundant expression of IL-1β that co-localized with GFAP in the GCL and INL of NMDA-injected WT mice compared with NMLA controls.
Figure 3.
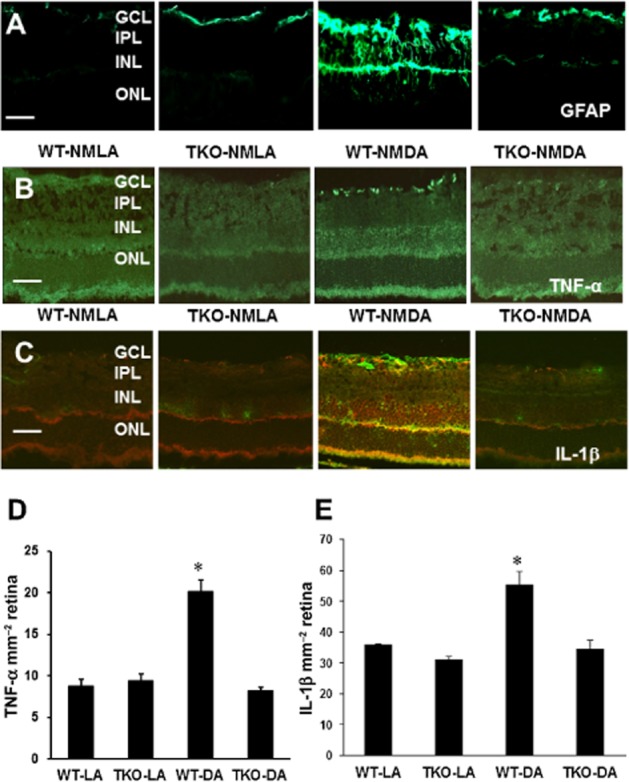
TXNIP deletion inhibits Müller cell activation and retinal inflammation. (A) Representative images showing a substantial increase in GFAP immunoreactivity in the filaments of Müller cells spanning across the inner portion of the neural retina in WT-NMDA mice as compared with NMLA controls. (B, D) Representative images and statistical analysis of prominent immunolocalization of TNF-α in the GCL and INL in WT-NMDA mice as compared with NMLA controls. WT-NMDA mice had ∼2-fold increase in TNF-α expression relative to all other groups. (C, E) Representative images and statistical analysis of immunolocalization of IL-1β (red) and GFAP (green) in NMDA-injected WT and TKO mice and corresponding controls. *Significant difference as compared with the rest of the groups at P < 0.05. IPL, inner plexiform layer; ONL, outer nuclear layer (n = 4–5 per group, 200× magnification, scale bar is 20 μm)
TXNIP is required for NLRP3 inflammasome activation and IL-1β release in Müller cultures
To determine the specific role of Müller cells in TXNIP-induced retinal inflammation, primary Müller cells were isolated from WT and TKO mice and cultured in the absence and presence of NMDA. The required NMDA concentration to stimulate TXNIP expression was determined with preliminary studies in rMC-1 cells (Supporting Information Figure S1B). TNF-α released into the cell-conditioned media following NMDA exposure was measured by elisa (Figure 4A). NMDA exposure increased TNF-α release by threefold in WT Müller cells whereas no change was seen in TKO Müller cells after NMDA treatment, indicating TXNIP is required for NMDA-induced retinal inflammation in Müller cells.
Figure 4.
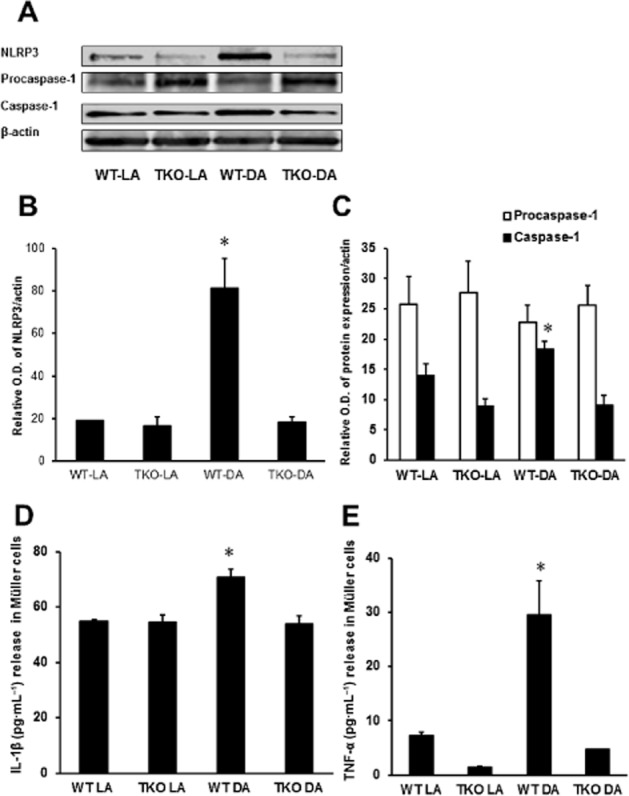
TXNIP deletion prevents NMDA-induced release of inflammatory mediators in primary Müller cell cultures. (A) Representative Western blot images of NLRP3, procaspase-1 and caspase-1 protein expression in primary mouse Müller cells isolated from WT and TKO mice and treated with 10 μM NMDA. β-actin served as the endogenous control. (B, C) Statistical analysis of NLRP3, procaspase-1 and caspase-1 protein levels in WT and TKO Müller cells treated with NMDA. NLRP3 and caspase-1 protein levels were significantly elevated in WT-DA Müller cells whereas TKO-DA Müller cells were similar to NMLA controls. (D) Quantification of IL-1β released by elisa from WT and TKO Müller cells treated with NMDA (10 μM). WT Müller cells demonstrated a significant increase in IL-1β released into the supernatant following NMDA treatment while no change was observed in TKO-DA Müller cells. (E) Quantification of TNF-α released from primary Müller cells isolated from WT and TKO mice and treated in the absence and presence of NMDA. Abundant release of TNF-α was observed in the supernatant from WT-DA Müller cells, ∼3-fold greater compared with WT-LA Müller cells. *Significant difference as compared with the rest of the groups at P < 0.05 (n = 4 cultures per group).
To investigate whether TXNIP is required for activation of the NLRP3 inflammasome in our NMDA model, we examined the expression of components in the NLRP3 inflammasome activation pathway. WT-NMDA Müller cells were observed to have elevated levels of NLRP3 (Figure 4B,C), which was not observed in TKO Müller cells. The increase in NLRP3 protein expression was accompanied by an increase in cleavage of caspase-1 from procaspase-1, expression in WT-NMDA Müller cells (Figure 4B,D). In addition, a significant up-regulation of IL-1β release (1.25-fold) was seen in WT-NMDA Müller cells (Figure 4E). The absence of TXNIP prevented the activation of NLRP3, cleavage of caspase-1 and the subsequent release of IL-1β.
Deletion of TXNIP prevents NMDA-induced early BRB breakdown, long-term neuro-and microvascular degeneration
To examine the effect of TXNIP deletion on BRB function, WT and TKO animals were injected with BSA-fluorescein. Vascular leakage was also observed after 24 h in WT-NMDA mice and the absence of TXNIP reversed the vascular leakage in TKO-NMDA mice (Figure 5A). Of note, retinas from TKO-NMLA mice had higher baseline for BSA-fluorescein compared with WT-NMLA controls. The number of acellular capillaries was counted to determine whether BRB dysfunction is a result of vascular cell death. Although some acellular capillaries were observed in WT mice (24 h post-injection), no significant difference was obtained among all animal groups (Figure 5B). However, microvascular degeneration was evident at the long term (14 days post-injection), where ∼2-fold increases in acellular capillary formation were observed in WT mice but not in TKO mice (Figure 5C,D). These effects were associated with neuronal cell death assessed by TUNEL assay. As shown in Figure 6A and C, NMDA induced several TUNEL-positive cells in all retina layers in WT and the largest number of apoptotic cells was seen in the GCL followed by INL which contains many cell types involved in neural retina function including bipolar, amacrine and horizontal cells. Deletion of TXNIP significantly prevented NMDA-induced apoptosis in the TKO-NMDA mice as compared with the WT-NMDA mice. Further neurotoxicity was also evident in the long-term study, in which WT mice showed 60% loss of nuclei in the GCL (Figure 6B,D) versus 40% loss observed after the short-term NMDA insult (Figure 1B–D). Deletion of TXNIP preserved neuronal cell viability and inhibited the long-term exacerbation of the initial neuronal damage induced by NMDA. Together, these results highlight the sustained neuronal and vascular insults and degeneration in the long term after suffering the initial neuronal excitotoxic effects of NMDA.
Figure 5.
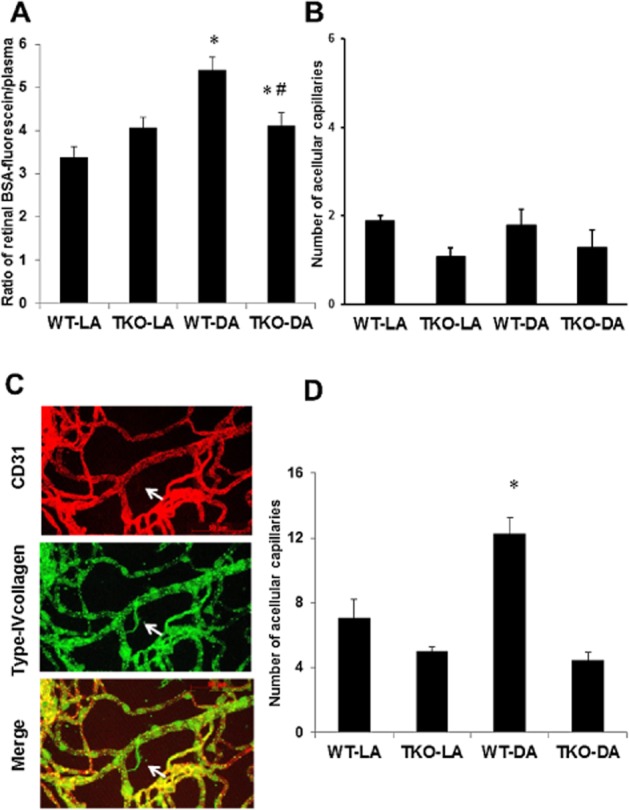
TXNIP deletion prevents BRB breakdown and retinal microvascular degeneration. (A) Statistical quantification of vascular permeability indicated that NMDA induced retinal vascular leakage, evident by extravasation of BSA-fluorescein (i.v. 10 mg·kg−1) in WT mice at 24 h post-injection but not in TKO mice. *Significant difference as compared with WT-LA at P < 0.05. #Significant differences as compared with WT-DA at P < 0.05 (n = 6). (B) Statistical analysis of the acellular capillary count in NMDA-injected mice showed no significant vascular cell death evident by number of acellular capillaries at 24 h post-injection. (C) Representative images of trypsin-digested retinas from animals that received NMDA for 2 weeks. Retinas were stained with extracellular matrix marker type IV collagen (green), endothelial marker CD31 (red) and their merge (yellow) acellular capillaries (red arrows). Arrow points to a typical acellular capillary that expressed extracellular matrix (green only) and lacks vascular marker. (D) Statistical analysis of the acellular capillary count, marker of microvascular degeneration in NMDA-injected mice at 2 weeks post-injection showed that deletion of TXNIP significantly reduced NMDA-induced capillary degeneration as compared with WT (n = 6–8 per group). *Significant difference as compared with the rest of the groups at P < 0.05.
Figure 6.
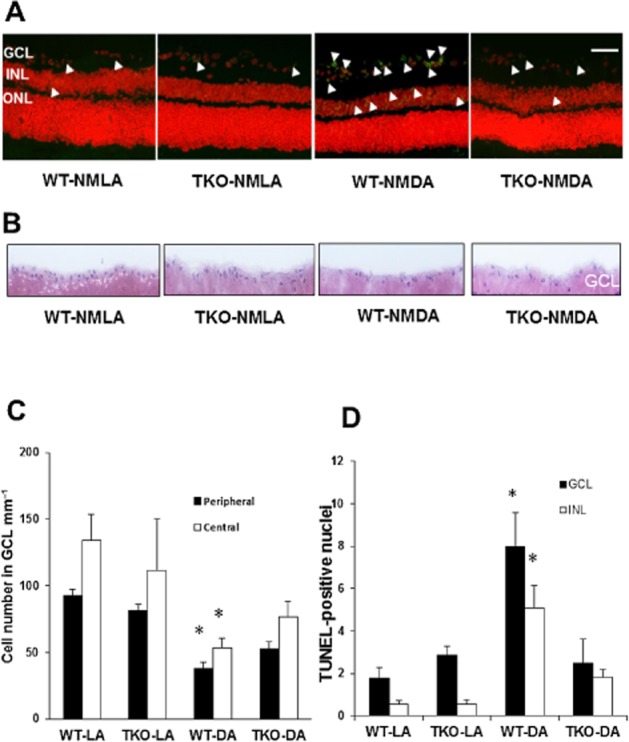
TXNIP deletion prevents long-term neuronal death and further RGC loss. (A, C) Representative images and statistical analysis of NMDA-induced neuronal death indicating significant increases in TUNEL-labelled cells (green nuclei, white arrowheads) 2 weeks post-injection in WT mice compared with TKO mice. Apoptotic cells were localized mostly to the GCL, and to a lesser extent INL and occasionally in the outer nuclear layer (ONL). Sections were counterstained with propidium iodide to label cell nuclei. Images are taken from the central region of the retina. *Significant difference as compared with the rest of the groups at P ≤ 0.05 (n = 5–7 per group, 200× magnification, scale bar is 20 μm). (B, D) Representative images and statistical analysis of the number of nuclei in the GCL from the long-term study (2 weeks post-injection) showing exacerbated cell loss (∼75%) in the GCL in WT but not in TKO mice after NMDA injection (n = 4–6 per group). *Significant difference as compared with the rest of the groups.
Deletion of TXNIP preserves retinal neuronal function
To determine the long-term effect of NMDA-induced neurotoxicity on retinal function, electroretinogram (ERG) measurements were performed in WT and TKO mice at 21 days (Figure 7). To compare scotopic responses across groups, b-wave amplitudes were normalized by a-wave amplitudes (see Supporting Information Figure S2 for more details). It was observed that the b-wave amplitude, originating largely from ON-bipolar cells, was reduced after NMDA injection. Figure 7A shows a significant decrease in the b/a wave ratio after NMDA injection in WT mice, which was partially restored in the TKO-NMDA mice. As shown in Figure 7B, averaged photopic flash responses were reduced by NMDA injection in WT mice, and they were again restored in the TKO-NMDA mice. Figure 7C shows kernels that were obtained to a pseudorandom noise stimulus, and again the TKO-NMDA mice had stronger responses than the WT-NMDA mice. These data suggest that TXNIP deletion preserves bipolar cell function to a large extent after NMDA insult.
Figure 7.
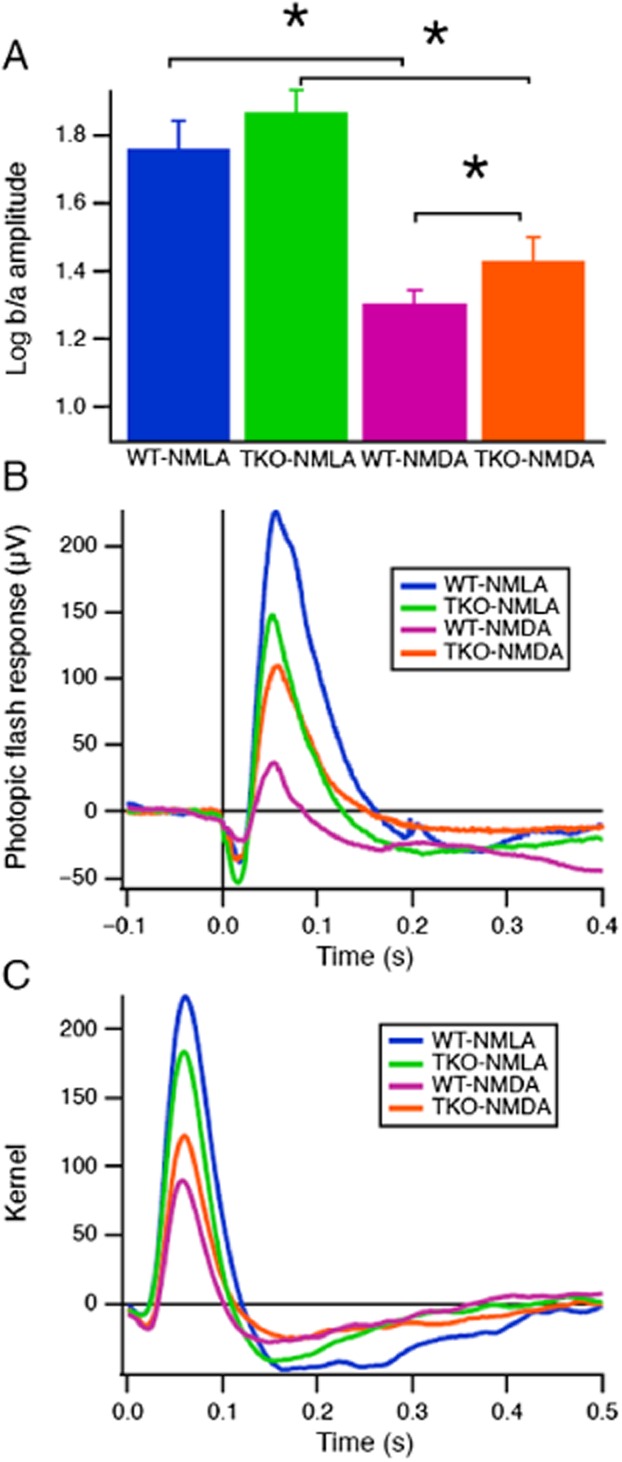
TXNIP deletion preserves retinal neuronal function. (A) To compare scotopic ERG responses across groups, b-wave amplitudes were normalized by a-wave amplitudes (see Supporting Information Figure S2 for more details). Means and SEMs of log ratios are shown for the 0.009 lumen intensity. Stars indicate significant differences (Wilcoxon test, P < 0.05). (B) Averaged photopic ERG flash responses are compared across groups. The flash had a peak luminance of 1.8 lumens on a background of 0.1 lumen, and a duration of 5 ms. (C) Kernels were obtained from a pseudorandom noise stimulus under photopic conditions. The noise had ‘natural’ statistics with equal power across log temporal frequency (n = 7–8 per group).
Expression of TXNIP and release of TNF-α and IL-1β in human glaucoma samples
We next examined whether the same increases in TXNIP and inflammatory end points can also be observed in humans. Therefore, we obtained post-mortem eye tissue samples of control and open-angle glaucoma patients from the Georgia Eye Bank; glaucoma is a disease well known for its retinal neuronal degeneration events. Glaucoma patients (n = 4) were observed to have elevated TXNIP expression in the retina, 1.9-fold higher than non-glaucoma patients (Figure 8A). The vitreous of glaucoma patients were found to have 2.5-and ∼3-fold higher levels of the pro-inflammatory cytokines TNF-α and IL-1β compared with control patients respectively (Figure 8B,C).
Figure 8.
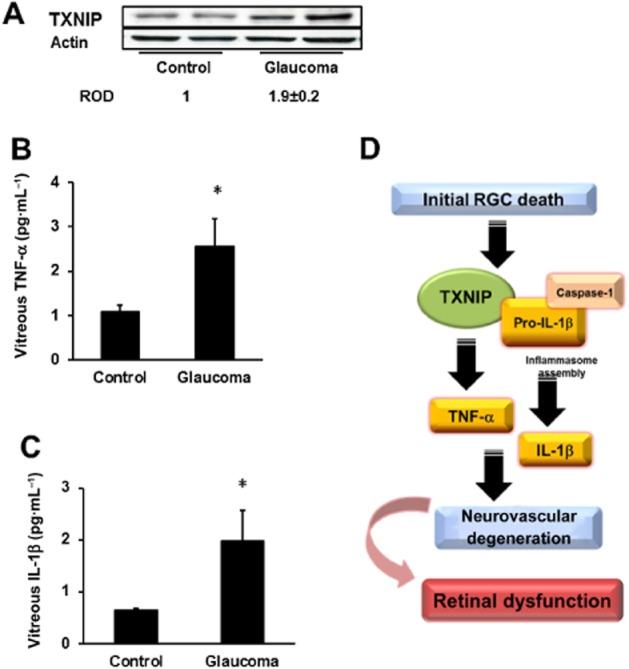
Human glaucoma patients have increased retinal TXNIP and release of TNF-α and IL-1β in vitreous. (A) Western blot analysis of TXNIP expression in retinal tissue of patients with open-angle glaucoma compared with controls, indicating a ∼2-fold increase of TXNIP expression in glaucoma patients. (B, C) elisa results showing 2.5-and ∼3-fold increase in pro-inflammatory cytokines TNF-α and IL-1β in the vitreous of glaucoma patients compared with control patients (n = 4, *P <.05). (D) Schematic representation of the proposed scheme of events by which TXNIP contributes to glial inflammation leading to sustained retinal neuro-and vascular degeneration and impairment of retinal function.
Discussion
Three important findings emerged from this study. Firstly, deletion of TXNIP prevents early neuronal cell death, oxidative stress, glial activation and blood–retina barrier breakdown in response to neurotoxic insult (Figures 5). Secondly, TXNIP is required for NMDA-induced inflammasome activation and release of IL-1β and TNF-α resulting in retinal inflammation (Figures 4). Thirdly, the long-term effects of NMDA-induced microvascular degeneration, exacerbated neurodegeneration and compromised retinal function were all prevented or reduced by TXNIP deletion (Figures 7). Furthermore, up-regulated retinal TXNIP expression was positively correlated with increases in TNF-α, IL-β levels in vitreous from patients with glaucoma compared with non-glaucoma patients (Figure 8). We believe that this is the first report to demonstrate the multiple roles of TXNIP in mediating and sustaining retinal injury in a model of neurotoxicity.
Neuronal death, particularly RGC death, occurs in multiple retinal diseases, including glaucoma, traumatic optic neuropathy, diabetic retinopathy and retinal ischaemia (reviewed in Osborne et al., 1999; Schmidt et al., 2008; Whitmire et al., 2011). Excitotoxicity due to the excess release of glutamate resulting from metabolic stress leading to death of neurons expressing ionotropic glutamate (NMDA) receptors is one of the established mechanisms. Therapeutic strategies that focused on targeting RGC death only have proven successful in experimental models (Chen et al., 1992; Dong et al., 2008; Wada et al., 2013), but have failed in clinical settings (Osborne, 2009; Danesh-Meyer, 2011). Therefore, there is a great need to examine other retinal cell types to develop more effective neuroprotective agents that can preserve retinal function at multiple levels. We and others have demonstrated that overexpression of thioredoxin, a major antioxidant and anti-apoptotic protein or inhibiting TXNIP, can counter oxidative stress and prevent RGC death in NMDA-induced neurotoxicity and in other models of experimental glaucoma and optic neuropathy (Inomata et al., 2006; Munemasa et al., 2008,2009; Al-Gayyar et al., 2010,2011; Spindel et al., 2012). Nevertheless, the impact of modulating the thioredoxin system on other retinal cell types and the subsequent effects on exacerbating RGC death remain unknown.
The cascade of molecular events leading to neuronal cell death in NMDA-induced neurotoxicity includes apoptotic cell death (Lam et al., 1999). In WT mice receiving short-term NMDA injections, we observed a large number of apoptotic cells in the GCL, containing RGCs and displaced amacrine cells. Some apoptosis was observed in the INL as well. In contrast, very few apoptotic cells were found in TKO mice injected with NMDA. Previous studies have established that PARP hyperactivation via NMDA receptor activity plays an important role in apoptotic cell death (Lam et al., 1999; Goebel and Winkler, 2006). WT-NMDA animals were found to have increased cleaved-PARP expression, whereas TKO-NMDA mice had similar expression to the NMLA controls. These results demonstrate that TXNIP plays a critical role in apoptotic cell death in NMDA-induced neurotoxicity. NMDA neurotoxicity models are documented to have oxidative and nitrative stress (El-Remessy et al., 2003; Nakajima et al., 2008; Inokuchi et al., 2009; Al-Gayyar et al., 2010). A significant decrease in Trx-R activity was observed in WT mice after NMDA insult, revealing a decreased antioxidant response. Of note, retinas from TKO mice demonstrated high levels of Trx-R reductase activity at baseline or in response to NMDA insult, in agreement with recent characterization of TKO model (Abdelsaid et al., 2013). Though a significant increase in nitrotyrosine was observed in WT animals with NMDA, baseline nitrotyrosine levels were lower in TKO mice, indicating better defence against nitrative stress in the absence of TXNIP.
In response to virtually all retinal insults, Müller glial cells are activated, evident by enhanced expression of intermediate filaments including GFAP (Bringmann et al., 2009). Here, we observed activation of Müller glial cells evident by increased GFAP expression that was associated with increases in TNF-α and IL-1β in WT but not in TKO mice. These results are consistent with previous findings showing that TXNIP plays a pivotal role in glial activation and release of pro-inflammatory cytokines (Al-Gayyar et al., 2011; Trueblood et al., 2011; Devi et al., 2012). TXNIP can contribute to retinal inflammation via multiple pathways including activation of NF-κB and transcription of inflammatory mediators (Perrone et al., 2009) and activation of the inflammasome assembly (Devi et al., 2012). Inflammasome is a multiprotein complex of the NLRP that activates an inflammatory cascade by binding procaspase-1 via caspase-recruitment domain of the adaptor protein ASC (Mariathasan et al., 2004). Once caspase-1 is activated, it can execute initial pro-inflammatory signals via caspase-1-dependent proteolytic maturation and secretion of IL-1β, IL-18 and interferon-γ. To examine the specific role of TXNIP in glial inflammation, we isolated primary Müller cells from both WT and TKO that were subjected to NMDA insult and examined release of inflammatory mediators. Pilot studies indicated that 10 μM NMDA exposure can induce TXNIP expression in a Müller cell line (Supporting Information Figure S1). Here, we demonstrated that NMDA induced significant expression of NLRP3, activated caspase-1, and released IL-1β and TNF-α into the cell culture medium of WT Müller cells. In TXNIP-null Müller cells this effect was prevented. These results lend further support to previous reports showing that TXNIP is integral to inflammasome assembly and release of IL-1β (Trueblood et al., 2011; Devi et al., 2012; Seki et al., 2010). Previous literature demonstrated mutual crosstalk between TNF-α and IL-1β in activating release of each other and exacerbating the inflammatory response (Dinarello et al., 1986; Ikejima et al., 1990). However, more elaborate work demonstrated that TNF-α can induce caspase-1 activation and IL-1β release in mouse macrophages (Franchi et al., 2009). Interestingly, in the latter study, ATP addition was required for the TNF-α-mediated effect. Previous reports demonstrated the ability of the NLRP3 inflammasome to sense extracellular ATP as one of the damage-associated molecular pattern molecule activators (Wen et al., 2011; Gombault et al., 2012; Savage et al., 2012). Our results suggest a scenario in which early RGC death may result in increased extracellular ATP release and exacerbate the pro-inflammatory action of TNF-α via activation of the NLRP3 inflammasome assembly and release of IL-1β that can affect other retinal cell types including vasculature and other neurons.
We next examined the effect of TXNIP-mediated release of inflammatory mediators on retinal vasculature. As shown in Figure 5, retinas from WT mice had compromised vascular integrity as indicated by a twofold increase in extravasation of BSA-fluorescein. Nevertheless, this BRB dysfunction was not due to vascular cell death, as indicated by a lack of acellular capillary formation 1 day after NMDA injection. Interestingly, retinas from WT mice demonstrated significant increases in vascular cell death (twofold) 14 days post initial NMDA insult. These effects were not observed in retinas from TKO mice. Having healthy retinal vasculature is critical to maintaining neuronal function. This notion is supported by results showing that neuronal cell counts in the GCL clearly showed exacerbated loss (60%) after 14 days compared with 40% after 1 day. Furthermore, the neurons in the GCL, INL and to a lesser extent outer nuclear layer clearly demonstrated abundant TUNEL-positive nuclei suggesting that NMDA-mediated initial RGC death contributed to long-term negative effects on bipolar and photoreceptor cells. The functional consequences of these changes were tested by ERG studies. In scotopic and especially in photopic tests, the NMDA-induced reduction in b-wave amplitudes was diminished in TKO mice. TXNIP deletion appears to preserve bipolar cell function after NMDA insult.
Retinal microvascular cell death can occur either directly, as a result of biochemical insults within endothelial cells, or indirectly, secondary to neuroglial activation (reviewed in Kern, 2007). Indeed, NMDA receptors were found to be expressed on retinal and brain vessels (Betzen et al., 2009; Nakazawa, 2009; LeMaistre et al., 2012). The findings that NMDA injection did not induce retinal microvascular cell death 1 day post-injection rule out the possibility of a direct effect. Thus, our data support a model where initial neuro-glial activation can result in the development of microvascular lesions, which in turn can sustain the long-term devastating effects of retinal neuronal death and decreased retinal sensory function.
In an effort to examine the translational impact of our work, we were able to obtain eyes from four patients with primary open-angle glaucoma. We found that glaucoma patients had elevated levels of TXNIP protein expression in retina and elevated levels of the pro-inflammatory cytokines TNF-α and IL-1β in the vitreous, compared with non-glaucoma patients. Although limited, these data suggest that TXNIP can play multiple roles in neurodegeneration seen in glaucoma patients. In conclusion, deletion of TXNIP prevented NMDA-induced neuronal cell death, glial activation and retinal inflammation. In addition, TXNIP deletion inhibited inflammasome activation, preserved BRB function and microvascular integrity, and preserved retinal function. With multiple pathways of action, TXNIP is a promising new target for the development of new therapies for the treatment of retinal neurodegenerative diseases.
Acknowledgments
We would like to thank Amber Still for her excellent technical assistance with the ERG studies. This work was supported by grants from EY-022408, JDRF (2-2008-149), and Culver Vision Discovery Institute to A. B. E. and K. E. B., postdoctoral fellowship from Islamic Development Bank, pre-doctoral fellowship from American Heart Association for M. A. A. and I. N. M., and postdoctoral fellowship from American Heart Association for B. A. M.
Abbreviations
- ARVO
association for research in vision and ophthalmology
- ASC
apoptosis-associated specklike adaptor protein
- BRB
blood retinal barrier
- CARD
caspase-recruitment domain of the adaptor protein ASC
- DAMPs
damage-associated molecular pattern molecules
- DTNB
5,5′-dithiobis(2-nitrobenzoic acid)
- ERG
electroretinogram
- GCL
ganglion cell layer
- GFAP
glial fibrillary acidic protein
- INL
inner nuclear layer
- NADPH
nicotinamide adenine dinucleotide phosphate
- NLRP3
NOD-like receptor protein
- NMLA
N-methyl-L-aspartate
- OCT
optimum cutting temperature compound
- ONL
outer nuclear layer
- PARP
poly-ADP-ribose polymerase
- PASH
periodic acid-Schiff and haematoxylin
- PBST
PBS plus 0.02% Tween 20
- RGC
retinal ganglion cell
- rMC-1
rat retinal Müller cell line
- Trx-R
thioredoxin reductase
- TKO
TXNIP-knockout
- TUNEL
terminal dUTP nick-end labeling
- TXNIP
thioredoxin interacting protein
Conflict of interest
We do not have any commercial associations that might create a conflict of interest in connection with our manuscript.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12535
(A) Representative images showing the colocalization of GFAP (green) and TNF-α (red) by immunohistochemical analysis in WT-NMDA mice. Co-localization was observed particularly in the ganglion cell layer and nerve fibre layer. (B) Western blot analysis of TXNIP expression in rMC-1 cells treated with a range of NMDA concentrations (1, 10 and 100 μM) to determine the dose needed to activate TXNIP expression for subsequent primary Muller cell experiments.
Scotopic ERG responses. (A) Average amplitudes of a-waves are plotted against flash luminance intensity. (B) Amplitudes of b-waves. (C) Log ratios of b- to a-wave amplitudes are plotted, normalizing data in panel B by the data in panel A to examine specific effects on inner nuclear layer function (n = 7–8 per group).
Showing the age, race and sex of patients as well as their medical history and glaucoma status (C, Caucasian; AF, Afro-American).
References
- Abdelsaid MA, Pillai BA, Matragoon S, Prakash R, Al-Shabrawey M, El-Remessy AB. Early intervention of tyrosine nitration prevents vaso-obliteration and neovascularization in ischemic retinopathy. J Pharmacol Exp Ther. 2010;332:125–134. doi: 10.1124/jpet.109.157941. [DOI] [PubMed] [Google Scholar]
- Abdelsaid MA, Matragoon S, El-Remessy AB. Thioredoxin Interacting Protein (TXNIP) expression is required for VEGF-mediated angiogenic signal in endothelial cells. Antioxid Redox Signal. 2013;19:2199–2212. doi: 10.1089/ars.2012.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Gayyar MM, Abdelsaid MA, Matragoon S, Pillai BA, El-Remessy AB. Neurovascular protective effect of FeTPPs in N-methyl-D-aspartate model: similarities to diabetes. Am J Pathol. 2010;177:1187–1197. doi: 10.2353/ajpath.2010.091289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Gayyar MMH, Abdelsaid M, Matragoon S, Pillai BA, El-Remessy AB. Thioredoxin interacting protein is a novel mediator of retinal inflammation and neurotoxicity. Br J Pharmacol. 2011;164:170–180. doi: 10.1111/j.1476-5381.2011.01336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali TK, Matragoon S, Pillai BA, Liou GI, El-Remessy AB. Peroxynitrite mediates retinal neurodegeneration by inhibiting NGF survival signal in experimental and human diabetes. J Diabetes. 2008;57:889–898. doi: 10.2337/db07-1669. [DOI] [PubMed] [Google Scholar]
- Ali TK, Al-Gayyar MM, Matragoon S, Pillai BA, Abdelsaid MA, Nussbaum JJ, et al. Diabetes-induced peroxynitrite impairs the balance of pro-nerve growth factor and nerve growth factor, and causes neurovascular injury. Diabetologia. 2011;54:657–668. doi: 10.1007/s00125-010-1935-1. [DOI] [PubMed] [Google Scholar]
- Bessero AC, Chiodini F, Rungger-Brandle E, Bonny C, Clarke PG. Role of the c-Jun N-terminal kinase pathway in retinal excitotoxicity, and neuroprotection by its inhibition. J Neurochem. 2010;113:1307–1318. doi: 10.1111/j.1471-4159.2010.06705.x. [DOI] [PubMed] [Google Scholar]
- Betzen C, White R, Zehendner CM, Pietrowski E, Bender B, Luhmann HJ, et al. Oxidative stress upregulates the NMDA receptor on cerebrovascular endothelium. Free Radic Biol Med. 2009;47:1212–1220. doi: 10.1016/j.freeradbiomed.2009.07.034. [DOI] [PubMed] [Google Scholar]
- Bonhomme-Faivre L, Forestier F, Auchere D, Soursac M, Orbach-Arbouys S, Farinotti R. Chronic administration of verapamil, ketoconazole and carbamazepine: impact on immunological parameters. Int J Pharm. 2002;238:133–137. doi: 10.1016/s0378-5173(02)00056-x. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Iandiev I, Pannicke T, Wurm A, Hollborn M, Wiedemann P, et al. Cellular signaling and factors involved in Muller cell gliosis: neuroprotective and detrimental effects. Prog Retin Eye Res. 2009;28:423–451. doi: 10.1016/j.preteyeres.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Chen HS, Pellegrini JW, Aggarwal SK, Lei SZ, Warach S, Jensen FE, et al. Open-channel block of N-methyl-D-aspartate (NMDA) responses by memantine: therapeutic advantage against NMDA receptor-mediated neurotoxicity. J Neurosci. 1992;12:4427–4436. doi: 10.1523/JNEUROSCI.12-11-04427.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danesh-Meyer HV. Neuroprotection in glaucoma: recent and future directions. Curr Opin Ophthalmol. 2011;22:78–86. doi: 10.1097/ICU.0b013e32834372ec. [DOI] [PubMed] [Google Scholar]
- Devi TS, Lee I, Huttemann M, Kumar A, Nantwi KD, Singh LP. TXNIP links innate host defense mechanisms to oxidative stress and inflammation in retinal Muller glia under chronic hyperglycemia: implications for diabetic retinopathy. Exp Diabetes Res. 2012;2012:438238. doi: 10.1155/2012/438238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA, Cannon JG, Wolff SM, Bernheim HA, Beutler B, Cerami A, et al. Tumor necrosis factor (cachectin) is an endogenous pyrogen and induces production of interleukin 1. J Exp Med. 1986;163:1433–1450. doi: 10.1084/jem.163.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong CJ, Guo Y, Agey P, Wheeler L, Hare WA. Alpha2 adrenergic modulation of NMDA receptor function as a major mechanism of RGC protection in experimental glaucoma and retinal excitotoxicity. Invest Ophthalmol Vis Sci. 2008;49:4515–4522. doi: 10.1167/iovs.08-2078. [DOI] [PubMed] [Google Scholar]
- El-Remessy AB, Khalil IE, Matragoon S, Abou-Mohamed G, Tsai NJ, Roon P, et al. Neuroprotective effect of (-)delta9-tetrahydrocannabinol and cannabidiol in N-methyl-D-aspartate-induced retinal neurotoxicity: involvement of peroxynitrite. Am J Pathol. 2003;163:1997–2008. doi: 10.1016/s0002-9440(10)63558-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi L, Eigenbrod T, Nunez G. Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol. 2009;183:792–796. doi: 10.4049/jimmunol.0900173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel DJ, Winkler BS. Blockade of PARP activity attenuates poly(ADP-ribosyl)ation but offers only partial neuroprotection against NMDA-induced cell death in the rat retina. J Neurochem. 2006;98:1732–1745. doi: 10.1111/j.1471-4159.2006.04065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gombault A, Baron L, Couillin I. ATP release and purinergic signaling in NLRP3 inflammasome activation. Front Immunol. 2012;3:414. doi: 10.3389/fimmu.2012.00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui ST, Andres AM, Miller AK, Spann NJ, Potter DW, Post NM, et al. Txnip balances metabolic and growth signaling via PTEN disulfide reduction. Proc Natl Acad Sci U S A. 2008;105:3921–3926. doi: 10.1073/pnas.0800293105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikejima T, Okusawa S, Ghezzi P, van der Meer JW, Dinarello CA. Interleukin-1 induces tumor necrosis factor (TNF) in human peripheral blood mononuclear cells in vitro and a circulating TNF-like activity in rabbits. J Infect Dis. 1990;162:215–223. doi: 10.1093/infdis/162.1.215. [DOI] [PubMed] [Google Scholar]
- Inokuchi Y, Imai S, Nakajima Y, Shimazawa M, Aihara M, Araie M, et al. Edaravone, a free radical scavenger, protects against retinal damage in vitro and in vivo. J Pharmacol Exp Ther. 2009;329:687–698. doi: 10.1124/jpet.108.148676. [DOI] [PubMed] [Google Scholar]
- Inomata Y, Nakamura H, Tanito M, Teratani A, Kawaji T, Kondo N, et al. Thioredoxin inhibits NMDA-induced neurotoxicity in the rat retina. J Neurochem. 2006;98:372–385. doi: 10.1111/j.1471-4159.2006.03871.x. [DOI] [PubMed] [Google Scholar]
- Kern TS. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp Diabetes Res. 2007;2007:95103. doi: 10.1155/2007/95103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. J Gene Med. 2010;12:561–563. doi: 10.1002/jgm.1473. [DOI] [PubMed] [Google Scholar]
- Koseki N, Kitaoka Y, Munemasa Y, Kumai T, Kojima K, Ueno S, et al. 17beta-estradiol prevents reduction of retinal phosphorylated 14-3-3 zeta protein levels following a neurotoxic insult. Brain Res. 2012;1433:145–152. doi: 10.1016/j.brainres.2011.11.034. [DOI] [PubMed] [Google Scholar]
- Lam TT, Abler AS, Kwong JMK, Tso MOM. N-methyl-D-aspartate (NMDA)-induced apoptosis in rat retina. Invest Ophthalmol Vis Sci. 1999;40:2391–2397. [PubMed] [Google Scholar]
- LeMaistre JL, Sanders SA, Stobart MJ, Lu L, Knox JD, Anderson HD, et al. Coactivation of NMDA receptors by glutamate and D-serine induces dilation of isolated middle cerebral arteries. J Cereb Blood Flow Metab. 2012;32:537–547. doi: 10.1038/jcbfm.2011.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–218. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- Matragoon S, Al-Gayyar MM, Mysona BA, Abdelsaid MA, Pillai BA, Neet KE, et al. Electroporation-mediated gene delivery of cleavage-resistant pro-nerve growth factor causes retinal neuro-and vascular degeneration. Mol Vis. 2012;18:2993–3003. [PMC free article] [PubMed] [Google Scholar]
- Munemasa Y, Kim SH, Ahn JH, Kwong JM, Caprioli J, Piri N. Protective effect of thioredoxins 1 and 2 in retinal ganglion cells after optic nerve transection and oxidative stress. Invest Ophthalmol Vis Sci. 2008;49:3535–3543. doi: 10.1167/iovs.08-1716. [DOI] [PubMed] [Google Scholar]
- Munemasa Y, Ahn JH, Kwong JM, Caprioli J, Piri N. Redox proteins thioredoxin 1 and thioredoxin 2 support retinal ganglion cell survival in experimental glaucoma. Gene Ther. 2009;16:17–25. doi: 10.1038/gt.2008.126. [DOI] [PubMed] [Google Scholar]
- Mysona B, Dun Y, Duplantier J, Ganapathy V, Smith SB. Effects of hyperglycemia and oxidative stress on the glutamate transporters GLAST and system xc-in mouse retinal Muller glial cells. Cell Tissue Res. 2009;335:477–488. doi: 10.1007/s00441-008-0742-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima Y, Inokuchi Y, Nishi M, Shimazawa M, Otsubo K, Hara H. Coenzyme Q10 protects retinal cells against oxidative stress in vitro and in vivo. Brain Res. 2008;1226:226–233. doi: 10.1016/j.brainres.2008.06.026. [DOI] [PubMed] [Google Scholar]
- Nakazawa T. Mechanism of N-methyl-D-aspartate-induced retinal ganglion cell death. Nippon Ganka Gakkai Zasshi. 2009;113:1060–1070. [PubMed] [Google Scholar]
- Osborne NN. Recent clinical findings with memantine should not mean that the idea of neuroprotection in glaucoma is abandoned. Acta Ophthalmol. 2009;87:450–454. doi: 10.1111/j.1755-3768.2008.01459.x. [DOI] [PubMed] [Google Scholar]
- Osborne NN, Ugarte M, Chao M, Chidlow G, Bae JH, Wood JP, et al. Neuroprotection in relation to retinal ischemia and relevance to glaucoma. Surv Ophthalmol. 1999;43(Suppl. 1):S102–S128. doi: 10.1016/s0039-6257(99)00044-2. [DOI] [PubMed] [Google Scholar]
- Perrone L, Devi TS, Hosoya K, Terasaki T, Singh LP. Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J Cell Physiol. 2009;221:262–272. doi: 10.1002/jcp.21852. [DOI] [PubMed] [Google Scholar]
- Savage CD, Lopez-Castejon G, Denes A, Brough D. NLRP3-inflammasome activating DAMPs stimulate an inflammatory response in glia in the absence of priming which contributes to brain inflammation after injury. Front Immunol. 2012;3:288. doi: 10.3389/fimmu.2012.00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt KG, Bergert H, Funk RH. Neurodegenerative diseases of the retina and potential for protection and recovery. Curr Neuropharmacol. 2008;6:164–178. doi: 10.2174/157015908784533851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science. 2010;327:296–300. doi: 10.1126/science.1184003. [DOI] [PubMed] [Google Scholar]
- Schroder K, Sagulenko V, Zamoshnikova A, Richards AA, Cridland JA, Irvine KM, et al. Acute lipopolysaccharide priming boosts inflammasome activation independently of inflammasome sensor induction. Immunobiology. 2012;217:1325–1329. doi: 10.1016/j.imbio.2012.07.020. [DOI] [PubMed] [Google Scholar]
- Seki M, Soussou W, Manabe S, Lipton SA. Protection of retinal ganglion cells by caspase substrate-binding peptide IQACRG from N-methyl-D-aspartate receptor-mediated excitotoxicity. Invest Ophthalmol Vis Sci. 2010;51:1198–1207. doi: 10.1167/iovs.09-4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazawa M, Suemori S, Inokuchi Y, Matsunaga N, Nakajima Y, Oka T, et al. A novel calpain inhibitor, ((1S)-1-((((1S)-1-Benzyl-3-cyclopropylamino-2,3-di-oxopropyl)amino)carbonyl)-3-me thylbutyl)carbamic acid 5-methoxy-3-oxapentyl ester (SNJ-1945), reduces murine retinal cell death in vitro and in vivo. J Pharmacol Exp Ther. 2010;332:380–387. doi: 10.1124/jpet.109.156612. [DOI] [PubMed] [Google Scholar]
- Spindel ON, World C, Berk BC. Thioredoxin interacting protein: redox dependent and independent regulatory mechanisms. Antioxid Redox Signal. 2012;16:587–596. doi: 10.1089/ars.2011.4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitt AW, Li YM, Gardiner TA, Bucala R, Archer DB, Vlassara H. Advanced glycation end products (AGEs) co-localize with AGE receptors in the retinal vasculature of diabetic and of AGE-infused rats. Am J Pathol. 1997;150:523–531. [PMC free article] [PubMed] [Google Scholar]
- Trueblood KE, Mohr S, Dubyak GR. Purinergic regulation of high glucose-induced caspase-1 activation in the rMC-1 rat retinal Muller cell line. Am J Physiol Cell Physiol. 2011;301:C1213–C1223. doi: 10.1152/ajpcell.00265.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada Y, Nakamachi T, Endo K, Seki T, Ohtaki H, Tsuchikawa D, et al. PACAP attenuates NMDA-induced retinal damage in association with modulation of the microglia/macrophage status into an acquired deactivation subtype. J Mol Neurosci. 2013;51:493–502. doi: 10.1007/s12031-013-0017-5. [DOI] [PubMed] [Google Scholar]
- Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12:408–415. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmire W, Al-Gayyar MMH, Abdelsaid MA, Yousufzai BK, El-Remessy AB. Alteration of growth factors and neuronal death in diabetic retinopathy: what we have learned so far. Mol Vis. 2011;17:300–308. [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Gong B, Hatala DA, Kern TS. Retinal ischemia and reperfusion causes capillary degeneration: similarities to diabetes. Invest Ophthalmol Vis Sci. 2007;48:361–367. doi: 10.1167/iovs.06-0510. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Representative images showing the colocalization of GFAP (green) and TNF-α (red) by immunohistochemical analysis in WT-NMDA mice. Co-localization was observed particularly in the ganglion cell layer and nerve fibre layer. (B) Western blot analysis of TXNIP expression in rMC-1 cells treated with a range of NMDA concentrations (1, 10 and 100 μM) to determine the dose needed to activate TXNIP expression for subsequent primary Muller cell experiments.
Scotopic ERG responses. (A) Average amplitudes of a-waves are plotted against flash luminance intensity. (B) Amplitudes of b-waves. (C) Log ratios of b- to a-wave amplitudes are plotted, normalizing data in panel B by the data in panel A to examine specific effects on inner nuclear layer function (n = 7–8 per group).
Showing the age, race and sex of patients as well as their medical history and glaucoma status (C, Caucasian; AF, Afro-American).